

Abstract
Circulating tumor DNA (ctDNA) is an emerging field of cancer research. For lung cancer, non‐invasive genotyping of EGFR is the foremost application. The activating mutations represent the ctDNA from all cancer cells, and the T790M‐resistant mutation represents that from resistant cells. We examined the ctDNA dynamics of EGFR mutations by using deep sequencing with a massively parallel DNA sequencer. We obtained 190 plasma samples from 57 patients at various times during the treatment course and classified them according to treatment status. The mutation detection rate of exon 19 deletion/L858R in plasma was high at the initiation of treatment with epidermal growth factor receptor tyrosine kinase inhibitor (EGFR‐TKI; P = 0.001), suppressed during EGFR‐TKI treatment before disease progression, and elevated after the onset of disease progression (P = 0.023). The mutation detection rate of T790M was low until the onset of disease progression and elevated thereafter (P = 0.01). Samples across the development of disease progression were obtained from 10 patients and showed a correlation between increased ctDNA level and disease progression. Decreased ctDNA level in response to the initiation of EGFR‐TKI was observed in 4 of 6 eligible patients. In two patients, the ctDNA dynamics suggested the presence of cancer cell populations only with the T790M mutation. In another patient, the T790M ctDNA represented cell subpopulations that respond to cytotoxic agents differently from the major population. Considering the high incidence, ctDNA could be a clinical parameter to complement information from image analyses.
Keywords: Carcinoembryonic antigen, circulating tumor DNA, epidermal growth factor receptor tyrosine kinase inhibitor, lung cancer, massively parallel DNA sequencer
Circulating tumor DNA (ctDNA) is the cell‐free DNA released from dying cancer cells into the bloodstream1 and is an emerging field in cancer research. Clinical applications of ctDNA are expected to be in the form of “liquid biopsy”,2 allowing for the replacement of tissue biopsy with non‐invasive blood tests. Circulating tumor DNA is a carrier that brings the genetic information of solid tumors to peripheral blood. In addition, the quantitative ctDNA levels are generally considered to correlate with tumor burden. These characteristics are the basis of its clinical applications.
In the case of lung cancer, due to the strong correlation between EGFR activating mutations and the efficacy of epidermal growth factor receptor tyrosine kinase inhibitors (EGFR‐TKIs),3, 4 detection of the mutations is indispensable for therapeutic decision making. Since the discovery of the correlation, the use of ctDNA has been extensively investigated for genotyping of EGFR. The recent introduction of next‐generation EGFR‐TKIs5, 6 targeted to the EGFR with a resistant mutation, that is, T790M,7 raises the need of genotyping after the onset of disease progression, which will considerably benefit from the use of ctDNA.
Another application of ctDNA is the monitoring of disease progression8 and drug resistance.9, 10, 11 From a technical viewpoint, this type of application requires quantitation of ctDNA and use of digital PCR12 or related technologies such as next‐generation sequencing is indispensable. In EGFR‐TKI treatment, activating and resistant mutations can be used as independent parameters, with activating mutation for the ctDNA from all cancer cells and resistant mutation (T790M) for the ctDNA from cells resistant to EGFR‐TKI.
We constructed a detection system for EGFR mutations in ctDNA by using deep sequencing with a massively parallel DNA sequencer.13 This system is one of the most intensively validated assay systems for ctDNA.14 In this report, we used clinical evaluation based on the Response Evaluation Criteria in Solid Tumors (RECIST)15 as a reference and examined ctDNA dynamics represented by activating and resistant mutations using the detection system.
Materials and Methods
Patient description
Lung cancer patients subjected to EGFR‐TKI treatment or with a history of EGFR‐TKI treatment were recruited in this study. Patients whose data were used as reference, that is, the data obtained in the multi‐institute study14 for the assessment of the detection system, were recruited as previously described. Written informed consent was obtained from all of the participants. This study was approved by the ethics committee of the Osaka Medical Center for Cancer and Cardiovascular Diseases (Osaka, Japan).
