

Summary
Kappa opioid receptors (KORs) are involved in a variety of aversive behavioral states, including anxiety. To date, a circuit based mechanism for KOR driven anxiety has not been described. Here we show that activation of KORs inhibits glutamate release from the basolateral amygdala (BLA) inputs to the bed nucleus of the stria terminalis (BNST), and occludes the anxiolytic phenotype seen with optogenetic activation of BLA-BNST projections. In addition, deletion of KORs from amygdala neurons results in an anxiolytic phenotype. Furthermore, we identified a frequency dependent optically-evoked local dynorphin-induced heterosynaptic plasticity of glutamate inputs in the BNST. We also find that there is cell type specificity to the KOR modulation of the BLA-BNST input, with greater KOR-mediated inhibition of BLA dynorphin-expressing neurons. Collectively, these results provide support for a model in which local dynorphin release can inhibit an anxiolytic pathway, providing a discrete therapeutic target for treatment of anxiety disorders.
Graphical abstract

Introduction
Anxiety disorders are a major health concern, with 7.3% of the global population suffering from an anxiety disorder at any given time (Baxter et al., 2013; Lepine, 2002). Despite the high expense of anxiety disorder treatments (Lepine, 2002) many of the most common treatments, including tricyclic antidepressants, monoamine oxidase inhibitors, benzodiazepines, and selective serotonin reuptake inhibitors, have side effects that limit their utility (Ravindran, 2010). In light of these limitations, there has been a greater effort to discover new modulatory systems for the treatment of anxiety disorders (Deisseroth, 2014; Holden, 2003; Johansen, 2013; Tye et al., 2011). In order to develop new and more efficacious therapeutics, however, a more thorough understanding of the circuitry underlying anxiety disorders is required.
Kappa opioid receptors (KORs) have been proposed as a potential target for stress and anxiety disorders, as well as substance abuse disorders (Wee and Koob, 2010). An abundance of behavioral pharmacological experiments have shown an anxiolytic effect of KOR antagonists are capable of overcoming the anxiogenic effects of a chronic or acute stressor, ethanol withdrawal, CRF, and KOR agonism (Bruchas et al., 2009; Knoll et al., 2007; Valdez and Harshberger, 2012). Findings have implicated recruitment of KOR signaling by its endogenous ligand dynorphin (Chavkin et al., 1982) as playing a key role in preclinical and clinical models of anxiety (Knoll et al., 2011); however the mechanism that underlies this effect and the circuitry involved has not yet been defined(Crowley and Kash, 2015). KOR modulation has been identified in key anxiety-related regions such as the dorsal raphe nucleus (Bruchas et al., 2010; Land et al., 2009), the ventral tegmental area (Spanagel et al., 1992), and the prefrontal cortex (Svingos and Colago, 2002; Tejeda et al., 2013). These regions interact with the bed nucleus of the stria terminalis (BNST), a key region involved in anxiety-related behaviors (Kash, 2012); however, thus far investigation of KORs in the BNST has so far been lacking. In addition, the BNST is known to express preprodynorphin (Poulin et al., 2009), and previous work from our lab has demonstrated KOR modulation of GABAergic transmission in the BNST (Li et al., 2012a). Together, these studies support the idea that KORs in the BNST could be a promising and important target for neuropsychiatric manipulations.
Based on the abundance of evidence implicating KORs in anxiety disorders, and the known role of the BNST in regulating anxiety-like behavior, we evaluated the role of KOR modulation of glutamate transmission in the BNST. We also examined the ability of local BNST dynorphin neurons to heterosynaptically modulate glutamatergic transmission. Here, we report that KORs in the BNST inhibit glutamatergic transmission on the BLA but not the PFC inputs. We also demonstrate alterations in anxiety-related behaviors through complementary manipulations of KORs and this circuit. Furthermore, we identify a technological approach to assessing peptidergic transmission in slice.
Materials and Methods
Subjects
All animal procedures were performed in accordance with the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill and the Animal Care and Use Committee of Washington University, conforming to US National Institutes of Health guidelines. Experiments were performed on adult male C57BL/6J mice and DBA/2J mice, both from Jackson Laboratory (Bar Harbor, ME). In addition, Preprodynorphin-IRES-Cre and R26-loxSTOPlox-L10-GFP (Al-Hasani et al., 2015; Krashes et al., 2014), and Floxed KOR conditional knock-out mice (Chefer et al., 2013) were generated as described previously, and bred in house at UNC. All mice were group housed in colony rooms with a 12:12hr light-dark cycle (lights on at 7 a.m.) with ad libitum access to rodent chow and water.
