

Abstract
Spectinamides are promising new semisynthetic anti-tubercular agents that are modified with a pyridyl side chain, which blocks native efflux from the tuberculosis cell. This study, describes the stability of an advanced panel of spectinamide analogs, with varying substitutions to the pyridyl side chain, to Phase-II conjugative metabolism by glucuronosyl transferase, sulfotransferase and glutathione-S-transferase enzymes using both human and rat S9 enzyme fractions. All solely 5-substituted pyridyl spectinamides exhibited complete stability towards Phase II conjugative enzymes. However, 4-chloro substituted pyridyl spectinamides were susceptible to glutathione conjugation with rates dependent on other substitutions to the pyridine ring. Electron donating 5-substitutions increased the propensity for glutathione conjugation and conversely the introduction of an electron withdrawing 5-fluoro group blocked all observed glutathione conjugation. Based on these Phase II metabolism studies, lead spectinamides 1329, 1445, 1599, 1661 and 1810 were found to have favorable properties for potential lead compounds with no Phase II liabilities.
Introduction
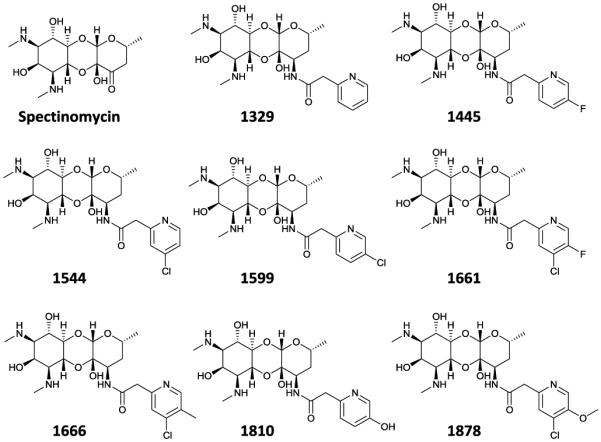
There is an urgent clinical need to develop new agents to treat drug resistant strains of tuberculosis.1, 2 Spectinamides are a new class of semi-synthetic antibiotics derived from spectinomycin (Fig 1.), which show impressive activity against multidrug and extensively drug resistant tuberculosis strains. Key to the success of this series is the chemical introduction of a pyridyl side chain, that blocks efflux of the spectinamides from M. tuberculosis by the efflux pump Rv1258c, while at the same time introducing some new favorable interactions with the mycobacterial ribosome. Substitutions to 4 and 5 positions of the pyridine ring were found to be favorable producing compounds with good MIC potency, impressive post-antibiotic effects and excellent potency in acute and chronic models of tuberculosis infection in mice.3
Figure 1. Structure of the lead spectinamides explored in this study.
The propensity of any new antibiotic for drug metabolism is an important determining factor for its therapeutic efficacy and potential toxicity.4 The major enzymatic reactions of drug metabolism are oxidation, reduction, hydrolysis (Phase-I), and conjugation (Phase-II).5 Our initial studies have shown that the spectinamides undergo little Phase-I metabolism, likely due to their hydrophilic character. In prioritizing which spectinamides to advance towards preclinical development, a Phase-II metabolism study was initiated to investigate the stability of lead spectinamides towards the three most frequent conjugation reactions for drugs6 mediated by uridine glucuronosyl transferases (UGTs), sulfotransferases (SULTs) and glutathione-S-transferases (GSTs), as there are several potential sites for conjugative metabolism within the spectinamide core.
Results and discussion
All compounds in this study were profiled for their susceptibility to Phase II metabolism using both rat and human enzymes. All the spectinamide compounds exhibited high stability towards glucuronide conjugation reactions, with above 93% of the parent compound remaining intact following incubation with rat and human liver S9 fractions and UDP glucuronic acid for 2h (Supplementary data figure S1, table S1). This includes 1810, which contains a phenolic group in the pyridine ring, a motif that is often subject to glucuronidation in other drugs. Next the propensity of the spectinamides for sulfoconjugation was tested. Only 1329 and 1544 underwent significant sulfoconjugation in rats with 89% of 1329 and 88% of 1544 remaining intact after 2h incubation (Supplementary data fig. S2, table S1). This activity was not observed with human enzymes with >96% of both compounds remaining intact after the same time period. Conversely, 1666 showed minor sulfoconjugation with human enzymes with 91% remaining, but no metabolism in rats. All other spectinamide compounds exhibited high stability towards sulfoconjugation reactions, with above 95% (rat) and 96% (human) remaining intact at the end of the incubation period. However, incubation of spectinamide compounds with liver S9 fractions and glutathione revealed that compounds 1544, 1666 and 1878 underwent significant glutathione conjugation with rat enzymes, with only 72, 13, 81% of parent compound remaining intact after 2h incubation, respectively. For human enzymes, only 1666 showed lower glutathione conjugation with 93.5% remaining, while 1544 and 1878 exhibited good metabolic stability with 98.7% remaining. All other compounds exhibited good stability with above 96% of parent compound remaining unchanged after 2h incubation in S9 fractions of both species (Fig. 2; Table 1).