Blood sampling and DNA preparation
Blood samples were collected between October 2010 and August 2013. Approximately 5 mL blood was taken from each patient, with EDTA as anticoagulant. Plasma was separated from the blood cells using low‐speed centrifugation. The plasma was transferred to a fresh tube and recentrifuged at 15 100 g for 10 min at room temperature and stored at −80°C until DNA extraction. Cell‐free DNA was purified as previously described.13
Clinical evaluation of disease progression
Evaluation of response to EGFR‐TKI treatment was carried out based on RECIST version 1.1,15 approximately 2 months after the initiation of EGFR‐TKI. Evaluation of disease progression during the EGFR‐TKI treatment was also based on the RECIST criteria. Additional criteria recommended in the Guideline for Treatment and Diagnosis of Lung Cancer (http://www.haigan.gr.jp/modules/guideline) were also applied.
Mutation detection system
The detection system searches mutations by deep sequencing, that is, sequencing a large number of gene fragments. Exons 19, 20, and 21 of the EGFR gene were independently amplified with PCR from patient plasma DNA, and deep sequencing was undertaken with the Ion Torrent PGM (Thermo Fisher Scientific, Waltham, MA, USA).16 More than 100 000 reads were obtained for each exon region. Because each read was of a single molecule, we were able to estimate the relative ratio of mutation alleles from the fraction of reads containing deletions/substitutions. A diagnostic score, termed the plasma mutation (PM) score, was defined as the number of reads with deletions (exon 19 deletions) or substitutions (exon 20, T790M; exon 21, L858R, L861Q) in 100 000 reads. Thresholds for mutation detection were set as previously described.13, 14
The laboratory procedures of the mutation detection were the same as previously described,13 except for the use of the latest versions of sequencing reagents at the time of assay. The assay was carried out on samples from various projects in order of arrival at the laboratory. The initial PCR amplification of EGFR exon fragments was successful in all of the samples, and mutation data were obtained from all the samples.
Results
Description of sample population
The samples were obtained from 57 patients at various times during their treatment course. The clinical characteristics of the patient population are summarized in Table 1. The number of samples from a single patient ranged from 5 to 13 samples in 16 patients, from 2 to 4 in 15 patients, and 1 in 26 patients. In total, 190 plasma DNA samples were obtained. Circulating tumor DNA data of 139 samples were used in the previous study.13 All clinical information was refined and updated for the following analysis.
Table 1.
Characteristics of patients with lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors (EGFR‐TKI)
| Patient characteristic | n |
|---|---|
| Sex | |
| Male | 17 |
| Female | 40 |
| Age, years | |
| <49 | 11 |
| 50–59 | 10 |
| 60–69 | 22 |
| 70–79 | 11 |
| 80–89 | 3 |
| Stage | |
| IIIA | 1 |
| IV | 56 |
| Histology | |
| Squamous cell carcinoma | 1 |
| Adenocarcinoma | 56 |
| Mutation in biopsy samples | |
| Exon 19 deletion | 31 |
| L858R | 20 |
| L861Q | 1 |
| G719A | 1 |
| Wild‐type | 1 |
| Unknown | 3 |
| Type of EGFR‐TKI treatment | |
| Gefitinib | 28 |
| Erlotinib | 13 |
| Gefitinib → erlotinib | 9 |
| Erlotinib → gefitinib | 1 |
| Erlotinib + bevacizumab | 2 |
| Erlotinib + gemcitabine | 2 |
| Cytotoxic agents | 2 |
| Initial effect of EGFR‐TKI | |
| Complete response | 3 |
| Partial response | 40 |
| Stable disease | 6 |
| Progressive disease | 2 |
| Not evaluable | 6 |
Analysis of plasma sample population
Data of all the 190 plasma samples, used as the analysis set, were pooled and subjected to the following analyses. We first compared the analysis set with the dataset obtained from the multi‐institute prospective study14 as the reference set. The patient population of the latter set is a uniform population for comparison of genotyping of plasma and biopsy samples. The numbers of samples that were mutation‐positive in plasma were grouped according to abundance (PM score range) in Table 2. To compare with the reference set, we focused on samples whose corresponding biopsy samples had exon 19 deletion or L858R for analysis of activating mutations. For T790M, all of the samples were used. The mutation detection rate in plasma was considerably lower in the analysis set. Samples with low PM scores were more frequent in the analysis set.