Slice electrophysiology
Coronal sections containing the BNST (300μM) were obtained from behaviorally-naïve mice rapidly decapitated under isoflurane. All experiments were conducted in the dorsolateral portion of the BNST. Lidocaine N-ethyl bromide (1 mg/ml) was included in the intracellular recording solution to prevent postsynaptic sodium spikes for all voltage-clamp experiments. For basal KOR pharmacological effects and characterization, picrotoxin (25 μM) was added to aCSF to isolate excitatory postsynaptic currents (EPSCs). Tetrodoxtoin (500nM) and picrotoxin (25μM) were added to the aCSF to isolate miniature EPSCs. Cells were held at -70mV to isolate AMPAR-mediated current (see the Supplemental Experimental Procedures).
For ex vivo optogenetic experiments, all brains were evaluated for light-evoked action potentials in the injection region (BLA, PFC, PVN, or BNST) using a potassium-gluconate-based internal recording solution. Brains were discarded and not used for further experimentation if action potentials were not obtained or injection sites were missed. A blue LED (470nm, CoolLed, Hampshire, United Kingdom) was used to optically stimulate release from channelrhodopsin (ChR2)-containing fibers (5 msec pulse for voltage-clamp experiments, 1 msec for current clamp experiments).
Stereotaxic surgery
Male mice were injected with viral constructs into regions of interest. A craniotomy was performed, and mice were bilaterally injected using a blunt needle (86200 and 65458-01, Hamilton Company, Reno, NV), with 400-500 nl of the vector into the BLA (stereotaxic coordinates from bregma: -1.30 anterior-posterior, +/−3.15 medial-lateral,–4.95 mm dorsal-ventral), 350 nl of the vector into the BNST (stereotaxic coordinates from bregma: +0.27 anterior-posterior, +/-0.90 medial-lateral, -4.25 dorsal-ventral), or 400-500 nl of the vector into the PFC (stereotaxic coordinates from bregma: +1.8 anterior-posterior, +/- 0.3-0.5 medial-lateral, -2.5 dorsal-ventral). For floxed KOR slice electrophysiology experiments, ChR2/CRE cocktail was prepared (250nL ChR2, 250nL CRE) and injected into the BLA.
Mice were allowed to recover for at least six weeks prior to behavioral experiments or electrophysiology (see the Supplemental Experimental Procedures).
Behavior
Anxiety-related behavioral assays were conducted as described previously (Pleil et al., 2015). Briefly, in the first series of experiments, floxed KOR knock-out mice with either ChR2-eYFP or Cre-GFP injected in the BLA were placed in the elevated plus maze for five minutes. For these experiments, a virus encoding ChR2 was injected in to control mice, but no in vivo illumination was used. In the second series of experiments, mice with either ChR2-eYFP or eYFP injected in the BLA with optical fibers in the BNST were systemically administered either saline or a KOR agonist. Thirty minutes later, they were tested in the open field.
Elevated Plus Maze
The elevated plus maze (EPM; Med Associates, St. Albans, VT) was made of white and black plastic and consisted of two open arms (75 × 7 cm) and two closed arms (75 × 7 × 25 cm) adjoined by a central area (7 × 7 × 25 cm). The arms were arranged in a plus configuration with arms of the same type (open or closed) opposite of each other. The maze was elevated 75cm with light levels maintained at 15 lux throughout the experiment. Mice were placed in the center of the EPM and allowed to explore freely. The EPM was cleaned with 70% ethanol between each trial. Movements were video recorded and analyzed using Ethovision 9.0 (Noldus Information Technologies, Leesburg, VA). The primary measures of reduced anxiety-like behavior were time spent in the open arm and number of entries into the open arm.
Open Field Test
Mice were injected with the KOR agonist U50,488 (5 mg/kg in saline, i.p.) or vehicle control (saline) and returned to their home cage for 30 min. The open field test was performed in a square enclosure (55 × 55 cm) with light levels maintained at 25 lux. Mice were placed in the center of the open field and allowed to roam freely for 21 min. Photostimulation alternated between off and on states in 3 min time segments, beginning with 3 min of no stimulation. For the photostimulated time segments, animals received 10 Hz (10 ms width) photostimulation (∼10 mW light power). The open field was cleaned with 70% ethanol between each trial. Movements were video recorded and analyzed using Ethovision 8.5 (Noldus Information Technologies, Leesburg, VA). The center was defined as a square comprised of 50% the total area of the OFT. Time in the center was the primary measure of anxiety-like behaviors.
