

Abstract
Osteosarcoma is the most common primary malignant bone tumor, and the frequent acquisition of chemoresistance is often an obstacle to achieving favorable outcomes during chemotherapy. Recently, Krüppel‐like factor 4 (KLF4) has been shown to be associated with chemotherapy resistance in a few tumors; however, the involvement of KLF4 in chemotherapy resistance in osteosarcoma cells remains unknown. In this study, quantitative real‐time PCR and western blot analysis revealed that KLF4 expression was significantly increased in response to cisplatin, methotrexate and doxorubicin treatment in osteosarcoma cells, and knockdown of KLF4 increased sensitivity to these anticancer drugs by decreasing cellular clonogenic ability and increasing apoptosis. Moreover, our data suggest that KLF4‐regulated drug resistance might, at least partially, positively regulate high‐mobility group box 1 (HMGB1), which was found to be a significant contributor to chemoresistance in osteosarcoma cells in our previous study. In summary, this study highlights the significance of KLF4/HMGB1 interaction in regulating chemotherapy resistance, and suggests that targeting KLF4/high‐mobility group box 1 may be a therapeutic strategy for osteosarcoma chemotherapy.
Keywords: Chemotherapy, drug resistance, high‐mobility group box 1, Krüppel‐like factor 4, osteosarcoma
Osteosarcoma is one of the most malignant bone tumors, with high incidence in children and adolescence.1, 2 Although the 5‐year survival rate has increased 60–70% within the past 10 years due to the systemic therapy development, osteosarcoma patients following treatment with the current therapy frequently develop drug resistance, which is still a major obstacle to achieving favorable outcomes. Thus, understanding the molecular mechanisms underlying the resistance of osteosarcoma cancer cells to chemotherapy is essential for the development of novel treatment strategies for this disease.
Krüppel‐like factor 4 (KLF4), known as gut‐enriched Krüppel‐like factor,3, 4 is a zinc finger‐containing transcription factor with essential functions, including cell proliferation, differentiation and stem cell reprogramming.5, 6 KLF4 plays opposing roles in different human cancers by transcriptionally regulating its downstream target genes.7, 8 Recently, KLF4 has been shown to be associated with chemotherapy resistance in a few tumors.9, 10, 11 However, the involvement of KLF4 in chemotherapy resistance in osteosarcoma cells remains unknown.
High mobility group box 1 (HMGB1) is a chromatin‐binding nuclear protein, and has been reported to be involved in human inflammatory diseases as well as several tumor types.12, 13, 14 Our previous study demonstrated that high HMGB1‐mediated autophagy is a significant contributor to multi‐drug resistance in osteosarcoma cells and that inhibition of HMGB1 increases the drug sensitivity of osteosarcoma cells.15, 16 However, the molecular mechanism underlying HMGB1 regulation during osteosarcoma cancer chemotherapy remains unknown. Of note, Liu et al. report that KLF4 promoted the expression, translocation and release of HMGB1 in mouse RAW264.7 macrophages in response to LPS treatment.17 Considering our previous findings of HMGB1 in chemotherapy resistance, we therefore proposed the hypothesis that KLF4 might be involved in HMGB1‐induced chemotherapy resistance in osteosarcoma cells.
In this study, to test this hypothesis, we treated osteosarcoma cells with commonly used anticancer reagents (cisplatin, methotrexate and doxorubicin) and then examined the expression of KLF4. We found that treatment with these anticancer reagents could induce high KLF4 expression, and suppression of KLF4 resulted in increased sensitivity to chemotherapy in vitro. In addition, KLF4 could positively regulate HMGB1 expression in osteosarcoma cells in vitro, at least partially, due to directly binding and transactivating HMGB1 promoter. Taken together, our results highlight the significance of KLF4/HMGB1 interaction in regulating chemotherapy resistance in osteosarcoma cells.
Materials and Methods
Cell culture and drug treatment
The osteosarcoma cell lines (MG‐63, SaOS‐2 and U‐2 OS) were originally obtained from the American Type Culture Collection (ATCC, Rockville, MI, USA), and cultured in Eagle's minimum essential medium supplemented with 10% FBS in a 5% CO2 and humidified atmosphere at 37°C. For drug treatment, cisplatin (Sigma‐Aldrich, Taufkirchen, Germany) was dissolved in N,N‐dimethylformamide at a concentration of 15 mg/mL and added to the culture medium in the indicated concentrations. Methotrexate was dissolved in 0.01N NaOH and further diluted in PBS to a concentration of 10 mg/mL. Doxorubicin (Sigma‐Aldrich) was dissolved in sterile water at a concentration of 20 mM and added to the culture medium in the indicated concentrations.
RNA preparation and quantitative real‐time PCR
Total RNA was extracted by Trizol reagent, and first strand cDNA was synthesized with SuperScript II (Invitrogen, Carlsbad, CA, USA). The transcript levels were detected by quantitative real‐time PCR analysis using SYBR Green Taq ReadyMix (Takara, Japan). The expression levels of HMGB1 and KLF4 were normalized to that of β‐actin mRNA, which served as an endogenous control. The following sequences of PCR primers were used: β‐actin: 5′‐AGGGGCCGGACTCGTCATACT‐3′ (forward), 5′‐GGCGGCACCACCATGTACCCT‐3′ (reverse); KLF4: 5′‐GCCGCTCCATTACCAAGAG‐3′ (forward), 5′‐ATCCACAGCCGTCCCAGTC‐3′ (reverse); HMGB1: 5′‐TCAAAGGAGAACATCCTGGCCTGT‐3′ (forward), 5′‐TCAAAGGAGAACATCCTGGCCTGT‐3′ (reverse). qRT‐PCR was performed on the ABI 7500 thermocycler (Applied Biosystems, Carlsbad, CA, USA). The relative expression levels were calculated using the 2−ΔΔct method.