Table 2.
Comparison of two datasets of plasma samples from lung cancer patients
| Reference data set exon 19 deletion (IIIB–IV) | Reference data set L858R (IIIB–IV) | Analysis data set exon 19 deletion | Analysis data set L858R | Analysis data set T790M | |
|---|---|---|---|---|---|
| Plasma‐positive samples | |||||
| <100 | 0 | 1 | 6 | 7 | 5 |
| 100–999 | 6 | 10 | 26 | 17 | 18 |
| 1000–9999 | 11 | 7 | 7 | 16 | 7 |
| >10 000 | 8 | 5 | 7 | 7 | 2 |
| Total | 25 | 23 | 46 | 47 | 32 |
| All samples | |||||
| Total | 36 | 30 | 83 | 92 | 190 |
| Detection rate in plasma, % | 69.4 | 76.7 | 55.4 | 51.1 | 16.8 |
Analysis set comprised 190 plasma samples from 57 patients taken during their treatment course. The reference dataset was obtained from a multi‐institute prospective study14 that comprised a uniform population for comparison of genotyping of plasma and biopsy samples. The plasma‐positive samples were grouped according to PM score range.
The samples of the analysis set may be classified into the following five categories: EGFR‐TKI treatment initiation (TKI initiation); EGFR‐TKI treatment before development of objective disease progression (non‐PD); EGFR‐TKI treatment with objective disease progression (PD); treatment other than EGFR‐TKI; and discontinued EGFR‐TKI treatment. The numbers of samples in the latter two groups were too small (n = 13 and 9, respectively) for a proper statistical analysis. The samples of TKI initiation were set as those before EGFR‐TKI treatment and those until 2 weeks after the initiation of the treatment based on previous observations.13 The samples of the first three categories are shown in the top sections of Table 3. Activating and T790M‐resistant mutations shared the same features with the non‐PD and PD samples: the mutation detection rate in plasma was low with non‐PD samples, and high with PD. To assess the significance of the difference in the mutation detection rates in the sample population against the variance between samples in the groups (non‐PD or PD), we used median values as the representative statistic for each group (non‐PD or PD) in each patient, determined as positive/negative in plasma (the bottom sections of Table 3), and calculated P‐values by using the Fisher's exact test. The statistical significance of exon 19 deletion/L858R and T790M were P = 0.023 and P = 0.010, respectively. We also applied this analysis for comparison of TKI initiation and non‐PD. The statistical significance of exon 19 deletion/L858R and that of T790M were P = 0.001 and P = 1.000, respectively. Thus, the mutation detection rate of exon 19 deletion/L858R in plasma was high in the initiation of EGFR‐TKI treatment, suppressed during EGFR‐TKI treatment before disease progression, and elevated after development of disease progression. The mutation detection rate of T790M was low until the onset of objective disease progression and elevated thereafter.
Table 3.
Numbers of plasma samples or cases from patients with lung cancer in each treatment status category
| TKI initiation | Non‐PD | PD | |
|---|---|---|---|
| Exon 19 deletion or L858R | |||
| Plasma‐positive samples | 18 | 32 | 31 |
| All samples | 24 | 92 | 41 |
| Detection rate in plasma, % | 75.0 | 34.8 | 75.6 |
| T790M | |||
| Plasma‐positive samples | 2 | 15 | 14 |
| All samples | 29 | 94 | 54 |
| Detection rate in plasma, % | 6.9 | 16.0 | 25.9 |
| Exon 19 deletion or L858R | |||
| Plasma‐positive cases | 13 | 7 | 12 |
| Plasma‐negative cases | 2 | 16 | 5 |
| T790M | |||
| Plasma‐positive cases | 1 | 2 | 8 |
| Plasma‐negative cases | 15 | 22 | 10 |
PD, disease progression; TKI, tyrosine kinase inhibitor.