Fig 2. Percent parent compound remaining after Glutathione conjugation reaction in rat liver S9 fraction (n=3).
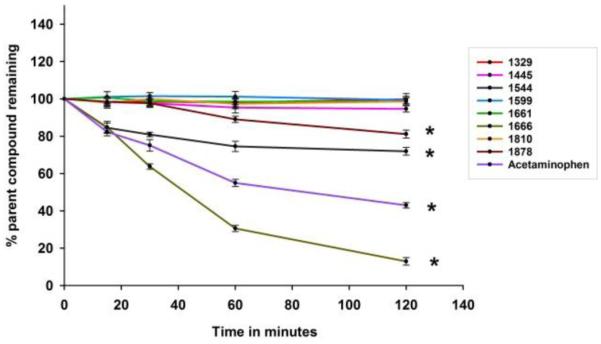
*indicates statistically significant differences from the initial concentration
Table 1.
Metabolic half-life (t½) of Spectinamide compounds in rat and human liver S9 fraction (mean (%CV) of 3 replicates)
| Glutathione Conjugation | ||||
|---|---|---|---|---|
| Rat | Human | |||
|
|
||||
| Compound | % parent remaining after 2 h |
t½ (h) | % parent remaining after 2 h |
t½ (h) |
| 1329 | 99.8 (3.1) | >50 | 96.7 (2.7) | 38.0 |
| 1445 | 96.6 (1.7) | 32.6 | 98.7 (2.9) | >50 |
| 1544 | 71.9 (2.1) | 3.35 | 98.7 (4.4) | >50 |
| 1599 | 99.4 (2.0) | >50 | 99.9(0.42) | >50 |
| 1661 | 98.6 (2.1) | >50 | 98.6 (2.8) | >50 |
| 1666 | 12.9 (1.9) | 0.66 | 93.5(1.6) | 23.0 |
| 1810 | 98.6 (2.0) | >50 | 99.9 (1.3) | >50 |
| 1878 | 81.1 (2.1) | 6.70 | 98.8 (1.7) | >50 |
| ACT | 43.0 (1.4) | 1.48 | 74.5 (3.9) | 4.40 |
Glutathione conjugation usually requires the substrate to have an electrophilic site that reacts with the nucleophile glutathione under the assistance of GST enzyme catalysis. These reactions usually convert the original drug to a form that is not able to bind to its target receptor, thus attenuating the biological response to the drug, yielding metabolites that are rapidly excreted. In the present study, using rat microsomes, all solely 5-substituted pyridiyl (1445, 1599, 1810) along with 4-chloro 5-fluoro pyridyl substituted (1661) spectinamides exhibited complete stability towards Phase II conjugative enzymes. Other 4-Chloro substituted spectinamides (1544, 1666 and 1878) formed glutathione conjugates. The speed of metabolic conversion varied amongst this subseries with 1666 showing a half-life of glutathione conjugation shorter than that of reference drug acetaminophen.