Western blot
Cells were lysed with 200 μL of lysis buffer and subjected to western blot analysis.16 Approximately 50 mg of total protein was separated by 10% SDS‐PAGE, transferred to a PVDF membrane and incubated with the appropriate antibodies. The antibodies to HMGB1 and KLF4 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and β‐actin antibody was obtained from Kangchen Biotechnology (Shanghai, China). The protein bands were visualized using ECL detection (Applygen, Beijing, China).
Krüppel‐like factor 4 expression plasmid and small interfering RNA transfection
The coding sequence of KLF4 was amplified and cloned into pcDNA3.1 vector to generate KLF4 expression plasmid (pcDNA3.1‐KLF4), and the empty pCDNA3.1 vector was used as control (pcDNA3.1‐Con). Primers used for KLF4 coding sequence are as follows: 5′‐GGATCCATGAGGCAGCCACCTGGC‐3′ (forward) and 5′‐GAATTCTTAAAAATGCCTCTTCATGTG‐3′ (reverse). Specific siRNA for KLF4 (si‐KLF4) and scrambled siRNA (negative control, si‐NC) were purchased from GeneChem (Shanghai, China). si‐KLF4: 5′‐CCUCCUGGACCUAGACUUUdTdTAAAGUCUAGGUCCAGGAGGdTdT‐3′; si‐NC: 5′‐UUCUCCGAACGUGUCACGUTTACGUGACACGUUCGGAGAATT‐3′. The siRNA was transfected into MG‐63 and SaOS‐2 cells at a final concentration of 20 nM in culture medium by using Lipofectamine RNAi Max (Invitrogen) according to the manufacturer's instructions.
ChIP assay
ChIP assays were performed as previously described.18 Briefly, 5 × 10*7 cells were crosslinked with 1% formaldehyde for 10 min at 37°C, and then sonicated under the condition of a Bioruptor™200, and fragmented chromatins were immunoprecipitated using an anti‐KLF4 antibody or control goat IgG antibody and a Chromatin Immunoprecipitation Kit (Millipore, Boston, MA, USA). Immunoprecipitated DNA were purified and quantitatively analyzed by real‐time PCR using a HMGB1 gene promoter‐specific primer set (5′‐TGTCGCCCTCACTTTTGAA‐3′ and 5′‐ATGGTGTATGTGTGCATGTG‐3′) to determine the enrichment. Values were presented as relative to DNA input. The assays were carried out in three replicates and relative fold change was calculated using the −∆∆Ct method.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was performed using nuclear extracts according to the protocol of the Light Shift Chemiluminescent EMSA Kit (Pierce, Carlsbad, CA, USA). Double‐stranded DNA probes (5′‐GTGAATGTGGGGCAAGAAGGGGGGGGGAGACCTGTGGGTGTCTCT‐3′ and 5′‐AGAGACACCCACAGGTCTCCCCCCCCCTTCTTGCCCCACATTCAC‐3′) were generated. Double‐stranded mutant DNA probes: 5′‐ATATAGAGCAAGAAGGGGGGGGGAGACCTATAGATG‐3′, 5′‐CATCTATAGGTCTCCCCCCCCCTTCTTGCTCTATAT‐3′. Nucleotides were labeled using biotin 3′‐end DNA labeling (Pierce). Detection of supershift mobility was assessed by reaction of KLF4 antibody (Santa Cruz Biotechnology) with nuclear extracts for 30 min before addition of the labeled nucleotides for the EMSA.
Reporter gene assays
Five different fragments of the promoter region of HMGB1 were amplified and ligated to pGL3 GFP reporter. MG‐63 cells were cultured to approximately 80% of the plates, and co‐transfected with HMGB1 promoter–reporter constructs and KLF4 expression plasmid using Lipofectamine 2000 (Invitrogen), according to the manufacturer's protocol. The cells were cultured for another 4 h, and the GFP expression levels were quantified by flow cytometry.
Cell proliferation, colony formation and apoptosis assays
Cell proliferation was evaluated by MTT assay. Cells were seeded in a 96‐well plate at 5000 cells per well and incubated for 24 h, then exposed to different doses of anticancer drugs for 48 h. Then, the cells were added to 10 mg/mL MTT (Sigma Chemicals, St. Louis, MO, USA). After incubation for 2 h, the reaction was terminated by removal of the supernatant followed by adding 200 μL of DMSO. The optical density at 570 nm was measured with a microplate reader (Bio‐Rad, Hercules, CA, USA). For colony formation assays, approximately 500 cells were placed in complete growth media in each 35‐mm dish and allowed to grow for 6 h. Then, with different doses of anticancer drugs, were added to each dish. After 24 h of treatment, the anticancer drugs were removed by adding fresh complete growth media, and cells were allowed to grow until visible colonies formed, and were stained and counted. For cell apoptosis assays, cells were collected and the cell apoptosis ratio was analyzed using the Annexin V‐FITC Apoptosis Detection Kit (BD Biosciences, San Diego, CA, USA), according to the manufacturer's instructions.
Statistical analysis
Data are expressed as the mean + SD. Differences between groups were analyzed by Student's t‐test or anova, and P < 0.05 was considered to be statistically significant.
Results
Krüppel‐like factor 4 is upregulated in response to chemotherapy in osteosarcoma cells
We first assayed the effects of the anticancer reagents cisplatin (Cis), methotrexate (Mtx) and doxorubicin (Dox) on the expression of KLF4 in osteosarcoma cells, respectively. As shown in Figure 1a, quantitative real‐time PCR (qRT‐PCR) revealed that these drugs significantly promoted increased mRNA expression levels of KLF4 in MG‐63, SaOS‐2 and U‐2OS cell lines after treatment with these anticancer agents for 48 h. In addition, western blot analysis showed similar results (Fig. 1b). These findings suggest that KLF4 is upregulated in response to chemotherapy in osteosarcoma cells, and, thus, may play an important role in regulating the chemoresistance in osteosarcoma cells.