We compared the distributions of the PM scores with the non‐PD and PD “mutation‐positive in plasma” samples (Fig. 1). The PD samples tended to be distributed in fractions of high PM scores compared with non‐PD samples.
Figure 1.
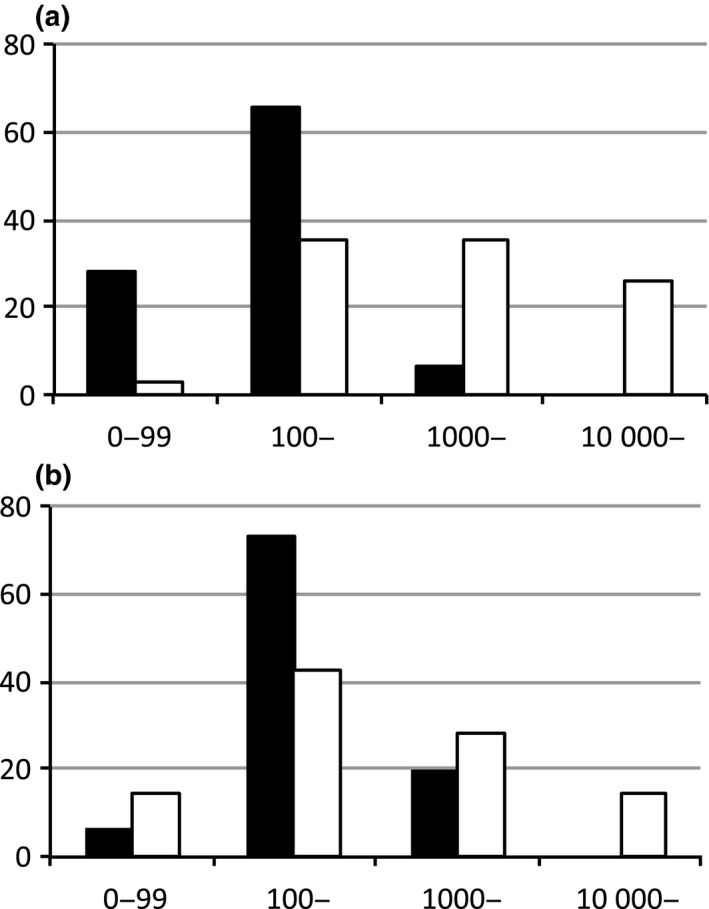
Distribution of plasma mutation (PM) scores of the “mutation‐positive in plasma” samples from lung cancer patients. Vertical axis, fraction of samples (%) within the PM score range; horizontal axis, PM score range; black bar, non‐disease progression samples; white bar, disease progression samples. (a) Exon 19 deletion/L858R. (b) T790M.
Temporal changes in ctDNA represented by the activating mutations
We then confirmed the above results with the dynamics of individual patients. Six patients, patients 1–3 and 6–8, had samples in TKI initiation, non‐PD, and PD. Another four patients, patients 4–5 and 9–10, had samples in non‐PD and PD (Figs 2,S1). The clinical information of the patients including details of objective disease progression is summarized in Table S1.
Figure 2.
