

SUMMARY
Biology has been revolutionized by tools that allow the detection and characterization of protein-protein interactions (PPI). Förster resonance energy transfer (FRET)-based methods have become particularly attractive as they allow quantitative studies of PPIs within the convenient and relevant context of living cells. We describe here an approach that allows the rapid construction of live-cell, FRET-based binding curves using a commercially-available flow cytometer. We illustrate a simple method for absolutely calibrating the cytometer, validate our binding assay against the gold-standard isothermal calorimetry (ITC), and use flow cytometric FRET to uncover the structural and functional effects of the Cushing syndrome-causing mutation (L206R) on PKA’s catalytic subunit. We discover that this mutation not only differentially affects PKAcat’s binding to its multiple partners, but also impacts its rate of catalysis. These findings improve our mechanistic understanding of this disease-causing mutation, while illustrating the simplicity, general applicability and power of flow cytometric FRET.
INTRODUCTION
Interactions between proteins underlie much of the richness and complexity of biology. Proteins rarely act in isolation; rather, their function is often modulated, regulated or localized by their interactions with other proteins (Robinson et al., 2007). Many biological processes, from signal transduction to synaptic vesicle release, require the sequential binding and unbinding of a network of proteins to achieve their intended effects. Consequently, techniques for detecting and characterizing protein-protein interactions (PPI) have been instrumental in advancing our understanding of biology.
These methods are broadly divided into techniques that probe protein-protein interactions under extracellular or intracellular conditions. The former group includes biochemical assays, surface plasmon resonance (SPR) and isothermal calorimetry (ITC), which are generally highly quantitative, but are relatively low-throughput and can be limited by the demands of isolating and purifying sufficient quantities of high-quality protein. The latter group includes techniques like two-hybrid assays, which are higher-throughput and have the advantage of interrogating proteins within an intracellular milieu, but are less quantitative with higher rates of false-positives and false-negatives (Bruckner et al., 2009). More recently, Förster resonance energy transfer (FRET)-based methods have gained popularity (Kenworthy, 2001), as they combine the ease and biological relevance of in-cell approaches with the specificity and quantitative power of biochemical assays.
For instance, quantitative FRET has been used successfully to characterize PPI through the construction of live-cell FRET-based binding curves (Chen et al., 2007; Erickson et al., 2003). Imagine a scenario where proteins A and B bind to form AB with dissociation constant Kd, as illustrated in Figure 1A. Traditional approaches to characterize their binding would do so by manually titrating one species against a fixed concentration of the other whilst measuring the amount of bound product formed. In the live-cell FRET-based approach, proteins A and B are first tagged with fluorescent proteins that have high FRET potential, such as the GFP-variants Cerulean (Rizzo et al., 2004) (Cer) and Venus (Nagai et al., 2002) (Ven), and expressed in live cells. The stochastic expression of A and B in different cells then results in a natural titration of the two species, with the genetically-tagged Ven and Cer reporting the concentrations of A, B and AB within each cell through their direct fluorescence and the amount of FRET. With an appropriate binding model, a binding curve can then be constructed, with each cell contributing a point on the graph (Figure 1B). As a result, a good estimation of binding affinity typically requires interrogation of a large number of cells expressing a wide range of concentrations around the Kd. Prior attempts to apply this methodology utilized wide-field fluorescence microscopy to measure fluorescences and FRET efficiencies cell-by-cell (Chen et al., 2007; Erickson et al., 2003). Although this approach enabled FRET-based binding studies to be done on most fluorescence microscopes, obtaining each binding curve was a slow and laborious task where fluorescent cells had to be measured one at a time. Moreover, the microscope required frequent and onerous calibrations to offset instrument drift. These challenges placed limits on the scope of problems addressable by this method.
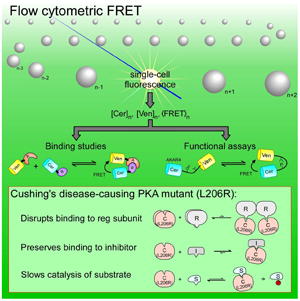
Figure 1. Flow cytometric FRET.
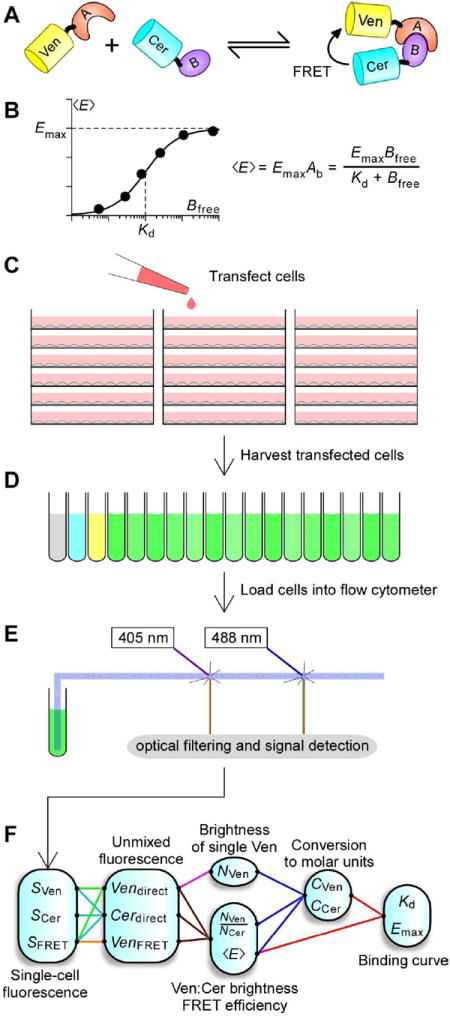
A. Cartoon depicting binding partners A and B tagged with fluorescent proteins Ven (monomeric Venus) and Cer (monomeric Cerulean). B. FRET binding curves are obtained by plotting 〈E〉 as a function of either Afree or Bfree. C.—E. HEK293(T) cells are cultured and transfected in 6 well plates, harvested into cytometer-compatible round bottom tubes, and loaded into the flow cytometer. F. Fluorescence signals are analyzed offline using custom software to yield the concentration of Cer, Ven, and 〈E〉, from which binding curves can be constructed.
The advent of the flow cytometer, an instrument capable of performing multichannel fluorescence measurements of single cells in a high-throughput manner, raised hopes that FRET-based protein interaction studies could be done more simply and efficiently. However, quantifying FRET with flow cytometry is challenging—acceptor photobleaching (Karpova et al., 2003) is impractical given the sub-millisecond excitation times, while flow cytometers capable of fluorescence lifetime measurements (Pinsky et al., 1993; Sun and Periasamy, 2015) are currently not widely available. Spectral methods for FRET determination predominate (Leavesley et al., 2013; Szaloki et al., 2013), though most groups only estimate FRET semi-quantitatively, tracking relative increases in donor quenching or sensitized acceptor emission, using this as a high-throughput approach to detect PPI’s (Banning et al., 2010; You et al., 2006). However, semi-quantitative FRET measurements are easily distorted by relative donor and acceptor expression levels (Berney and Danuser, 2003), potentially leading to erroneous estimations of binding. Moreover, proteins that bind weakly but have a favorable FRET conformation (e.g. aligned dipoles, shorter bound distance) might have a similar FRET efficiency to proteins that bind strongly albeit with unfavorable FRET conformations. Thus, the capability to measure binding in a dose-dependent, high-throughput manner is important, as it not only results in higher confidence of detecting actual binding, but also enables large scale studies of protein binding as with drug design and discovery, and structure-function analyses.