Figure 1.
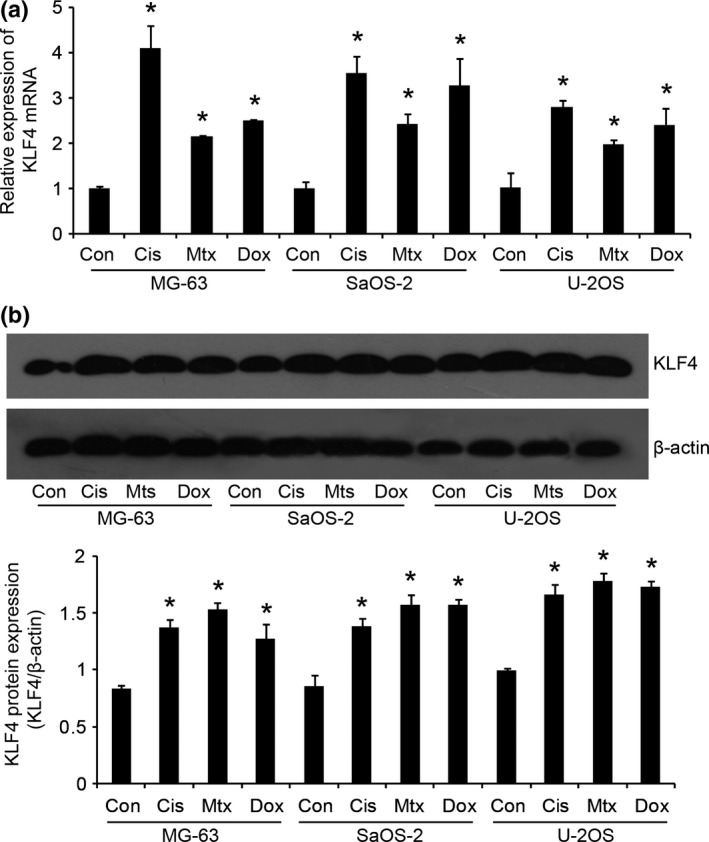
Krüppel‐like factor 4 (KLF4) is upregulated in response to chemotherapy in osteosarcoma cells. Osteosarcoma cell lines (MG‐63, SaOS‐2 and U‐2OS) were treated with 50 μM cisplatin (Cis), 50 μM methotrexate (Mtx) and 0.2 μg/mL doxorubicin (Dox) for 48 h, respectively. Then, the expression levels of KLF4 were examined by quantitative real‐time PCR (a) and western blot (b). *P < 0.05 versus the control (Con).
Suppression of Krüppel‐like factor 4 increases sensitivity to chemotherapy in vitro
To examine the potential role of KLF4 in the regulation of anticancer drug sensitivity of osteosarcoma cells, we established KLF4 knocked down osteosarcoma cells by KLF4‐specific siRNA (MG‐63/si‐KLF4, SaOS‐2/si‐KLF4) and the corresponding control cells by scrambled siRNA (MG‐63/si‐Con and SaOS‐2/si‐Con). qRT‐PCR and western blot analysis validated that MG‐63/si‐KLF4 and SaOS2/si‐KLF4 cells expressed significantly lower mRNA and protein levels of KLF4 than in the corresponding control cells. Using the MTT assays, we found that knockdown of KLF4 in these cells rendered them significantly more sensitive to Dox‐induced, Cis‐induced and Mtx‐induced cell injury, respectively (Fig. 2a), and this was also associated with drastically decreased clonogenic ability (Fig. 2b) and high levels of apoptotic cell death (Fig. 2c). In contrast, when KLF4 was overexpressed in MG‐63 and SaOS‐2 cells, we observed an opposite effect on Dox‐induced, Cis‐induced and Mtx‐induced cell injury (Fig. 3a), clonogenic ability (Fig. 3b) and apoptotic cell death (Fig. 3c). Taken together, these data suggest that targeted suppression of KLF4 increases sensitivity to chemotherapy in osteosarcoma cells in vitro.
Figure 2.
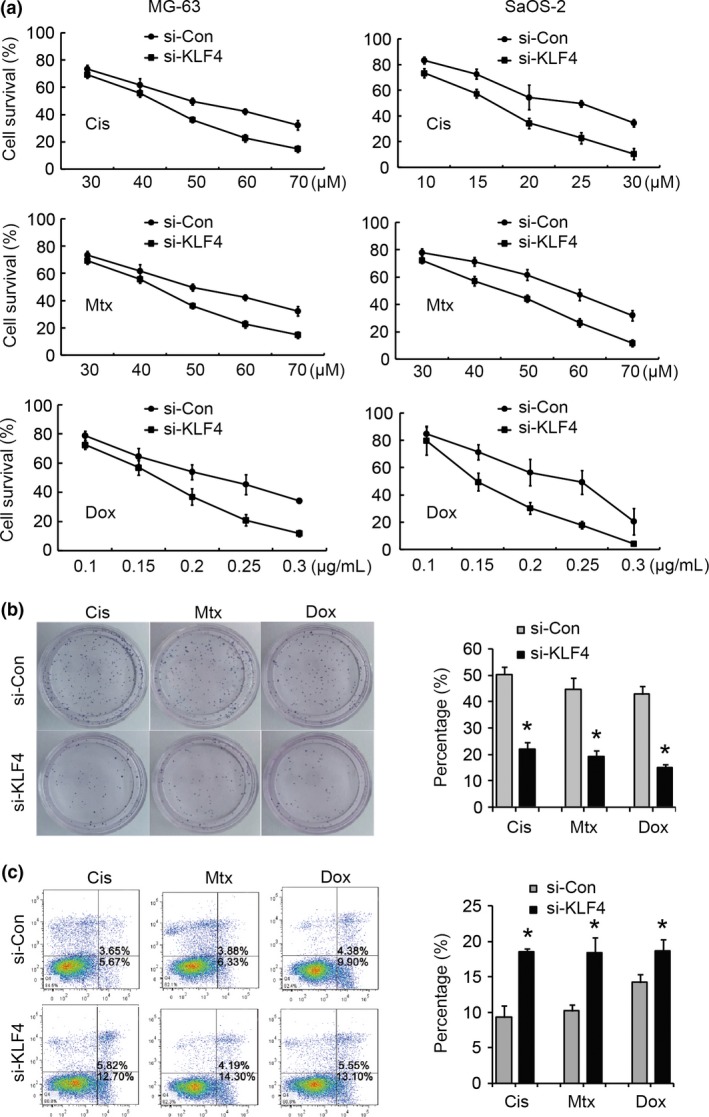
Suppression of Krüppel‐like factor 4 (KLF4) increases sensitivity to chemotherapy in vitro. KLF4 was knocked down by KLF4‐specific siRNA in MG‐63 and SaOS‐2 cells, respectively, and scrambled siRNA was taken as control. The impacts of knockdown of KLF4 on drug sensitivity at different doses of cisplatin (Cis), methotrexate (Mtx) and doxorubicin (Dox) were determined by MTT assays (a), colony formation assays (b) and FCM‐based cell apoptosis analysis (c). For colony formation assays and cell apoptosis analysis, MG‐63 and SaOS‐2 cells were treated with a final concentration of 50 μM cisplatin, 50 μM methotrexate and 0.2 μg/mL doxorubicin, respectively. Apoptotic cells consist of both early and late apoptotic cells. *P < 0.05 versus the control.