Reactivity of substituted 4-Chloro Pyridines to Phase II glutathione conjugation
The high solubility, lack of metabolism by other Phase I or II enzymes and varying rates of metabolism make this an excellent series to study how different substitution to 4-chloro pyridines dictates their propensity for glutathione conjugation, for which there are few reports in the medicinal chemistry literature. The inherent chemical reactivity of 4-chloro pyridines to nucleophilic aromatic substitution is well known chemically. Biochemically, they are typically stable to co-incubation with thiols under ambient conditions but under enzyme catalysis can be selectively activated to irreversibly inactivate enzymes such as dimethylarginine dimethylaminohydrolase (DDAH).7, 8 The enzyme mediated mechanism for the glutathione inactivation of 4-chloropyridyl spectinamides can be envisioned as shown in Figure 3, similar to the proposed DDAH mechanism which has been explored in detail. In this study, the modulating effect of chemical substitution at the 5-position was explored in regards to hepatic glutathione mediated conjugation. The order of stability 1661>1878>1544>1666 correlates to the calculated pKa values of pyridyl nitrogen (1661, 2.0; 1878, 3.1; 1544, 3.6, 1666, 3.9), which supports the mechanism outlined in Figure 3, where enzyme mediated protonation of the pyridyl ring is likely the key initiating step of this degradatory process. This order, in which electronic donation groups are favored to enhance degradation is also consistent with the ability of the groups to stabilize an intermediate cationic state rather than anionic intermediate state produced by direct nucleophilic aromatic substitution reaction. Indeed, the electron withdrawing 5-F atom in 1661 is seen to block all glutathione conjugation. Interestingly, this SAR contrasts with the study of Inoue et. al on the stability of 3-substituted 2-chloropyridine derivatives in rat liver microsomes fortified with glutathione, in which substrate degradation increased with the introduction stronger electron withdrawing groups in the 3-position.9 Therefore the stability of 1661 is interesting from a mechanistic standpoint and may warrant further exploration to see if this effect is specific to the fluorine substitution or if other electron drawing groups also stabilize this 4-chlorospectinamide to glutathione conjugation.
Figure 3. Proposed mechanism of Glutathione S-transferase mediated Glutathione Conjugation.

Conclusions
We have already demonstrated that the spectinamides are stable to Phase-I metabolism. Herein based on these Phase II metabolism studies, lead spectinamides 1329, 1445, 1599, 1661 and 1810 were found to have favorable properties for potential lead compounds with no Phase II liabilities. Compounds 1544, 1666 and 1878 formed glutathione conjugates. Formation of conjugated products likely removes their anti-tubercular activity and also increases the risk for adverse pharmacological events, thus 1544, 1666 and 1878 have been deprioritized for lead advancement.
Experimental
Phase-II conjugation reactions
All conjugation reactions were performed in triplicate. For glucuronide conjugation, 2 mg/mL of rat or human liver S9 (BD Biosciences, Woburn, MA) was prepared in 0.1 M potassium phosphate buffer (pH 7.4), mixed with 50 μg of alamethicin (Sigma, St. Louis, MO) and placed on ice for 15 min to stimulate the UGT enzymes for maximal activity. To 250 μL of the above mixture, 50 μL of spectinamide test compound solution (100 μM in incubation), 50 μL of MgCl2 (1 mM in incubation), and 100 μL of 0.1 M potassium phosphate buffer were added and preincubated for 5 minutes at 37° C at 150 rpm. To initiate the reaction, 50 μL of uridine diphosphoglucuronic acid (UDPGA, 5 mM in incubation; Sigma, St. Louis, MO) was added to give a 500 μL final volume. 7-Hydroxycoumarin (7-HC) was used as the positive control. Negative control reactions were performed without UDPGA. For sulfoconjugation, 2 mg/mL of rat or human liver S9 was prepared in 0.1 M potassium phosphate buffer (pH 7.4) and placed on ice. To 250 μL of the above mixture, 50 μL of spectinamide test compounds solution (100 μM in incubation), 50 μL of MgCl2 (1 mM in incubation), and 100 μL of 0.1 M potassium phosphate buffer were added and preincubated for 5 minutes at 37° C at 150 rpm. To initiate the reaction, 50 μL of PAPS (Sigma, St. Louis, MO; 0.2 mM in incubation) was added to give a 500 μL final volume. 7-Hydroxycoumarin was used as the positive control. Negative control reactions were performed without PAPS. For glutathione conjugation, 10 mg/mL of rat or human liver S9 was prepared in 0.1 M potassium phosphate buffer (pH 7.4) and placed on ice. To 250 μL of the above mixture, 50 μL of spectinamide test compounds solution (100 μM in incubation), 50 μL of MgCl2 (1 mM in incubation), and 100 μL of 0.1 M potassium phosphate buffer and preincubate the mixture for 5 minutes at 37° C at 150 rpm. To initiate the reaction, 50 μL of glutathione (Sigma Aldrich, St. Louis, MO; 0.2 mM in incubation) was added to give a 500 μL final volume. Acetaminophen (ACT) was used as the positive control. Negative control reactions were performed without glutathione.