In this study, we describe a simple method to calibrate a commercially-available flow cytometer to perform quantitative FRET, using this approach both to construct FRET-based binding curves of fluorescent protein-tagged proteins and to exploit a FRET-based activity sensor. We validate our FRET-based binding assay by comparing our binding affinity estimates with those obtained from ITC, and use our method to discern the mechanism by which a recently discovered point mutation (L206R) in the PKA catalytic subunit results in adrenal Cushing’s syndrome. Our results demonstrate the ease, efficiency and broad utility of flow cytometric FRET, raising expectations that FRET-based binding assays will contribute to a new era of biological discovery.
RESULTS
Quantitative FRET with flow cytometry
With proper calibration and selection of fluorescent molecules, fluorescence signals from the flow cytometer can reveal the concentrations of each fluorescent species and the average FRET efficiency within each cell. We chose for our experiments Cer and Ven bearing monomeric A206K mutations (Zacharias et al., 2002) because of its favorable FRET properties, and compatibility with a dual violet and blue laser-equipped flow cytometer. A brief outline of our workflow is outlined in Figure 1C-F: HEK293(T) cells within 6 well plates are transfected with constructs containing Cer and/or Ven, and harvested 16-48 hours after transfection into round bottom tubes. Each round bottom tube is sequentially loaded onto the flow cytometer, where cells are focused into single stream and interrogated one-at-a-time by the two lasers. The resultant fluorescence and laser scatter are optically filtered into multiple channels, and detected, amplified and digitized. The recorded data is then analyzed using custom MATLAB code according to the sequence diagrammed in Figure 1F. Fluorescence signals from single cells (Figure S1) are converted through linear unmixing (see Experimental Procedures and Figure S2) into the three fundamental signals of the Cer/Ven system—direct Cer emission (Cerdirect), direct Ven emission (Vendirect), and sensitized Ven emission from FRET (VenFRET). Vendirect is directly proportional to the number of Ven molecules (NVen), while Cerdirect is proportional to the number of Cer molecules (NCer) not quenched by FRET as follows,
| (1) |
| (2) |
where g = γε with γ encompassing the instrument-dependent, and ε the fluorophore-dependent, aspects of fluorophore excitation, including laser power, attenuation by optical components, and the extinction coefficient at the laser wavelength. F = ξϕ with ϕ representing the fluorophoredependent, and ξ the instrument-dependent, terms of fluorophore emission, such as the quantum efficiency, optical filtering, as well as linear photodetection, amplification and digitization of fluorescence. NVen can be determined once the brightness of a single Ven molecule (gVen fVev) is known, but determination of NCer further requires an estimation of the average FRET efficiency 〈E〉. The average FRET efficiency is a function of g and f, and depending on whether it is measured with respect to the donor (Cer) or acceptor (Ven), can be expressed as (Supp. A):
| (3) |
| (4) |
Thus, absolute calibration of the flow cytometer depends on determination of g and f for both Ven and Cer. Once obtained, total concentrations of Cer (CCer) and Ven (CVen) are calculated from NCer and NVen after accounting for cellular volume (Figure S1F). Achieving precise accounting of the fluorescent species expressed in live cells then allows the construction of FRET-based binding curves.
Absolute calibration of the flow cytometer
Absolute calibration is achieved when fluorescence signals can be converted to an absolute number/concentration of fluorescent proteins with equations (1) and (2). Whereas the instrument-dependent terms γ and ξ are difficult to determine, the fluorophore-dependent terms ε and ϕ are readily accessible (Nagai et al., 2002; Rizzo et al., 2004). Fortunately, calibration standards for flow cytometry with known numbers of fluorochromes are commercially available, allowing determination of γ and ξ. We obtained beads conjugated with predetermined numbers of FITC molecules, as FITC is spectrally close to Venus. We can then absolutely calibrate our flow cytometer to the brightness of a single FITC molecule (gFITC fFITC) from the slope of the fluorescence vs number of FITC molecules relation (Figure 2A). Since the instrument is unchanged, γ and ξ are constant, and we can directly calculate for the brightness of a single Ven using known values of ε and ϕ for fully-matured Ven and FITC (Seybold et al., 1969; Shaner et al., 2005):
| (5) |
Figure 2. Calibrating the flow cytometer for FRET.
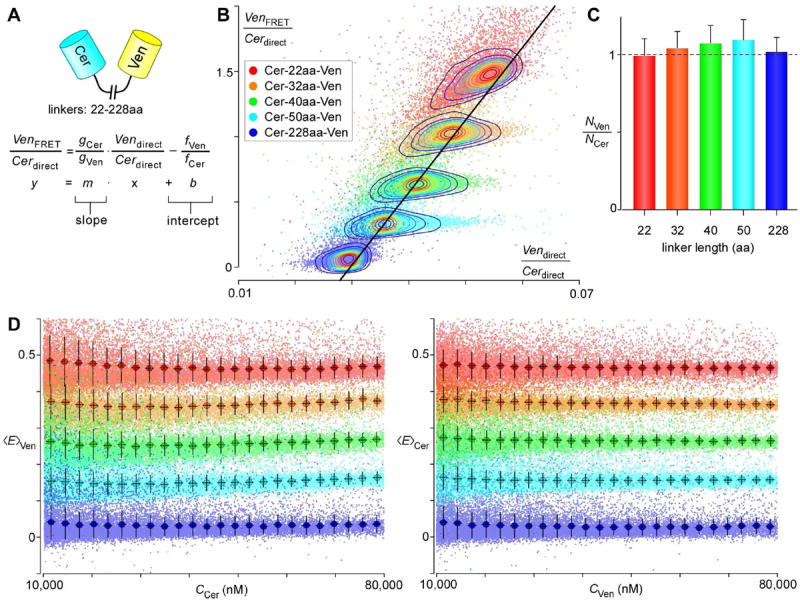
A. The ratios gCer/gVen and fVen/fCer can be discerned from Cer-Ven dimers of varying linker lengths using the following linear equation. B. Each cell expressing a particular Cer-Ven dimer is plotted as a single colored dot on the graph. Contour lines are overlaid on top of each Cer-Ven dimer to show the areas of highest density. C. The mean ± SD of NVen/NCer for each dimer is plotted. D. Plots of 〈E〉Ven and 〈E〉Cer for the five Cer-Ven dimers, with each point representing data from a single cell.