Figure 3.
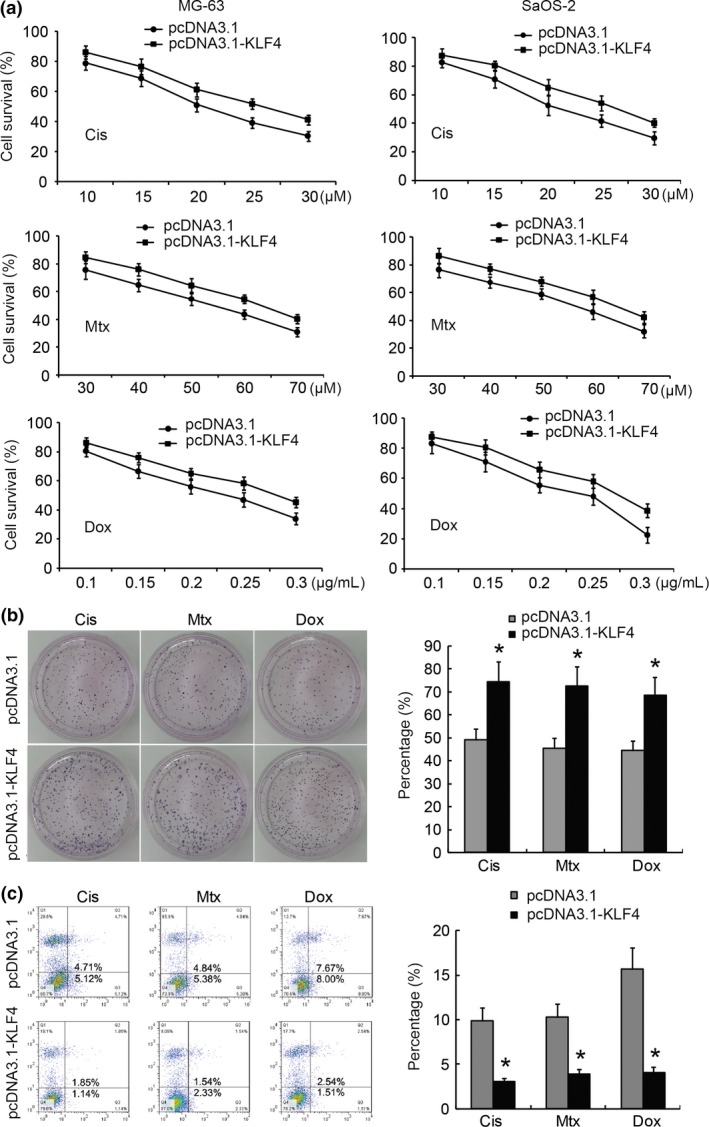
Overexpression of Krüppel‐like factor 4 (KLF4) increases sensitivity to chemotherapy in vitro. KLF4 was overexpressed in MG‐63 and SaOS‐2 cells, respectively, and the impacts of overexpression of KLF4 on drug sensitivity at different doses of cisplatin (Cis), methotrexate (Mtx) and doxorubicin (Dox) were determined by MTT assays (a), colony formation assays (b) and FCM‐based cell apoptosis analysis (c). For colony formation assays and cell apoptosis analysis, MG‐63 and SaOS‐2 cells were treated with a final concentration of 50 μM Cis, 50 μM Mtx and 0.2 μg/mL Dox, respectively. Apoptotic cells consist of both early and late apoptotic cells. *P < 0.05 versus the control.
Krüppel‐like factor 4 positively regulates high‐mobility group box 1 expression in osteosarcoma cells
Our previous report revealed that chemotherapy agents, including Cis, Mtx and Dox, induced HMGB1 expression in osteosarcoma cells.16 To determine whether KLF4 is responsible for HMGB1 transcriptional regulation, we examined the effect of KLF4 knockdown or overexpression on the expression of HMGB1 in MG‐63 and SaOS‐2 cells. qRT‐PCR and western blot analysis both revealed that knockdown of KLF4 inhibited mRNA and protein expression levels of HMGB1 in MG‐63 and SaOS‐2 cells, while overexpression of KLF4 resulted in increased HMGB1 expression (Fig. 4). This suggests that KLF4 may positively regulate HMGB1 expression in osteosarcoma cells.