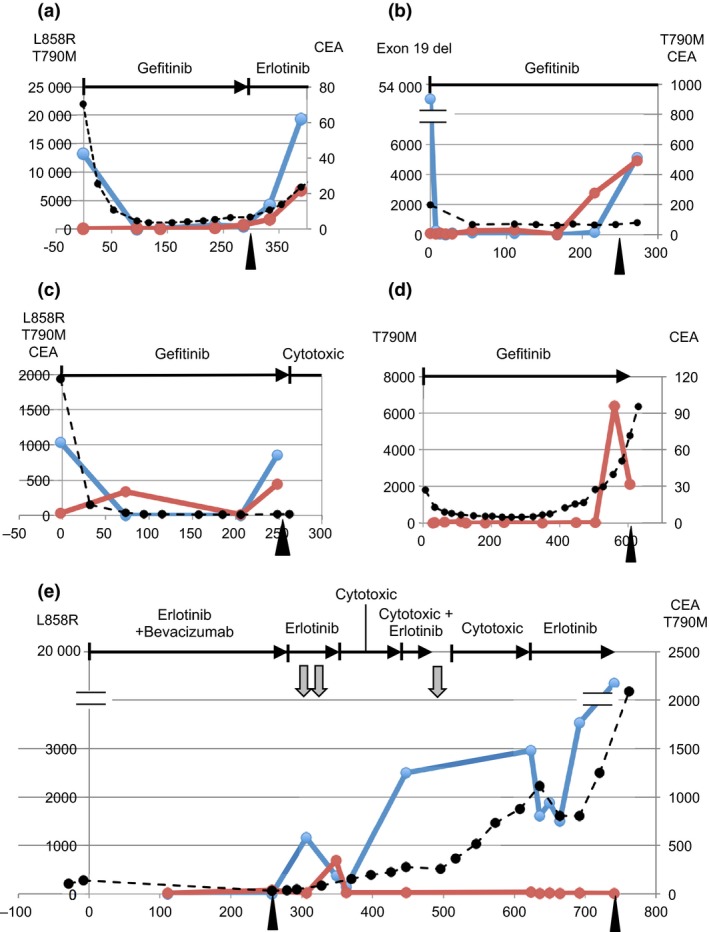
Circulating tumor DNA dynamics in the course of epidermal growth factor receptor tyrosine kinase inhibitor treatment of lung cancer patients. Vertical axis, plasma mutation score (exon 19 deletion, L858R, and T790M) or protein concentration (carcinoembryonic antigen [CEA], μg/mL). Horizontal axis, days from initiation of treatment. The horizontal lines at the top of each panel indicate treatment: vertical bar, initiation of therapy; arrowhead, termination of therapy. Gray arrows below the horizontal lines indicate radiotherapy. Arrowheads in the bottom of each panel indicate disease progression time points. Blue lines indicate activating mutations (exon 19 deletion or L858R). Red lines indicate T790M. Black broken lines indicate CEA. In the patients who had no data exceeding the threshold of detection (plasma mutation score) or whose data were within the normal range (CEA), data were not presented in graphs. (a) Patient 1, female, aged 65 years, with stage IV disease. (b) Patient 2, female, aged 71 years, with stage IV disease. (c) Patient 3, male, aged 68 years, with stage IV disease. (d) Patient 4, male, aged 76 years, with stage IV disease. (e) Patient 5, female, aged 59 years, with stage IV disease.
First, we examined the dynamics of the activating mutations. The activating mutations were detected, except patient 4 (Fig. 2d). Patients 1–3 represented the typical pattern (Fig. 2a–c). Before the initiation of EGFR‐TKI, the ctDNA level of the activating mutations was high, but after the initiation of EGFR‐TKI, the level decreased. Other patients also showed dynamics concordant to the results of the above‐mentioned analysis of the sample population. In patient 1, the dynamic pattern of carcinoembryonic antigen (CEA) was similar to that of ctDNA. However, in patients 2 and 3, CEA showed an initial decrease in response to EGFR‐TKI but did not show an increase reflecting disease progression.