As seen with equations (3) and (4), calculating average FRET efficiencies requires the ratios of g and/or f for both Cer and Ven. These absorption and emission ratios can be determined (Chen et al., 2006; Nagy et al., 2005) with the following rationale: with Cer-Ven fusion proteins, the FRET efficiency should be the same regardless of the perspective from which it is measured, as long as there is one acceptor for every donor molecule (Supp. A). That is, since 〈E〉Cer = 〈E〉Ven in this scenario, equations (3) and (4) can be rearranged to obtain:
| (6) |
Equation (6) predicts a linear relationship from which the slope and intercept yields our desired ratios (Figure 2A). Plotting the appropriate quantities for five Cer-Ven dimers with varying linker lengths (from 22-228 amino acids), we initially obtained scatter plots with significant tails towards the origin (Figure S3A). Upon further examination, we found curiously that 〈E〉Cer, but not 〈E〉Ven, had unexpectedly large variance (Figure S3B,C). We hypothesized that this asymmetric result could be explained by the presence of a significant fraction of non-fluorescent immature Ven molecules, since near-invisible Ven would result in low 〈E〉Cer without affecting 〈E〉Ven. This result highlighted for us the importance of reducing this immature fraction for all our experiments, as the presence of immature fluorescent proteins leads to the underestimation of both the true FRET efficiency and the actual number of tagged proteins. We thus applied the protein synthesis inhibitor cycloheximide for 2 hours prior to flow cytometric interrogation, reasoning that we could reduce the immature fraction by halting new synthesis of fluorescent proteins and allowing existing Ven molecules to fully mature. Indeed, doing so dramatically reduced the variance of 〈E〉Cer, resulting in the graphs shown in Figure 2. The plot in Figure 2B enabled us to obtain estimates of 0.0178 and 1.65 for gVen/fCer and fVen/fCer. These quantities when multiplied also give the relative brightness of Cer and Ven, thus furnishing sufficient constraints to achieve absolute calibration. We tested our estimates of the absorption and emission ratios both by examining the NCer/NVen ratio, and by plotting 〈E〉Cer and 〈E〉Ven, of these dimers. Reassuringly, NCer/NVen is near 1 (Figure 2C), and 〈E〉Cer = 〈E〉Ven for each dimer, with dimers bearing shorter linker lengths having higher FRET efficiencies (Figure 2D).
Live cell FRET-based binding curves
We then sought to construct FRET-based binding curves with a test set of peptides whose binding had previously been well-characterized by ITC (Harkiolaki et al., 2003). We created constructs tagged with either Cer or Ven (Figure 3A) using the exact sequence of their peptides. After expressing them in HEK293 cells, we obtained single cell measurements of CCer, CVen and 〈E〉 using flow cytometry. Because proteins that bind to one another do so in hand-and-glove fashion, the bound complex adopts a conformation with FRET efficiency Emax. Since unbound proteins have negligible FRET,
| (7) |
| (8) |
where Ab and Db refer to the fraction of acceptor (Ven) and donor (Cer) molecules bound. Furthermore, the 1:1 binding model has the following analytic solution:
| (9) |
| (10) |
Thus, our approach is to iteratively minimize the difference between Ab as calculated using equations (7) and (9), or Db using equations (8) and (10), with the parameters Emax and Kd (Figure S4). Binding curves of Ven-SH3 (Gads, aa.256-322) with five binding partners (SLP-76, aa.231-243; Gab1, aa.515-527; USP8, aa.403-415; SLP-76 (D236K), aa.231-243; SLP-76 (K240R), aa.231-243), are presented in Figure 3B. Each binding curve is constructed from a single trial where >100,000 fluorescent cells were collected, with each cell contributing a dot on the plot. With each trial taking 3-5 minutes, data for all 5 constructs were collected in less than 30 minutes. Data from this large number of cells form a cloud charting the course of each binding curve with high resolution. Protein pairs with binding affinities within an order of magnitude of 1 μM can be maximally resolved, with points decorating the entirety of the binding curve (Figure 3B, green and blue). On the other hand, binding curves of weakly-binding (Figure 3B, cyan and red) or strongly-binding (Figure 3B, black) protein pairs would miss points at both extremes (Ab or Db near 0 or 1), because of the difficulty of expressing proteins at concentrations upwards of several hundred μM in live cells, or because of the difficulty of quantifying faintly fluorescent cells when proteins are weakly expressed. Nonetheless, binding curves that capture a significant fraction of the full range of binding can still yield good estimates of binding affinities through the minimization procedure described above (Figure S4). Analyzing binding data from all peptide pairs assessed, we obtained a relation between affinity estimates using flow cytometry and those using ITC (Figure 3C). The results impressively demonstrate that affinities estimated with both methods are highly correlated, though estimations with flow cytometry yielded affinities that were on average ~3-fold lower than those with ITC (Figure 3C, red line). The discrepancies between these two methods might be the result of small alterations in the energetics of binding due to the steric influence of the comparatively large fluorescent protein tags. They might also reflect the presence of invisible inhibitors of the binding reaction, due to endogenous inhibitor proteins and/or non-fluorescent immature fluorophores. Regardless of the cause, these results show that high-resolution FRET binding curves can be constructed quickly and efficiently with flow cytometry, yielding estimates of binding affinity comparable to those by in vitro studies.
Figure 3. Flow cytometric FRET binding curves.
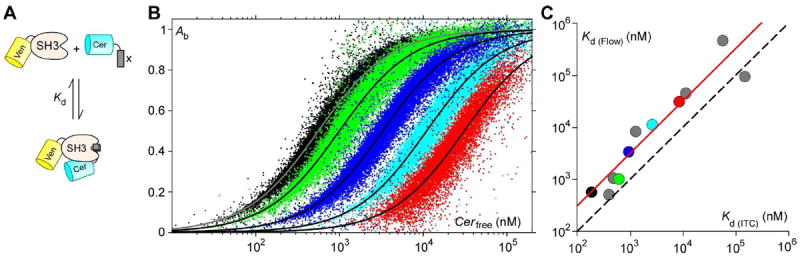
A. Diagram of the binding reaction between Ven-SH3 (Mona/Gads) and Cer-X, where X represents one of several binding partners of Mona/Gads. B. FRET binding curves for Ven-SH3 in complex with SLP-76, aa.231-243 (black), Gab1, aa.515-527 (green), USP8, aa.403-415 (blue), SLP-76 (D236K), aa.231-243 (cyan), and SLP-76 (K240R), aa.231-243 (red). Their respective binding affinities were Kd = 0.8, 1, 4, 7 and 24 μM, with Emax = 0.39, 0.4, 0.37, 0.35 and 0.35 respectively. C. Comparison between dissociation constants measured by flow cytometry (y-axis) and those measured by ITC (x-axis). On average, affinities measured by flow cytometry are ~3-fold less.
Regulation of Protein Kinase A—implications of a catalytic subunit mutant (L206R)
Recently, a number of papers were published describing the remarkable discovery that a large subset of patients with adrenal Cushing’s syndrome harbored the same point mutation (L206R) in the P+1 loop of the catalytic subunit of PKA (PKAcat) (Beuschlein et al., 2014; Cao et al., 2014; Di Dalmazi et al., 2014; Goh et al., 2014; Sato et al., 2014). This mutation was found to increase baseline PKA activity, with this gain of function largely thought to be due to disruption of binding between PKAcat and its regulatory subunit (PKAreg). However, there remained doubt as to whether some residual binding was preserved, given that PKI, whose binding interface with PKAcat structurally resembles PKAreg, is still able to bind to and inhibit the mutant PKAcat (Cao et al., 2014). We thus sought to resolve this question, using flow cytometric FRET to perform both binding and functional studies to understand how the L206R mutation affects PKAcat’s ability to interact with its various partners and thus alter its function.