Histological verification
Nissl staining was performed in cohorts of mice used for in vivo optogenetics to confirm injections and fiber placements (see the Supplemental Experimental Procedures).
Viral vectors
The viral constructs AAV2-CaMKIIα-ChR2-eYFP, AAV2-CaMKIIα-eYFP, AAV5-EF1α-DIO-ChR2-eYFP, AAV5-EF1α-DIO-eYFP, AAV5-EF1α-DIO-ChR2-mCherry, AAV5-CaMKIIα-ChR2-eYFP, AAV5-CaMKIIα-Cre-GFP, and AAV2-CaMKIIα-Cre-eYFP, described elsewhere (Kim et al., 2013) were obtained from the UNC Viral Vector Core (Chapel Hill, NC).
Data analysis and statistics
Data are expressed as means ± SEM for all figures. For all experiments, 2-way ANOVAs, paired t-tests, unpaired t-tests, and linear regression were used where appropriate, as described in figure captions. Statistical analyses were conducted using Prism 6.0 (GraphPad, La Jolla, CA), and figures were made in Illustrator CC 2015 (Adobe, San Jose, CA).
Results
KORs signal via a presynaptic, p38- and calcium-dependent mechanism to inhibit glutamate release in the BNST
An extensive body of literature links glutamate signaling in the BNST to both anxiolytic and anxiogenic behavior (Davis et al., 2010; Hubert and Muly, 2014; Kim et al., 2013). Therefore, we first examined whether KOR activation could alter glutamate function in the BNST. We found that multiple KOR-selective agonists (Dynorphin-A and U69,593) inhibited electrically-evoked excitatory post synaptic currents (eEPSCs) in the BNST, both of which were blocked by pre-application of the selective KOR antagonist norBNI (Figure 1A-D). This KOR-mediated inhibition was not reversed by norBNI (Figure 1E-F), suggesting that this is a form of long lasting plasticity. We next assessed the intracellular signaling mechanism of this KOR modulation, as KORs have been shown to signal through both p38 (Bruchas et al., 2007) and MEK/ERK (Li et al., 2012a). p38 signaling in particular has been shown to be necessary for the dysphoria associated with KOR activation (Bruchas et al., 2007). The KOR agonist effect was blocked in the presence of the p38 inhibitor SB203580 but not the MEK/ERK inhibitor SL-327 (Figure 1G) or the PKA inhibitor RP-Camps (Figure 1H). Together, these results suggest that KOR mediated inhibition of glutamate function in the BNST involves p38, but not ERK or PKA signaling.
Figure 1. KOR activation inhibits glutamate transmission in the BNST.

(A) Representative experiment demonstrating KOR-mediated inhibition of eEPSC amplitude. Inset, eEPSC trace from the same neuron showing pre (black) and post (red) 1μM U69,593 application. Scale bar represents 200pA by 20msec. (B) KOR activation by U69,593 inhibited eEPSC amplitude (red circles, paired t-test, baseline v. min 21-25, t4 = 30.70, P < 0.001) and was blocked by continuous application of the KOR antagonist norBNI, 100nM (yellow circles, paired t-test, baseline v. min 21-25, t4 = 0.003, P > 0.05); the KOR agonist U69,593 significantly inhibited eEPSC amplitude as compared to the norBNI block effect (unpaired t-test, acsf v. norBNI, min 16-20, t8 = 10.22, P < 0.001). (C) Representative experiment demonstrating KOR inhibition by 300nM Dynorphin-A. (D) KOR activation by 300nM Dynorphin-A produces a robust inhibition of eEPSCs (red circles, paired t-test, baseline v. min 21-25, t4 = 18.65, P < 0.001) that is blocked by the KOR antagonist norBNI (yellow circles, paired t-test, baseline v. min 21-25, t4 = 2.783, P = 0.05) mimicking the results seen with U69,593 (Fig. 1B). Both U69,593 (E) and Dynorphin-A (F) activation of KORs are non-reversible forms of inhibition. Post U69,593 application of the KOR antagonist norBNI (100nm) failed to reverse the inhibition by either KOR agonist (U69,593, paired t-test, baseline v. min 21-25, t4 = 13.88, P < 0.001; Dynorphin-A, paired t-test, baseline v. min 21-25, t4 = 14.30, P < 0.001). (G) The p38 inhibitor SB203580 (10μM) but not the MEK/ERK inhibitor SL-327 (20μM) blocked KOR-mediated inhibition of eEPSCs (SB203580 effect, baseline v. min 16-20, t4 = 2.619, P > 0.05; SL-327 effect, baseline v. min 