On the whole, the increase in ctDNA level represented by the activating mutations at the onset of objective disease progression was observed in 9 of 10 patients. Including patient 4, whose T790M ctDNA exhibited the same pattern, all 10 patients showed increased ctDNA levels. By contrast, increased CEA level was present in only 4 (40%) of 10 patients. Decreased ctDNA at the initiation of EGFR‐TKI was observed in 4 (67%) of 6 patients eligible for analysis. In this timing, decreased CEA level was observed in 6 (60%) of 10 patients.
Temporal changes in ctDNA represented by the T790M‐resistant mutation
We then examined the ctDNA dynamics represented by the T790M‐resistant mutation. In general, as indicated by the above analysis of the sample population, the ctDNA level of T790M increased in response to disease progression but was consistently smaller than that of the activating mutations (patients 1, 3, 9, and 10; Figs 2a,c,S1d,e). The T790M ctDNA level indicated the fraction of the mutation allele or cancer cells with the resistant mutation.
Patients 3 and 4 (Fig. 2c,d) had positive data points only with T790M, suggesting the presence of cancer cell populations carrying the T790M mutation but no activating mutations. In patient 3, the T790M ctDNA appeared transiently during the non‐PD period. In patient 4, the T790M ctDNA appeared in parallel with disease progression. Because the primary lesion of patient 4 had exon 19 deletion, EGFR‐TKI would have eliminated the original cancer cell population and a unique cell population with T790M only would have appeared in PD. Otherwise, alleles with exon 19 deletion would have been lost during the treatment.
Patient 5 (Fig. 2e) had a considerable number of samples in PD. After the initial onset of disease progression, ctDNA appeared. During therapy with cytotoxic agents, the T790M ctDNA was suppressed, but the activating mutation was constantly increased. This suggests that cytotoxic agents successfully suppressed the T790M‐positive cell populations, but the agents could not control the growth of other cancer cells with the activating mutation. A subsequent rechallenge of EGFR‐TKI suppressed the ctDNA level with the activating mutation and without T790M, reflecting a transient improvement in disease status. The dynamic pattern of ctDNA coincided with the treatment status and may explain the mechanism of successful rechallenge.
The results of the multi‐institute study for the assessment of diagnostic accuracy14 indicated that the possibility of false‐positive results was negligible for data points with PM scores exceeding 300 (limit of quantitation [LOQ]). False‐positive rates for exon 19 deletion, L858R, and T790M were estimated as 2%, 0%, and 1%, respectively, from the diagnostic specificities calculated by using the genotyping results from biopsy samples as a comparator. These figures are the maximum estimates, and the real figures would have been lower because the calculation of the specificities assumed that the biopsy samples had no false negatives. All data points relevant for the above‐mentioned cases sufficed the criterion: their PM scores exceeded the LOQ. Because the PM score of the T790M ctDNA at day 216 of patient 2 did not exceed the LOQ (Fig. 2b), it was retained to judge it as a distinct cancer cell population. Owing to the high reproducibility of data points over the LOQ,13 the possibility of false negatives of the data points potentially over the LOQ is also negligible.
Discussion
In our series of studies on non‐invasive genotyping of EGFR, we prioritized the validation of the detection system, performing a multi‐institute prospective study.14 We then proceeded to analyze other datasets referring to the performance revealed by the study. Our ctDNA data provide firm confidence, eliminating the need for parallel experiments such as analysis of biopsy samples.
Most studies on ctDNA dynamics have been restricted to the analysis of individual patients, and statistical analysis of a population is sparse. We showed a statistically significant difference in the mutation detection rate in plasma samples between the disease and treatment conditions. The ctDNA dynamics of individual patients were generally consistent with the results of the analysis of the sample population and showed that ctDNA is a possible clinical parameter to complement image analysis. A major characteristic of ctDNA dynamics is a wider dynamic range of responses compared to the serum biomarker CEA. From a scientific viewpoint, identification of the T790M ctDNA representing distinct cancer cell subpopulations is particularly interesting. We observed this phenomenon in another prospectively collected dataset (manuscript in preparation). Another study that used a different technique also observed this phenomenon.17
The PM score represents the relative ratio of ctDNA to plasma cell‐free DNA. Other studies, for example, a study using droplet digital PCR carried out by Oxnard et al.,18 have used the absolute amount of ctDNA rather than the relative ratio. Currently, both parameters are likely to exhibit good correlation with the disease status. However, processing raw data for medical interpretation is an important component of technologies, and a detailed comparison might be necessary.