Figure 4.
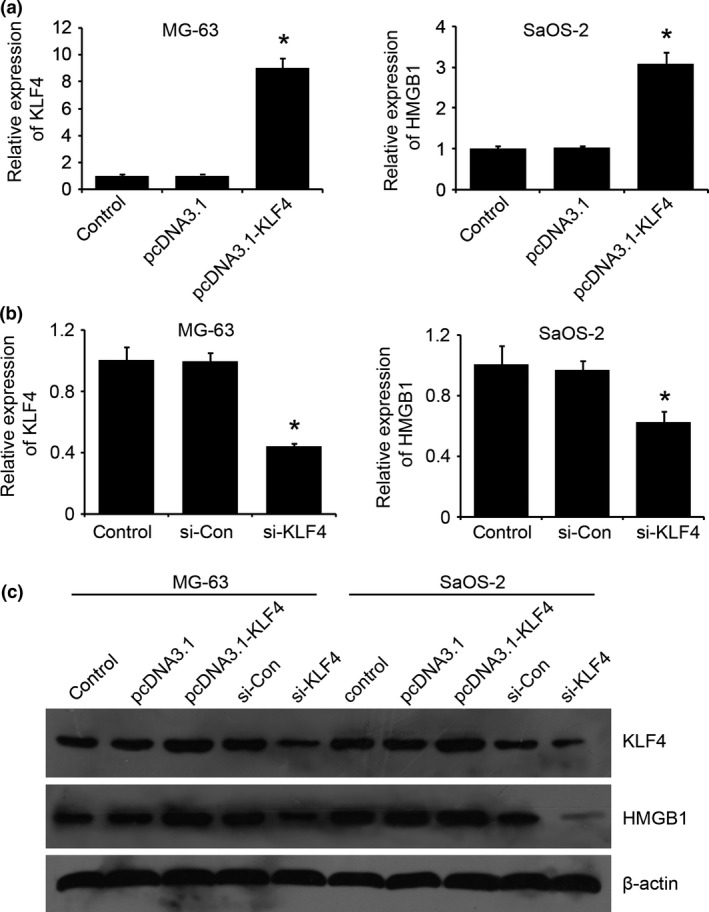
Krüppel‐like factor 4 (KLF4) positively regulates high‐mobility group box 1 (HMGB1) expression in osteosarcoma cells. (a,b) KLF4 was overexpressed by transfecting with KLF4 expression plasmids (pcDNA3.1‐KLF4) or knocked down by KLF4‐specific siRNA (si‐KLF4) in MG‐63 and SaOS‐2 cells, and qRT‐PCR was performed to validate the KLF4 mRNA levels. (c) The expression levels of KLF4 as well as HMGB1 protein were examined by western blot, which revealed that KLF4 positively regulates HMGB1 expression in osteosarcoma cells. *P < 0.05 versus the control.
Krüppel‐like factor 4 binds directly to the high‐mobility group box 1 promoter and regulates its transcription
To determine whether KLF4 directly regulates HMGB1 expression, we firstly performed promoter reporter assays to identify the KLF4‐responsive region on the HMGB1 promoter. MG‐63 cells were co‐transfected with pcDNA3.1‐KLF4 expression plasmid and pGL3 GFP reporter with five different fragments of the promoter region of HMGB1 (Fig. 5a). As shown in Figure 5b, progressive deletion of the promoter sequence from −1899 to −456 did not affect the HMGB1 promoter activity, but deletion between −456 and −240 significantly attenuated the promoter activity. The data indicate that the region between −456 and −240 is critical for responding to KLF4. In addition, EMSA also revealed that KLF4 could directly bind to the region between −456 and −240 on the HMGB1 promoter. As shown in Figure 6a, preincubation of nuclear extracts from Cis‐treated MG‐63 cells with MHGB1 probes resulted in DNA/protein complex formation, which was competed on with the addition of unlabeled (cold) probes (Fig. 6a, lanes 2–3), whereas preincubation with mutant unlabeled probes had no affect on DNA/protein complex formation (Fig. 6a, lane 4). Moreover, addition of KLF4 antibody resulted in the formation of a shift band (Fig. 6a, lane 5). These data suggest that KLF4 is able to bind to the KLF4‐responsive core region on the HMGB1 promoter. Finally, ChIP assays combined with quantitative PCR (ChIP‐qPCR) were performed in MG‐63 and SaOS‐2 cells treated with 50 uM Cis for 48 h and it was found that KLF4 could directly bind to the KLF4‐responsive core region on the HMGB1 promoter in MG‐63 and SaOS‐2 cells in vivo. As shown in Figure 6b, immunoprecipitation of the chromatin/protein complex with KLF4 resulted in the enrichment of KLF4 protein at the HMGB1 promoter region near the region between −456 and −240. The promoter sequence of HMGB1 was also analyzed using Matinspector Professional (www.genomatix.de) and TESS (www.cbil.upenn.edu), The KLF4‐binding site was existed at region −379 to −335 bp upstream the TSS of HMGB1. Based on our recent study and previous work,16 here we provide a model of KLF4‐induced chemotherapy in osteosarcoma cells, in which increased expression of KLF4 during chemotherapy activates HMGB1, thus further promoting autophagosome maturation and autophagy (Fig. 7). Therefore, KLF4/HMGB1 interaction is a potential therapeutic target for use in osteosarcoma.
Figure 5.