We first tagged PKAcat and PKAreg with Ven and Cer, and performed our flow cytometric live cell FRET binding assay as before. In this case however, a 2:2 binding model (Supp. B) was used because the PKA holoenzyme forms a tetrameric complex (Taylor et al., 2013). The results indicate clear binding between the two wild-type subunits, with cells lining the binding curve (Figure 4A). The binding affinity was lower than anticipated (Cheng et al., 2001), likely because of the abundance of endogenous PKA in HEK293 cells. Nevertheless, flow cytometric analysis of the mutant L206R catalytic subunit demonstrated complete disruption of binding with the R1α regulatory domain throughout the entire range of expression (Figure 4B). Experiments with the R2β regulatory subunit yielded similar results (Figure S5A-B). We then assessed binding between PKAcat and its natural inhibitor PKI. A FRET binding study of PKI-Ven with the wildtype Cer-PKAcat was obtained (Figure 4C), which showed clear binding (Kd = 3.5 μM), though again with lower affinity than in vitro values(Scott et al., 1985) due to competition from endogenous proteins. Assessing binding of PKI with L206R PKAcat (Figure 4D), we found surprisingly that binding was preserved, though with weaker affinity (Kd = 10 μM). Thus, these experiments show that the interfacial L206R mutation differentially affects PKAcat’s interactions with two of its natural binding partners.
Figure 4. Differential effects on binding by PKAcat L206R.
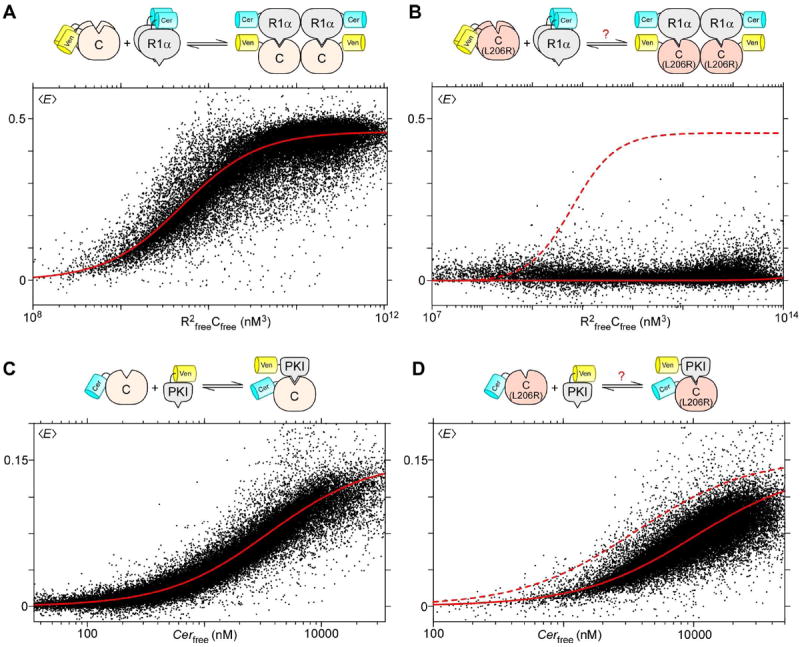
A. Fluorescent-protein tagged PKAcat and PKAreg fall on a FRET binding curve, with Emax = 0.46 and Kd ≈ 1.7 μM. B. Binding curves between PKAcat (L206R) and PKAreg show complete lack of binding. C. and D. Similarly, these scatter plots show binding curves between PKI and either PKAcat or PKAcat L206R, both of which bind with Emax = 0.16, and Kd = 3.5 and 10 μM respectively.
The partner-specific effects of L206R on PKAcat binding suggested that the rate of phosphorylation of its targets might also be affected, as transient binding of PKAcat to its substrate is a prerequisite to catalysis. We thus sought to examine the functional consequence of this mutant PKAcat using the FRET-based PKA activity sensor AKAR4 (Depry et al., 2011). Figure 5A diagrams a kinetic scheme of AKAR4, with AKAR4 phosphorylation requiring an intermediate enzyme-substrate bound state reached through dissociation constant k-1/k1 and departed through catalytic rate constant k2. Dephosphorylation by endogenous phosphatases is represented by a single rate constant k3. Because AKAR4 exists primarily in 2 states—unphosphorylated (Emin) or phosphorylated (Emax), the average FRET efficiency of a cell expressing AKAR4 is linearly related to the fraction of AKAR4 phosphorylated (P) by the following equation:
| (11) |
Emin can be approximated by expressing AKAR4 in live cells, and applying the PKA inhibitor H-89 (thus driving k2 to 0) whilst measuring FRET with flow cytometry (Figure 5B, black). On the other hand, we can get close to Emax by over-expressing PKAcat (Figure 5B, red). Surprisingly, 〈E〉, which should be independent of substrate concentration (equation 11), appeared to increase with AKAR4 expression. We reasoned this was likely because cells expressing more AKAR4 tend to have higher PKAcat expression as well. Indeed, PKAcat levels should influence P with the following relation (Supp. C):
| (12) |
where ymax = k2/(k2 + k3), Ke = Km(1 − ymax), and Km = (k−1 + k2)/k1. We estimated k3 with flow cytometry by examining the time-dependent decay of the mean population of cells co-expressing AKAR4 and PKAcat after exposure to 100 μM H-89 (Figure 5C). We examined cells expressing >4 μM AKAR4 to ensure all cells probed started with saturating PKA activity, and estimated from the exponential decay that k3 ~ 0.03 sec-1.
Figure 5. Enzyme kinetics of PKA L206R revealed through FRET-based PKA sensor.