In two patients, the ctDNA dynamics suggested the presence of cancer cell populations only with the T790M mutation. Investigation of their origin is interesting, but there are two concerns. Circulating tumor DNA is considered as a correlate of tumor burden, but it is not necessarily true, as the direct correlation is with dying cancer cells. Thus, for example, when cells with high turnover rate yield more ctDNA than those with low turnover rate, their population sizes might not be parallel with ctDNA levels. Another concern is technical. Such cells would exist as a minor population in tumor tissues, and their detection is technically demanding. The most refined experiment on EGFR so far collected 50 cells for each sample.19 Recent developments of single cell genomics have enabled genotyping of single cells, which would provide accurate information.
Monitoring of acquired resistance has considerable clinical benefits. Several agents have resistant mutations that can serve as markers to detect ctDNA. Studies on the dynamics were reported with anti‐EGFR antibodies and aromatase inhibitors, as well as EGFR‐TKI using mutations of KRAS,9, 10 ESR1,20 and EGFR,11, 13, 18 respectively. In EGFR‐TKI treatment, the activating and T790M‐resistant mutations can be used as independent parameters. In the case of the other agents, usually only resistant mutations were used because additional procedures are required to identify marker mutations for the entire cancer cell population. As indicated above, in most cases, the activating mutations served as a better marker for disease progression owing to acquired resistance. The merit of the T790M ctDNA may be, besides that it is a direct cause of resistance, that it represents cell subpopulations that respond to therapies differently from the major populations. A similar example was reported with rociletinib.21
Several new EGFR‐TKIs targeted to T790M such as AZD92915 and rociletinib6 are emerging, and their use requires detection of T790M. Because rebiopsy is technically demanding, and biopsy samples do not necessarily represent the entire cancer cell population, detection of T790M in plasma DNA has considerable clinical benefits. A recent study reported discordant genotypes between tumor biopsy and blood‐based analyses.22 As shown earlier, therapies with cytotoxic agents may suppress the T790M ctDNA. It did not appear before the onset of acquired resistance either. Thus, the timing of sampling should be precisely determined. Detection of T790M is among the most intensively studied in this field, and applications of various technologies are reported.11, 18, 23, 24 Because quantitative approaches may not be necessary for this purpose, evaluation of various technologies might be beneficial.
The present study clearly establishes the correlation between ctDNA dynamics and disease progression/response to EGFR‐TKI. However, for clinical applications, prospective studies are essential. In particular, detailed descriptions of responses to various therapies are needed. Moreover, the findings on the T790M ctDNA need confirmation with additional samples.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Circulating tumor DNA dynamics in the course of epidermal growth factor receptor tyrosine kinase inhibitor treatment. These patients are not shown in Fig. 2.
Table S1. Patient information for cases in Figs 2,S1.
Acknowledgments
The authors thank Ms. Shiho Sasaki for her excellent technical assistance. This work was partially supported by grants from the Osaka Foundation for the Prevention of Cancer and Cardiovascular Diseases.