For all preparations, 50 μL samples were withdrawn at 0, 15, 30, 45, 60, 75, 90 and 120 minutes of incubation. The reactions were stopped by the addition of 200 μL of ice-cold methanol containing an internal standard. After sitting on ice for 30 min, stopped reactions were centrifuged to pellet precipitated protein, and the supernatant was collected for LC-MS/MS analysis. Disappearance of the parent compound was monitored during the incubation period. The percentage of parent compound remaining intact after incubation was estimated with respect to the amount of the compound at time zero.
LC-MS/MS assay
The collected supernatants were analyzed for drug concentrations by an LC-MS/MS assay. Chromatographic separation of the supernatant was carried out on a Luna 3 μm hydrophobic interaction liquid chromatography (HILIC) 100 × 4.6 mm column (Phenomenex, Torrance, CA) using a gradient mobile phase of methanol and 10 mM ammonium formate, pH ~2.75, at a flow rate of 0.4 ml min−1. Detection was performed with a SCIEX 4500 or 5500 triple-quadruple mass spectrometer (AB Sciex, Framingham, MA) with electrospray ionization in multiple reaction monitoring mode, using the compound specific mass transfer. A calibration curve ranging from 1.95 to 5,000 μg l−1 was constructed for each test compound and validated. The peak area ratios of analyte to the internal standard were linear over the concentration range tested for all compounds, with all correlation coefficients (weighted least-square linear regression analyses) >0.996. Accuracy (deviation of the analyzed quality-control samples from nominal values) was within ±3% over the entire range of the calibration curve, and the precision (coefficient of variation of repeated measurements of the quality-control samples) was <2% for all compounds.
Data analysis
Disappearance of the parent compound was monitored during the incubation period. The percentage of parent compound remaining intact after 120 min incubation was estimated with respect to the amount of the compound at time zero.
Statistical analysis
All values are expressed as their mean ± coefficient of variation (%CV). Statistical differences of the means were assumed to be significant when P < 0.05 by one-way ANOVA followed by Tukey’s test.
Supplementary Material
Acknowledgements
This work was supported by research grants R01AI090810 and S10OD016226 by the National Institutes of Health and the American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
Electronic Supplementary Information (ESI) available. See DOI: 10.1039/x0xx00000x
References
- 1.Casali N, Nikolayevskyy V, Balabanova Y, Harris SR, Ignatyeva O, Kontsevaya I, Corander J, Bryant J, Parkhill J, Nejentsev S, Horstmann RD, Brown T, Drobniewski F. Nature genetics. 2014;46:279–286. doi: 10.1038/ng.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mdluli K, Kaneko T, Upton A. Cold Spring Harbor perspectives in medicine. 2015;5 doi: 10.1101/cshperspect.a021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee RE, Hurdle JG, Liu J, Bruhn DF, Matt T, Scherman MS, Vaddady PK, Zheng Z, Qi J, Akbergenov R, Das S, Madhura DB, Rathi C, Trivedi A, Villellas C, Lee RB, Rakesh, Waidyarachchi SL, Sun D, McNeil MR, Ainsa JA, Boshoff HI, Gonzalez-Juarrero M, Meibohm B, Bottger EC, Lenaerts AJ. Nature medicine. 2014;20:152–158. doi: 10.1038/nm.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park BK, Boobis A, Clarke S, Goldring CE, Jones D, Kenna JG, Lambert C, Laverty HG, Naisbitt DJ, Nelson S, Nicoll-Griffith DA, Obach RS, Routledge P, Smith DA, Tweedie DJ, Vermeulen N, Williams DP, Wilson ID, Baillie TA. Nature reviews. Drug discovery. 2011;10:292–306. doi: 10.1038/nrd3408. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez FJ, Coughtrie M, Tukey RH. Goodman and Gilman's The Pharmacological Basis of Therapeutics. 12 McGraw-Hill; 2011. [Google Scholar]
- 6.Evans WE, Relling MV. Science. 1999;286:487–491. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- 7.Johnson CM, Linsky TW, Yoon DW, Person MD, Fast W. Journal of the American Chemical Society. 2011;133:1553–1562. doi: 10.1021/ja109207m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson CM, Monzingo AF, Ke Z, Yoon DW, Linsky TW, Guo H, Robertus JD, Fast W. Journal of the American Chemical Society. 2011;133:10951–10959. doi: 10.1021/ja2033684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inoue K, Ohe T, Mori K, Sagara T, Ishii Y, Chiba M. Drug metabolism and disposition: the biological fate of chemicals. 2009;37:1797–1800. doi: 10.1124/dmd.109.027698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.