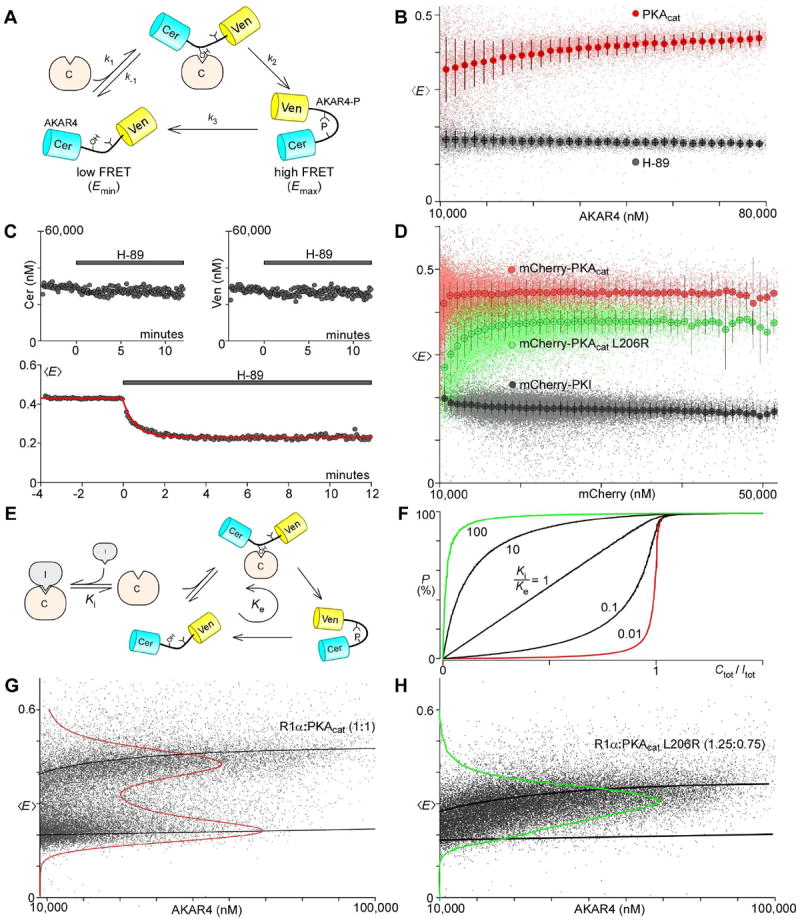
A. Diagram illustrating the different states of the PKA activity sensor AKAR4. In its unphosphorylated state, it has low FRET (Emin). Phosphorylation through a Michaelis-Menten scheme (k-1, k1, k2) results in a state of high FRET (Emax). Dephosphorylation (k3) occurs through endogenous phosphatases. B. Scatter plot of single cell FRET efficiency as a function of AKAR4 expression, either with PKAcat overexpressed (red) or with H-89 (100 μM) in the solution (black). C. H-89 (100 μM) was added to cells expressing AKAR4 and PKAcat at time zero. While measurements of Cer and Ven concentrations were unaffected by addition of H-89 (top), 〈E〉 declined exponentially with τ = 33 sec (red curve). D. Scatter plot of single cell FRET efficiency vs concentration of mCherry-tagged proteins. PKI-expressing cells (black) have 〈E〉 near Emin. Expression of mCherry-PKAcat (red) resulted in 〈E〉 increasing to near Emax, while expression of mCherry-PKAcat(L206R) resulted in a gentler ascent of 〈E〉 to a lower plateau. E. Scheme outlining the addition of an inhibitor. KI represents the dissociation constant of the inhibitor, whereas Ke represents the “aggregate dissociation constant” for the AKAR4 system. F. Solution to the model diagrammed in E. where P (percent of AKAR4 phosphorylated) is plotted versus Ctot/Itot (PKAcat to inhibitor ratio) for different values of KI/Ke. G. and H. Single cell FRET efficiencies of cells transfected with PKAreg together with either PKAcat or PKAcat(L206R). Mean results of either PKAcat (L206R in H.) or PKAreg alone are represented by thick black lines, with their kernel density estimates overlaid in red.
Equation (12) suggested a revealing relationship between AKAR4 phosphorylation and PKAcat, and we thus sought to acquire a means of tracking cellular PKAcat levels, while continuing to monitor PKA activity with Cer-Ven FRET. We found that fully-matured mCherry fulfilled this role beautifully, as it was excitable with the 488 nm laser with insignificant bleedthrough into other channels (Figure S2I-K). Additionally, mCherry levels can be estimated after accounting for Ven spectral overlap, and single fluorophore brightness calibration with a Cer-mCherry dimer (Figure S2L). Having added additional bandwidth to our flow cytometric system, we first co-expressed in live cells mCherry-tagged PKI with AKAR4 and obtained a largely flat FRET vs mCherry-PKI curve, where we find that increasing expression of mCherry-PKI depressed 〈E〉 to Emin (Figure 5D, black). On the other hand, co-expression of mCherry-tagged PKAcat and AKAR4 yielded a curve (Figure 5D, red) that satisfies equation (12) with Pmax = 0.445 and Ke = 0.1 μM. A similar experiment with mCherry-tagged mutant (L206R) PKAcat instead yielded a markedly different curve (Figure 5D, green) with Pmax,mut = 0.385 and Ke,mut = 12 μM. The reduction in Pmax,mut and 100-fold increase in Ke,mut suggests that the L206R mutation not only weakens the catalytic subunit’s interaction with AKAR4 (k1,mut ≪ k1), but also impairs catalysis (k2,mut < k2). Since k2 ≫ k-1, our estimates of k3 allow us to approximate k1 ≈ k3/Ke = 1×105 M-1sec-1 and k1,mut ≈ 0.8×103 M-1sec-1. On the other hand, precise quantification of k2 is more difficult, with Emax too close to Pmax (ymax ~ 1) to be resolved. Nonetheless, based on prior biochemical studies (Moore et al., 2003) we can assume k2 ~ 20 sec-1, which yields: k2,mut ~ 0.1 sec-1.
Addition of an “inhibitor”, such as PKAreg or PKI, to the PKAcat + AKAR4 system results in a scheme diagrammed in Figure 5E. The inhibitor competitively binds to PKAcat, effectively reducing the number of catalytic molecules that can phosphorylate AKAR4. The solution to this system reveals an interesting feature of the PKA regulatory system (Figure 5F)—the fraction of AKAR4 phosphorylated depends not only on the relative number of catalytic and regulatory subunits (Ctot/Itot), but also on the relative rates of PKAreg-PKAcat binding and AKAR4 cycling (KI/Ke) (Supp. D). It is intuitive that Ctot/Itot < 1 is required for baseline suppression of PKAcat activity since there has to be at least one regulatory subunit for every catalytic subunit. On the other hand, KI/Ke reflects the competition between PKAreg and substrate for PKAcat, describing the predilection of PKAcat for either participating in the phosphorylation or quiescent pathway. When KI/Ke ≪ 1 (Figure 5F, red), PKAcat strongly prefers PKAreg, and so is mostly inactive even when Ctot/Itot is just under 1. On the other hand, if KI/Ke ≫ 1 (Figure 5F, green), as would be the case with PKAcat and (cAMP)2-PKAreg, PKAcat is mostly active, even when outnumbered by regulatory subunits. Furthermore, this model predicts that examining AKAR4 phosphorylation when both PKAcat and R1α are expressed in HEK293 cells would yield data that allows interpretation of the relative binding affinities of PKAcat. For instance, if KI/Ke ≪ 1, we would expect a bimodal FRET distribution with most cells displaying either high or low FRET, since the steepness of the P vs Ctot/Itot curve (Figure 5F, red) indicates that only a small fraction of cells would lie within the narrow range of Ctot/Itot necessary for intermediate AKAR4 activity. Indeed, when we transfect untagged PKAcat and R1α with AKAR4, we observe a bimodal distribution of FRET activity (Figure 5G), suggesting that KI/Ke ≪ 1. Similarly, co-expression of PKI with wild-type PKAcat yields a bimodal FRET distribution (Figure S5C), again consistent with KI/Ke ≪ 1. In contrast, co-expression of R1α with mutant L206R PKAcat produced a unimodal distribution (Figure 5H), suggesting that even though the L206R mutation weakened PKAcat association with AKAR4, disruption to R1α binding was much more severe, resulting in KI/Ke > 1 and an inability to restrain PKAcat activity. In contrast, co-expression of PKI with mutant L206R PKAcat, recapitulates a bimodal distribution (Figure S5D), showing that PKI remains capable of blocking L206R PKAcat activity. This is consistent with the earlier findings of weakened yet persistent binding between mutant PKAcat and PKI (Figure 4D). Thus, these functional experiments reveal that the mutation L206R in the critical P+1 loop of PKAcat is consequential not only with regards to its interactions with its regulators and inhibitors, but also its substrates.