Cancer Sci 107 (2016) 353–358
Funding Information
Osaka Foundation for the Prevention of Cancer and Cardiovascular Diseases
References
- 1. Schwarzenbach H, Hoon DS, Pantel K. Cell‐free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 2011; 11: 426–37. [DOI] [PubMed] [Google Scholar]
- 2. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 2014; 32: 579–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lynch TJ, Bell DW, Sordella R et al Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 4. Paez JG, Janne PA, Lee JC et al EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 5. Cross DA, Ashton SE, Ghiorghiu S et al AZD9291, an irreversible EGFR TKI, overcomes T790M‐mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014; 4: 1046–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sequist LV, Soria JC, Goldman JW et al Rociletinib in EGFR‐mutated non‐small‐cell lung cancer. N Engl J Med 2015; 372: 1700–9. [DOI] [PubMed] [Google Scholar]
- 7. Pao W, Miller VA, Politi KA et al Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2: e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Diehl F, Schmidt K, Choti MA et al Circulating mutant DNA to assess tumor dynamics. Nat Med 2008; 14: 985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diaz LA Jr, Williams RT, Wu J et al The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012; 486: 537–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Misale S, Yaeger R, Hobor S et al Emergence of KRAS mutations and acquired resistance to anti‐EGFR therapy in colorectal cancer. Nature 2012; 486: 532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taniguchi K, Uchida J, Nishino K et al Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res 2011; 17: 7808–15. [DOI] [PubMed] [Google Scholar]
- 12. Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci USA 1999; 96: 9236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kukita Y, Uchida J, Oba S et al Quantitative identification of mutant alleles derived from lung cancer in plasma cell‐free DNA via anomaly detection using deep sequencing data. PLoS ONE 2013; 8: e81468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uchida J, Kato K, Kukita Y et al Diagnostic accuracy of noninvasive genotyping of EGFR in lung cancer patients by deep sequencing of plasma cell‐free DNA. Clin Chem 2015; 61: 1191–6. [DOI] [PubMed] [Google Scholar]
- 15. Eisenhauer EA, Therasse P, Bogaerts J et al New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45: 228–47. [DOI] [PubMed] [Google Scholar]
- 16. Rothberg JM, Hinz W, Rearick TM et al An integrated semiconductor device enabling non‐optical genome sequencing. Nature 2011; 475: 348–52. [DOI] [PubMed] [Google Scholar]
- 17. Sueoka‐Aragane N, Katakami N, Satouchi M et al Monitoring EGFR T790M with plasma DNA from lung cancer patients in a prospective observational study. Cancer Sci 2016. doi:10.1111/cas.12847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oxnard GR, Paweletz CP, Kuang Y et al Noninvasive detection of response and resistance in EGFR‐Mutant lung cancer using quantitative next‐generation genotyping of cell‐free plasma DNA. Clin Cancer Res 2014; 20: 1698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taniguchi K, Okami J, Kodama K, Higashiyama M, Kato K. Intratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinib. Cancer Sci 2008; 99: 929–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sefrioui D, Perdrix A, Sarafan‐Vasseur N et al Short report: Monitoring ESR1 mutations by circulating tumor DNA in aromatase inhibitor resistant metastatic breast cancer. Int J Cancer 2015; 137: 2513–9. [DOI] [PubMed] [Google Scholar]
- 21. Piotrowska Z, Niederst MJ, Karlovich CA et al Heterogeneity Underlies the Emergence of EGFRT790 Wild‐Type Clones Following Treatment of T790M‐Positive Cancers with a Third‐Generation EGFR Inhibitor. Cancer Discov 2015; 5: 713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sundaresan TK, Sequist LV, Heymach JV et al Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood‐based analyses. Clin Cancer Res 2015. doi:10.1158/1078‐0432.CCR‐15‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakamura T, Sueoka‐Aragane N, Iwanaga K et al A noninvasive system for monitoring resistance to epidermal growth factor receptor tyrosine kinase inhibitors with plasma DNA. J Thorac Oncol 2011; 6: 1639–48. [DOI] [PubMed] [Google Scholar]
- 24. Sakai K, Horiike A, Irwin DL et al Detection of epidermal growth factor receptor T790M mutation in plasma DNA from patients refractory to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci 2013; 104: 1198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Circulating tumor DNA dynamics in the course of epidermal growth factor receptor tyrosine kinase inhibitor treatment. These patients are not shown in Fig. 2.
Table S1. Patient information for cases in Figs 2,S1.