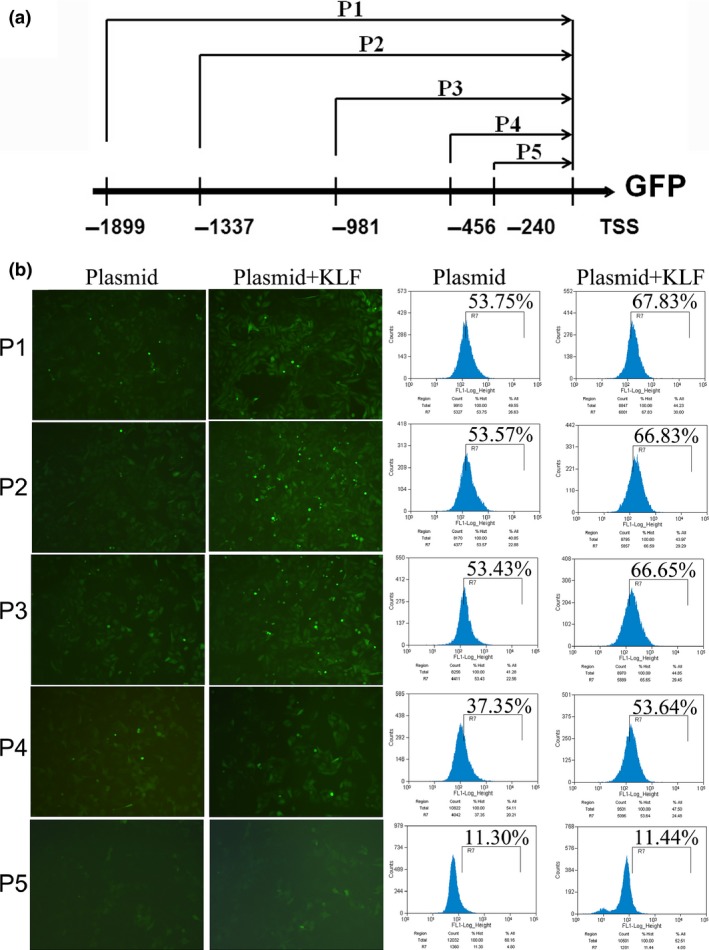
Identification of the Krüppel‐like factor 4 (KLF4)‐responsive core region on the high‐mobility group box 1 (HMGB1) promoter. (a) A diagram showing pGL3 GFP reporter with five different fragments of the promoter region of HMGB1. The translation starting point ATG is defined as position +1. (b) Effect of KLF4 on the HMGB1 promoter activity. MG‐63 cells were co‐transfected pGL3 GFP reporter with five different fragments of the promoter region of HMGB1 (P1‐P5) and pcDNA3.1‐KLF4 expression plasmid, and the GFP expression levels were quantified by flow cytometry.
Figure 6.
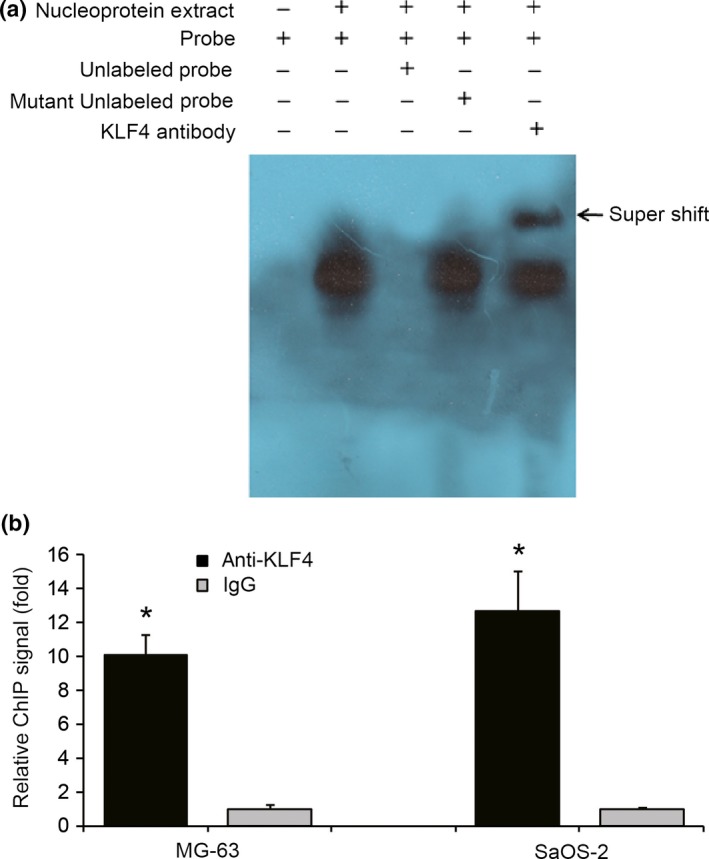
Krüppel‐like factor 4 (KLF4) binds to the promoter of high‐mobility group box 1 (HMGB1). (a) EMSA demonstrated KLF4 binding to HMGB1 promoter. Nucleoprotein extracts from cisplatin‐treated MG‐63 were prepared. Unlabeled (cold) probe and mutant unlabeled (cold) probe were added at an excess concentration (100×). Addition of KLF4 antibody resulted in the formation of shift band. (b) MG‐63 and SaOS‐2 cells were treated with 50 uM cisplatin for 48 h, then crosslinked and sonicated, and fragmented chromatins were immunoprecipitated using an anti‐KLF4 antibody as well as IgG antibody as controls. Immunoprecipitated DNA were purified and quantitatively analyzed by qRT‐PCR to determine the enrichment of KLF4 protein at the HMGB1 promoter region. *P < 0.05 versus the control.
Figure 7.