DISCUSSION
This study reports the use of the flow cytometer for quantitative live-cell FRET, demonstrating its utility both in providing a high-throughput assay for measuring protein-protein binding affinities, and also in revealing biological mechanisms through population-based analyses with FRET-based sensors. We show a simple procedure for absolute calibration of a commercially-available flow cytometer, using it to construct live-cell FRET binding curves that yield binding affinities comparable to those from ITC. With flow cytometric FRET, we demonstrate that a mutation in PKAcat differentially affects its interaction with its binding partners, resulting in unregulated but slowed catalysis of certain substrates. These results demonstrate that flow cytometric FRET represents a simple, fast and powerful approach to interrogating protein binding and function, providing an important adjunct to traditional biochemical tools. The increasing affordability, accessibility and capabilities of flow cytometry in recent years raise expectations that this methodology will find widespread use and development for years to come.
Strengths and Limitations of Flow Cytometric FRET
The major advantages of this strategy over in vitro approaches stem from the use of living mammalian cells as biochemical reactors. Proteins expressed in living mammalian cells receive the support of chaperones and accessory proteins, maturing with appropriate post-translational modifications, and thus tend to remain more stable within their native cellular contexts than without. With lengthy and costly protein purifications rendered unnecessary, binding assays can be accomplished with greater ease and speed, making larger scale studies of protein-protein interactions under various pharmacologic or genetic manipulations more practical. In addition to protein binding, flow cytometric FRET also permits population-level analyses of biological function in living cells via the ever-growing list of FRET-based biosensors. All in all, it is the strict accounting of every fluorescent species in addition to the FRET efficiency within each cell afforded by this approach that enables the engagement of biological problems from a quantitative and model-based perspective. In this light, our work here represents a few of many possible applications of flow cytometric FRET, and we anticipate that uses beyond those suggested here will continue to surface, especially when coupled with cytometers with sorting capabilities, or automated flow cytometric platforms.
Assaying binding with flow cytometric FRET is not without limitations. These include the necessity of altering proteins with fluorescent protein tags, and the interference by unaccounted endogenous proteins. In many cases, the former problem is surmountable as protein tags can be designed such that they do not affect native protein function, in accordance with the modular nature of many proteins. The unwanted contribution of endogenous proteins, on the other hand, typically have their highest impact on high affinity interactions, as their effects can usually be minimized by the overexpression of fluorescently-tagged recombinant proteins. If necessary, genetic strategies for knocking down endogenous protein levels can also be employed. Finally, the high-throughput measurement of single-cell fluorescence imposes additional challenges on flow cytometric FRET. When compared to microscope-based FRET, flow cytometric FRET performs more poorly with dimly fluorescent cells due to the cytometer’s lower signal-to-noise ratio at low fluorescent intensities. This causes the flow cytometric measurement of FRET from poorly-expressing proteins, such as might be the case in some subcellularly-localized proteins, to be enveloped in noise. However, this is a technological hurdle that we anticipate will be overcome with brighter fluorescent reporters and more sensitive and precise instruments. A more fundamental limitation of flow cytometric FRET relates to the need to detach cells from their substrates for spectroscopic interrogation. Not only are unique cell features such as cell morphology and local density lost when cells are dissociated, but resuspending naturally-adherent cells in solution also alters aspects of a cell’s biology, potentially affecting the proteins under study. Although these issues likely affect only a minority of projects, they should be seriously considered before any flow cytometric experiment.
Implications of L206R for PKA function
We determined that the Cushing’s disease-causing L206R mutation within the P+1 loop of PKAcat completely disrupts its interaction with PKAreg, while largely preserving its binding to PKI. The differential effect on binding of L206R is consistent with prior studies showing the ability of PKI but not PKAreg to suppress the activity of L206R PKAcat (Cao et al., 2014; Cheung et al., 2015). Additionally, monitoring PKA activity using the FRET-based PKA sensor AKAR4 further reveals that this mutant PKAcat has not only weakened binding to AKAR4, but also reduced catalytic activity. These findings are not unexpected, given that mutagenesis of other residues within the P+1 loop have been shown to interfere with catalytic activity (Moore et al., 2003). However, they do conflict with reports that showed equal (Calebiro et al., 2014) or elevated (Cheung et al., 2015) catalysis by L206R PKAcat with the synthetic substrate Kemptide. The reason for this discrepancy is not clear, but may be due to the differential effects of the L206R mutation on different PKA substrates. Finally, we described a model of AKAR4 phosphorylation incorporating both substrate and inhibitor that allowed us to assess the relative “affinities” for substrate vs inhibitor (KI/Ke) through the distribution of FRET efficiencies. These experiments independently confirmed the disparate effects on binding of PKAcat L206R, and also illustrated the importance of an intermediary Ke for optimal switching of PKA activity. Overall, these findings hold key implications for understanding the pathogenesis of PKAcat L206R mediated disease, and guiding future therapeutic strategies.
EXPERIMENTAL PROCEDURES
Molecular biology
All cDNA plasmid constructs used in this study have transcription driven by the CMV promoter. Plasmids encoding monomeric Cerulean (Cer-C1) and Venus (Ven-C1) as well as dimers C32V, C40V, C50V and CTV were gifts from Steven Vogel (NIH). Cer-N3, Ven-N3 and mCherry-N3, with multiple cloning sites N-terminal to the fluorescent proteins (FP), were generated through PCR amplification of the FPs (startless) using primers P01/P02 (primers listed in Supp. E) from Cer-C1, Ven-C1 and mCherry-C1 (Clontech) and insertion into EGFP-N3 (Clontech) with BamHI/BsrGI. These C1 and N3 plasmids serve as the backbones to which other proteins can be fused to Cer or Ven at either terminus. C22V and C22R were generated by cutting and inserting from Ven-N3 or mCherry-N3 into Cer-C1 with BglII/XbaI. The carboxy-terminal SH3 domain from Mona/Gads was PCR amplified from mouse tail DNA with P03/P04, and inserted into Ven- C1 with BglII/HindIII. Its various Cer-tagged 13 amino-acid binding partners were synthesized by annealing, cutting and inserting complementary DNA primer pairs (P05-P28) bearing the desired sequence into Cer-C1 with XhoI/HindIII. Plasmids containing human PKAcat (pDONR223-PRKACA), PKAreg (pDONR223-PRKAR1A and pDONR223-PRKAR2B) and PKI (pRSV-PKIv2) were obtained from addgene.org, while AKAR4 was a gift from Jin Zhang (Hopkins). PKAcat was PCR amplified with P29/P30 and cut with BamHI/HindIII before insertion into Cer-C1, Ven-C1 and mCherry-C1 using BglII/HindIII to make Cer-PKAcat, Ven- PKAcat and mCherry-PKAcat. The L206R mutation in PKAcat was made using overlap-extension PCR with overlap primers P31/P32 and flank primers P29/P30, with reinsertion using EcoRV/XhoI. PKAreg-Cer and PKAreg-Ven were made by PCR amplification of PKAreg R1α (P33/P34) or PKAreg R2β (P35/P36) and insertion into Cer-N3 and Ven-N3 with KpnI/BamHI or KpnI/BamHI/BglII respectively. PKI-Cer, PKI-Ven and PKI-mCherry were constructed by PCR amplification using P37/P38 and insertion into Cer-N3, Ven-N3 or mCherry-N3 using KpnI/BamHI. Untagged PKAcat was made by PCR amplification using P39/P40 and insertion into pcDNA3.1 (Invitrogen) using KpnI/XbaI, while untagged PKAreg (R1α) was made by cutting PKAreg-Cer (R1α) and inserting into Cer-C1 with KpnI/BamHI. All constructs were verified by restriction digest and Sanger sequencing.
Cell culture, transfection and preparation for flow cytometry
HEK293 and HEK293T cells were cultured using sterile techniques on 6 well plates at 37°C with 5% CO2. Our culture media contained Dulbecco’s modified Eagles’ medium (DMEM; Invitrogen) supplemented with 10% FBS, 5 mM L-glutamine, 50 units/ml penicillin and 50 μg/ml gentamicin. Cells within each well were transiently transfected at 75% confluency using polyethylenimine (Polysciences) with a 3:1 (PEI:DNA) ratio. 16-48 hours after transfection, cells were trypsinized and transferred to 1.6 ml microcentrifuge tubes where they were washed twice and then resuspended in 500 μl of HEPES-buffered Tyrode’s solution. Cells were then transferred to 5 mL round bottom polystyrene tubes for use in the flow cytometer. In most experiments, 100 μM cycloheximide was added to cells 2-4 hours prior to flow cytometric interrogation to halt new protein synthesis, allowing immature fluorescent proteins to fully mature.
Flow cytometry
We used an Attune® acoustic focusing flow cytometer (Life Technologies) equipped with violet (405 nm) and blue (488 nm) lasers. Forward and side scatter signals were developed from the violet laser and used to gate for single, healthy cells. SCer, and SFRET channels were taken from the violet laser, while SFRET and Sred were off the blue laser. Optical filters used for SCer, SVen, SFRET and Sred were 480/50, 530/30, 530/30 and 600LP respectively. Flow cytometric signals were collected using “high-sensitivity” mode at a flow rate of 100 μl/min. Cells are illuminated for 40 μs by each of the two lasers, with a time-of-flight of 1.2 ms from the violet to the blue laser. We confirmed that there was no appreciable photoactivation of Venus by violet (405nm) light (data not shown). Maintenance and performance tracking of the flow cytometer were performed with Attune® performance tracking beads (Life Technologies) in accordance with manufacturer’s specifications. FITC beads used for absolute calibration and for calibration across PMT voltages were Quantum™ FITC-5 MESF (Bangs Laboratories, Inc). Fluorescence data was exported as FCS files for further processing with custom MATLAB® (Mathworks) software.
Several control experiments were prepared every time the flow cytometer was used: cells exposed to PEI but without plasmids served as our blank controls, and used to define the background level for each channel (BGCer, BGVen, BGFRET, BGRed); Cer, Ven and mCherry each expressed by itself gives the proportionality constants (RD1, RD2, RD3, RA1, RA2, RC) for spectral unmixing; Cer and Ven expressed together provides an estimate of the concentration-dependent collisional FRET (Figure S2M-R). The series of Cer-Ven dimers for gA/gD and fA/fD calibration were performed monthly, while calibration with Quantum™ beads were used only when the flow cytometer has drifted significantly from baseline, as determined by serial performance testing.
Data processing
Our custom software serves 3 functions: 1) as a means to organize experimental constructs by name, type and date, 2) to determine and store the necessary instrument- and fluorophoredependent constants, and 3) to transform the raw fluorescence values into FRET efficiencies and binding curves, as outlined in Figure 1F. For all samples, single cells are first selected through sequential gating with forward- and side-scatter plots (Figure S1A-C). Background levels are determined from blank cells by taking the mean fluorescence levels of each channel after discarding 1% of outliers, and used for background subtraction in all subsequent analysis (Figure S1D-E). RA1 and RA2 were taken from Ven-only cells (Figure S2A) by taking the slope of a linear regression of SFRET vs SVen, and SFRET vs SRed (Figure S2C-D). Similarly, RD1, RD2 and RD3 are determined Cer-only cells with SFRET vs SCer, SVen vs SCer, and SRed vs SCer respectively (Figure S2E-H). Importantly, these relations between channels become supralinear at very high fluorescence intensities (Figure S2B), likely due to inner filter effects, and so highly expressing cells were excluded from analysis. Moreover, to minimize the influence of outliers, binned median fluorescences were used to calculate the linear regressions from which the proportionality constants (RD1, RD2, RD3, RA1, RA2) are determined. The unmixed signals (Cerdirect, Vendirect, VenFRET and Cherry) were then obtained by inverting the following relation:
These unmixed quantities allow calibration of gA/gD and fA/fD using a series of Cer-Ven dimers and equation (6), calculation of mean cellular FRET efficiencies according to equations (3) or (4), as well as determination of NVen and NCer using equations (1) and (2). As detailed in the main text, the absolute brightness of Ven (gAfA) was determined via comparison with FITC beads, and the brightness of Cer (gDfD) assessed relative to that of Ven through the Cer-Ven dimer series. We also estimated the brightness of mCherry (gChfCh) relative to Cer using a Cer-mCherry dimer (Figure S2L), which enabled us to calculate NCherry from Cherry. It is especially important to determine experimentally the brightness of Cer relative to that of Ven because an accurate gauge of the brightness ratio is so crucial to calculating the FRET efficiency. Finally, we estimated the volume of an average HEK293 cell by pipetting trypsinized HEK293 cells onto a glass coverslip, and measuring the radii of these loosely attached spherical cells with confocal microscopy (Figure S1F).
Supplementary Material
HIGHLIGHTS.
Absolute calibration of commercially-available flow cytometer for quantitative FRET
Flow cytometry can generate FRET-based binding curves, validated by ITC
Mutant PKAcat (L206R) associated with Cushing’s syndrome cannot bind PKAreg
L206R differentially affects PKAcat binding to its partners, and slows catalysis
Acknowledgments
We thank S. Vogel for his gift of the Cer-Ven constructs, J. Zhang for her gift of AKAR4, and T. Inoue for helpful discussions and comments on the manuscript. We are grateful to Wanjun Yang for her assistance with cell culture, and other members of the Calcium Signals Lab for insightful contributions while this work was at an early stage. We are forever indebted to our mentor, the late D.T. Yue, for his leadership and for showing us how to approach science with grace and humility. This work is supported by MSTP funding (S.R.L.) and NIH grant R01 MH065531 (D.T.Y.).
Footnotes
AUTHOR CONTRIBUTIONS
S.R.L, L.J.S. and D.T.Y. conceived and designed the study; S.R.L. and L.J.S. performed all experiments; S.R.L. processed and analyzed all data and wrote the manuscript with input from L.J.S.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four appendices, list of primers, and five figures, and can be found with this article online.
We have no conflicts of interests to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Shin-Rong Lee, Email: slee222@jhmi.edu.
Lingjie Sang, Email: lsang1@jhmi.edu.
David T. Yue, Email: dyue@jhmi.edu.