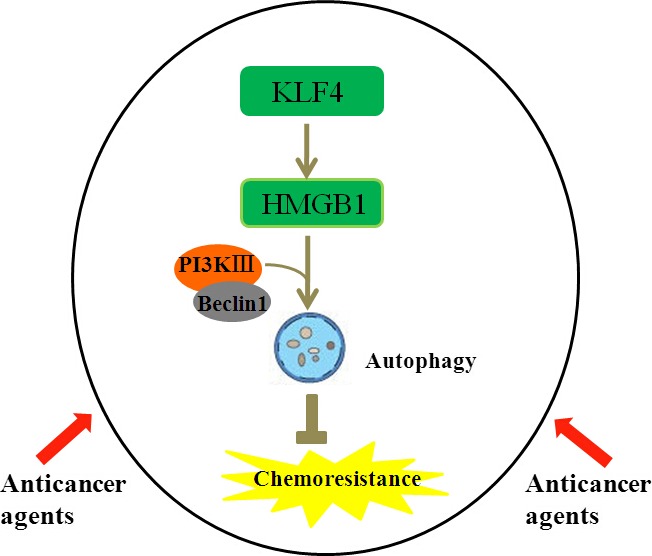
Krüppel‐like factor 4 (KLF4) promotes high‐mobility group box 1 (HMGB1)‐induced chemotherapy resistance in osteosarcoma cells. Anticancer agents such as cisplatin, doxorubicin and methotrexate are commonly used in the treatment of patients with osteosarcoma. These drugs increase expression of KLF4 in osteosarcoma cells by an unknown mechanism. Upregulated KLF4 increases HMGB1 expression by binding and activating the promoter of HMGB1. Increased HMGB1 binds BECN1 to promote the formation of the PI3K III‐Beclin 1 complex and stimulates autophagosome maturation and autophagy.
Discussion
Osteosarcoma is a high‐grade malignant bone tumor. Although the introduction of chemotherapy has reduced its mortality, the clinical effectiveness is limited by the emergence of drug resistance, which ultimately leads to poor therapeutic outcomes. Consequently, it is necessary to discover novel molecules regulating drug resistance to develop more effective targeted therapies. In the present study, we found that KLF4 expression was significantly increased in response to treatment with commonly used anticancer drugs in osteosarcoma cells. In addition, we demonstrated for the first time that knockdown of KLF4 increased sensitivity to these anticancer drugs in osteosarcoma cells in vitro, by decreasing clonogenic ability and increasing apoptosis. Finally, our data suggested that KLF4‐mediated drg resistance might be through directly binding to the HMGB1 promoter and positively regulating its transciption, which highlighted the significance of KLF4/HMGB1 interaction in regulating chemotherapy resistance in osteosarcoma cells.
Krüppel‐like factor 4 contains C2H2‐type zinc fingers at the C‐terminal and regulates the expression of target genes by binding to GC‐rich or CACCC promoters.5, 6 Recent studies have shown that the expression of KLF4 is altered in certain types of cancer. For example, KLF4 was downregulated in gastrointestinal (GI) tumors and negatively‐regulated GI cancer EMT,7 while the expression of KLF4 was upregulated in other types of cancers, such as breast cancer,19, 20 and head and neck squamous cell cancer,11 and functioned as an oncogene which enhanced tumor development and progression. Increasing evidence has revealed that KLF4 is associated with cancer cell chemotherapy resistance. To address the involvement of KLF4 in chemotherapy resistance in osteosarcoma cells, we first examined the expression alteration of KLF4 in osteosarcoma cells in response to anticancer reagents (Cis, Mtx and Dox), and found that these drugs significantly promoted increased KLF4 mRNA and protein expression levels. Further in vitro study revealed that suppression of KLF4 by siRNA increases sensitivity to chemotherapy. In contrast, when KLF4 was overexpressed in MG‐63 and SaOS‐2 cells, we observed an opposite effect on sensitivity to chemotherapy. Our data are consistent with previous reports that increased KLF4 contributed to drug resistance. For example, with a higher level of KLF4, hepatocarcinoma cell line T3A‐A3 was found to be more resistant to Cis than HepG2 cells, which have lower levels of KLF4 expression, and KLF4 knockdown reduced Cis resistance in T3A‐A3 cells.9 Tai et al. report that enforced KLF4 expression in head and neck squamous cell carcinoma (HNSCC) SAS cells significantly increased multi‐drug resistance.11 Taken together, KLF4 may have a potential role in the regulation of chemotherapy sensitivity of reported cancers and other cancers.
Our previous study revealed that HMGB1 promotes drug resistance in osteosarcoma;16 however, the regulation on HMGB1 expression remains unclear. As HMGB1 contains putative KLF4‐binding elements in its promoter region, we hypothesized that KLF4 might affect the expression of HMGB1. In this study, through KLF4 knockdown or overexpression, we found that KLF4 could positively regulate HMGB1 expression in osteosarcoma cells. HMGB1 promoter reporter assays, electrophoretic mobility shift assay and chromatin immunoprecipitation assays validated that KLF4 could directly bind to KLF4‐binding elements (three “CACCC” elements) on the region between −456 and −240 of the HMGB1 promoter in osteosarcoma cells. Our data are consistent with a previous report that KLF4 could bind to HMGB1 in mouse RAW264.7 macrophages when encounted LPS treatment.17
Autophagy is a catabolic process critical to maintaining cellular homeostasis and responding to various nutrient starvation or metabolic stress. Many anticancer reagents result in enhanced autophagy, which facilitates the cancer cells' resistance to chemotherapy treatment, and the abrogation of autophagy potentiates the re‐sensitization of therapeutic‐resistant cancer cells to the anticancer treatment.21, 22 Recent reports have demonstrated that HMGB1 is a critical regulator of autophagy in fibroblasts,23 leukemia,24, 25, 26 colon,27 pancreatic cancer28 and osteosarcoma cells.15, 16 Our previous study also demonstrated that knockdown of HMGB1 or inhibition of autophagy increases apoptosis, and reverses drug resistance in osteosarcoma cells.16 In this study, we validated that KLF4, upregulated during chemotherapy treatment in osteosarcoma cells, is a positive regulator of HMGB1.
In summary, our results demonstrate that KLF4 functions as an inducer of chemotherapy resistance in osteosarcoma, at least partially, by binding to and activating the HMGB1 promoter, which suggests that targeting KLF4/HMGB1 may be a therapeutic strategy for osteosarcoma chemotherapy.