References
- Banning C, Votteler J, Hoffmann D, Koppensteiner H, Warmer M, Reimer R, Kirchhoff F, Schubert U, Hauber J, Schindler M. A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PloS one. 2010;5:e9344. doi: 10.1371/journal.pone.0009344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berney C, Danuser G. FRET or no FRET: a quantitative comparison. Biophysical journal. 2003;84:3992–4010. doi: 10.1016/S0006-3495(03)75126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuschlein F, Fassnacht M, Assie G, Calebiro D, Stratakis CA, Osswald A, Ronchi CL, Wieland T, Sbiera S, Faucz FR, et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. The New England journal of medicine. 2014;370:1019–1028. doi: 10.1056/NEJMoa1310359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruckner A, Polge C, Lentze N, Auerbach D, Schlattner U. Yeast two-hybrid, a powerful tool for systems biology. International journal of molecular sciences. 2009;10:2763–2788. doi: 10.3390/ijms10062763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D, Hannawacker A, Lyga S, Bathon K, Zabel U, Ronchi C, Beuschlein F, Reincke M, Lorenz K, Allolio B, et al. PKA catalytic subunit mutations in adrenocortical Cushing’s adenoma impair association with the regulatory subunit. Nature communications. 2014;5:5680. doi: 10.1038/ncomms6680. [DOI] [PubMed] [Google Scholar]
- Cao Y, He M, Gao Z, Peng Y, Li Y, Li L, Zhou W, Li X, Zhong X, Lei Y, et al. Activating Hotspot L205R Mutation in PRKACA and Adrenal Cushing’s Syndrome. Science. 2014 doi: 10.1126/science.1249480. [DOI] [PubMed] [Google Scholar]
- Chen H, Puhl HL, 3rd, Ikeda SR. Estimating protein-protein interaction affinity in living cells using quantitative Forster resonance energy transfer measurements. Journal of biomedical optics. 2007;12:054011. doi: 10.1117/1.2799171. [DOI] [PubMed] [Google Scholar]
- Chen H, Puhl HL, 3rd, Koushik SV, Vogel SS, Ikeda SR. Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophysical journal. 2006;91:L39–41. doi: 10.1529/biophysj.106.088773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Phelps C, Taylor SS. Differential binding of cAMP-dependent protein kinase regulatory subunit isoforms Ialpha and IIbeta to the catalytic subunit. The Journal of biological chemistry. 2001;276:4102–4108. doi: 10.1074/jbc.M006447200. [DOI] [PubMed] [Google Scholar]
- Cheung J, Ginter C, Cassidy M, Franklin MC, Rudolph MJ, Robine N, Darnell RB, Hendrickson WA. Structural insights into mis-regulation of protein kinase A in human tumors. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:1374–1379. doi: 10.1073/pnas.1424206112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depry C, Allen MD, Zhang J. Visualization of PKA activity in plasma membrane microdomains. Molecular bioSystems. 2011;7:52–58. doi: 10.1039/c0mb00079e. [DOI] [PubMed] [Google Scholar]
- Di Dalmazi G, Kisker C, Calebiro D, Mannelli M, Canu L, Arnaldi G, Quinkler M, Rayes N, Tabarin A, Laure Jullie M, et al. Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study. The Journal of clinical endocrinology and metabolism. 2014;99:E2093–2100. doi: 10.1210/jc.2014-2152. [DOI] [PubMed] [Google Scholar]
- Erickson MG, Liang H, Mori MX, Yue DT. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 2003;39:97–107. doi: 10.1016/s0896-6273(03)00395-7. [DOI] [PubMed] [Google Scholar]
- Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, Kuntsman JW, Korah R, Suttorp AC, Dietrich D, et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nature genetics. 2014 doi: 10.1038/ng.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkiolaki M, Lewitzky M, Gilbert RJ, Jones EY, Bourette RP, Mouchiroud G, Sondermann H, Moarefi I, Feller SM. Structural basis for SH3 domain-mediated high-affinity binding between Mona/Gads and SLP-76. The EMBO journal. 2003;22:2571–2582. doi: 10.1093/emboj/cdg258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova TS, Baumann CT, He L, Wu X, Grammer A, Lipsky P, Hager GL, McNally JG. Fluorescence resonance energy transfer from cyan to yellow fluorescent protein detected by acceptor photobleaching using confocal microscopy and a single laser. Journal of microscopy. 2003;209:56–70. doi: 10.1046/j.1365-2818.2003.01100.x. [DOI] [PubMed] [Google Scholar]
- Kenworthy AK. Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods. 2001;24:289–296. doi: 10.1006/meth.2001.1189. [DOI] [PubMed] [Google Scholar]
- Leavesley SJ, Britain AL, Cichon LK, Nikolaev VO, Rich TC. Assessing FRET using spectral techniques. Cytometry Part A : the journal of the International Society for Analytical Cytology. 2013;83:898–912. doi: 10.1002/cyto.a.22340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Adams JA, Taylor SS. Structural basis for peptide binding in protein kinase A. Role of glutamic acid 203 and tyrosine 204 in the peptide-positioning loop. The Journal of biological chemistry. 2003;278:10613–10618. doi: 10.1074/jbc.M210807200. [DOI] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nature biotechnology. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Nagy P, Bene L, Hyun WC, Vereb G, Braun M, Antz C, Paysan J, Damjanovich S, Park JW, Szollsi J. Novel calibration method for flow cytometric fluorescence resonance energy transfer measurements between visible fluorescent proteins. Cytometry Part A : the journal of the International Society for Analytical Cytology. 2005;67:86–96. doi: 10.1002/cyto.a.20164. [DOI] [PubMed] [Google Scholar]
- Pinsky BG, Ladasky JJ, Lakowicz JR, Berndt K, Hoffman RA. Phase-resolved fluorescence lifetime measurements for flow cytometry. Cytometry. 1993;14:123–135. doi: 10.1002/cyto.990140204. [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nature biotechnology. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- Robinson CV, Sali A, Baumeister W. The molecular sociology of the cell. Nature. 2007;450:973–982. doi: 10.1038/nature06523. [DOI] [PubMed] [Google Scholar]
- Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y, Yoshida K, Nagata Y, Sato-Otsubo A, Yoshizato T, et al. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science. 2014;344:917–920. doi: 10.1126/science.1252328. [DOI] [PubMed] [Google Scholar]
- Scott JD, Fischer EH, Demaille JG, Krebs EG. Identification of an inhibitory region of the heat-stable protein inhibitor of the cAMP-dependent protein kinase. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:4379–4383. doi: 10.1073/pnas.82.13.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seybold PG, Gouterman M, Callis J. Calorimetric, photometric and lifetime determinations of fluorescence yields of fluorescein dyes. Photochemistry and photobiology. 1969;9:229–242. doi: 10.1111/j.1751-1097.1969.tb07287.x. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nature methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Sun Y, Periasamy A. Localizing protein-protein interactions in living cells using fluorescence lifetime imaging microscopy. Methods in molecular biology. 2015;1251:83–107. doi: 10.1007/978-1-4939-2080-8_6. [DOI] [PubMed] [Google Scholar]
- Szaloki N, Doan-Xuan QM, Szollosi J, Toth K, Vamosi G, Bacso Z. High throughput FRET analysis of protein-protein interactions by slide-based imaging laser scanning cytometry. Cytometry Part A : the journal of the International Society for Analytical Cytology. 2013;83:818–829. doi: 10.1002/cyto.a.22315. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Zhang P, Steichen JM, Keshwani MM, Kornev AP. PKA: lessons learned after twenty years. Biochimica et biophysica acta. 2013;1834:1271–1278. doi: 10.1016/j.bbapap.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You X, Nguyen AW, Jabaiah A, Sheff MA, Thorn KS, Daugherty PS. Intracellular protein interaction mapping with FRET hybrids. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18458–18463. doi: 10.1073/pnas.0605422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.