Disclosure Statement
The authors have no conflict of interest to declare.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81272947 and 81302338).
Cancer Sci 107 (2016) 242–249
Funding Information
National Natural Science Foundation of China (Grant / Award Number: ‘81272947’, ‘81302338’).
References
- 1. Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology, and End Results Program. Cancer 2009; 115: 1531–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Siclari VA, Qin L. Targeting the osteosarcoma cancer stem cell. J Orthop Surg Res 2010; 5: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Garrett‐Sinha LA, Eberspaecher H, Seldin MF, de Crombrugghe B. A gene for a novel zinc‐finger protein expressed in differentiated epithelial cells and transiently in certain mesenchymal cells. J Biol Chem 1996; 271: 31384–90. [DOI] [PubMed] [Google Scholar]
- 4. Shields JM, Christy RJ, Yang VW. Identification and characterization of a gene encoding a gut‐enriched Kruppel‐like factor expressed during growth arrest. J Biol Chem 1996; 271: 20009–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bieker JJ. Kruppel‐like factors: Three fingers in many pies. J Biol Chem 2001; 276: 34355–8. [DOI] [PubMed] [Google Scholar]
- 6. Philipsen S, Suske G. A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res 1999; 27: 2991–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cui J, Shi M, Quan M, Xie K. Regulation of EMT by KLF4 in gastrointestinal cancer. Curr Cancer Drug Targets 2013; 13: 986–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ding B, Liu P, Liu W, Sun P, Wang CL. Emerging roles of kruppel‐like factor 4 in cancer and cancer stem cells. Asian Pac J Cancer Prev 2015; 16: 3629–33. [DOI] [PubMed] [Google Scholar]
- 9. Jia Y, Zhang W, Liu H, Peng L, Yang Z, Lou J. Inhibition of glutathione synthesis reverses Kruppel‐like factor 4‐mediated cisplatin resistance. Cancer Chemother Pharmacol 2012; 69: 377–85. [DOI] [PubMed] [Google Scholar]
- 10. Park JT, Chen X, Trope CG, Davidson B, Shih Ie M, Wang TL. Notch3 overexpression is related to the recurrence of ovarian cancer and confers resistance to carboplatin. Am J Pathol 2010; 177: 1087–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tai SK, Yang MH, Chang SY et al Persistent Kruppel‐like factor 4 expression predicts progression and poor prognosis of head and neck squamous cell carcinoma. Cancer Sci 2011; 102: 895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kang R, Zhang Q, Zeh HJ 3rd, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res 2013; 19: 4046–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tang D, Kang R, Zeh HJ 3rd, Lotze MT. High‐mobility group box 1 and cancer. Biochim Biophys Acta 2010; 1799: 131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tang D, Kang R, Zeh HJ 3rd, Lotze MT. High‐mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal 2011; 14: 1315–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang J, Liu K, Yu Y et al Targeting HMGB1‐mediated autophagy as a novel therapeutic strategy for osteosarcoma. Autophagy 2012; 8: 275–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang J, Ni J, Liu K et al HMGB1 promotes drug resistance in osteosarcoma. Cancer Res 2012; 72: 230–8. [DOI] [PubMed] [Google Scholar]
- 17. Liu J, Liu Y, Zhang H, Chen G, Wang K, Xiao X. KLF4 promotes the expression, translocation, and release of HMGB1 in RAW264.7 macrophages in response to LPS. Shock 2008; 30: 260–6. [DOI] [PubMed] [Google Scholar]
- 18. Zhang J, Chen YL, Ji G et al Sorafenib inhibits epithelial–mesenchymal transition through an epigenetic‐based mechanism in human lung epithelial cells. PLoS ONE 2013; 8: e64954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Foster KW, Frost AR, McKie‐Bell P et al Increase of GKLF messenger RNA and protein expression during progression of breast cancer. Cancer Res 2000; 60: 6488–95. [PubMed] [Google Scholar]
- 20. Pandya AY, Talley LI, Frost AR et al Nuclear localization of KLF4 is associated with an aggressive phenotype in early‐stage breast cancer. Clin Cancer Res 2004; 10: 2709–19. [DOI] [PubMed] [Google Scholar]
- 21. Chen S, Rehman SK, Zhang W, Wen A, Yao L, Zhang J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim Biophy Acta 2010; 1806: 220–9. [DOI] [PubMed] [Google Scholar]
- 22. Sehgal AR, Konig H, Johnson DE et al You eat what you are: Autophagy inhibition as a therapeutic strategy in leukemia. Leukemia 2015; 29: 517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amornsupak K, Insawang T, Thuwajit P, O‐Charoenrat P, Eccles SA, Thuwajit C. Cancer‐associated fibroblasts induce high mobility group box 1 and contribute to resistance to doxorubicin in breast cancer cells. BMC Cancer 2014; 14: 955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu L, Yang M, Kang R et al HMGB1‐induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 2011; 25: 23–31. [DOI] [PubMed] [Google Scholar]
- 25. Yang L, Yu Y, Kang R et al Up‐regulated autophagy by endogenous high mobility group box‐1 promotes chemoresistance in leukemia cells. Leuk Lymphoma 2012; 53: 315–22. [DOI] [PubMed] [Google Scholar]
- 26. Zhao M, Yang M, Yang L et al HMGB1 regulates autophagy through increasing transcriptional activities of JNK and ERK in human myeloid leukemia cells. BMB Rep 2011; 44: 601–6. [DOI] [PubMed] [Google Scholar]
- 27. Livesey KM, Kang R, Zeh HJ 3rd, Lotze MT, Tang D. Direct molecular interactions between HMGB1 and TP53 in colorectal cancer. Autophagy 2012; 8: 846–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kang R, Tang D, Schapiro NE et al The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ 2010; 17: 666–76. [DOI] [PMC free article] [PubMed] [Google Scholar]