16-20, t4 = 14, P < 0.0001). (H) The PKA inhibitor RpCamps (5μM) does not alter eEPSCs (paired t-test, baseline v. min 16-20, t4 = 10, P = 0.0004) (I) Representative mEPSC traces (top and bottom) pre (left) and post (right) U69,593 application, conducted in 500nM TTX and 5μM picrotoxin. No significant changes in mEPSC decay kinetics were seen, (not shown, paired t-test, t6 = 0.8170, P > 0.1). (J) mEPSC frequency (paired t-test, t5 = 5.567, P < 0.001) but not amplitude (K) (paired t-test, t5 = 0.2141, P > 0.1) was reduced following application of the KOR agonist U69,593 Raw values for mEPSC frequency (not shown, baseline frequency mean = 3.406, SEM = 3.475; post-U69,593 frequency mean = 2.179, SEM = 1.118) and amplitude (not shown, baseline amplitude mean = -29.36, SEM = 1.944, post-U69,593 amplitude mean = -25.71, SEM = 1.465). This inhibition is abolished in zero calcium aCSF, where both frequency (L) (paired t-test, t5 = 1.959, P > 0.05) and amplitude (M) (paired t-test, t5 = 2.017 P > 0.05) of mEPSCs remain unaltered by U69,593.
We next determined the impact of KOR agonists on miniature excitatory synaptic transmission to more clearly understand the mechanism of inhibition. KOR activation reduced miniature EPSC frequency but not amplitude, with no alterations in miniature EPSC kinetics (Figure 1I-K). This KOR modulation of mEPSCs was absent when recordings were conducted in zero calcium conditions similar to those previously published (Figure 1L-M). Taken together, this demonstrates a presynaptic, p38- and calcium-dependent form of glutamate inhibition, mechanistically similar to a form of long-term depression (LTD) that has been seen in the dorsal striatum following activation of opioid receptors (Atwood et al., 2014).
KORs inhibit pathway-specific glutamate inputs to the BNST
Glutamatergic innervation to the BNST arises from multiple cortical and subcortical nuclei (Dong et al., 2001; Kim et al., 2013). We next probed KOR inhibition of two of these pathways, the prefrontal cortex (PFC) and the basolateral amygdala (BLA). We injected ChR2 (AAV2-CaMKIIα-ChR2-eYFP) in either the BLA or PFC and conducted slice electrophysiology experiments in the BNST. Independent activation of these pathways produced a robust, light-evoked EPSC, consistent with other studies examining BLA projections (Felix-Ortiz et al., 2013) (Figure 2A-C). The light-evoked BLA-BNST EPSC was inhibited by KOR activation, while the light-evoked PFC-BNST EPSC not altered by KOR agonist (Figure 2D), demonstrating that KOR modulation of glutamate transmission is pathway specific. Optogenetic activation of the BLA input to the BNST produced action potentials in BNST neurons reliably up to 20-40 hz (Figure 2E). As we have previously shown that KOR activation can inhibit GABA release (Li et al., 2012b), we wanted to test whether activation of KOR signaling would lead to a net reduction of BLA-induced action potentials in the BNST. Consistent with a net inhibitory action of KOR on this pathway, the KOR agonist significantly reduced the fidelity of these BLA-BNST induced action potentials. This demonstrates that presynaptic KORs show specificity in inhibition of glutamate inputs in the BNST.
Figure 2. KOR-mediated inhibition of eEPSCs is pathway specific.

(A) Left, representative injections of ChR2 to the BLA and expression in the BNST. Right, representative traces for light-evoked EPSCs from the BLA (top, blue) and PFC (bottom, green). (B) Both the PFC and BLA send robust glutamatergic projections to the BNST, which do not differ in amplitude (unpaired t-test, t8 = 1.901, P > 0.05). (C) The BLA sends a monosynaptic EPSC and significantly time-delayed polysynaptic IPSC (paired t-test, t7 = 5.232, P < 0.01), comparable to BLA projections to other outputs. (D) Similarly, the PFC sends a monosynaptic EPSC and a significantly time-delayed polysynaptic IPSC (paired t-test, t4 = 4.138, P < 0.05). (E) KOR activation inhibits BLA-BNST light-evoked EPSC amplitude (paired t-test, baseline v. min 16-20, t4 = 14.86, P < 0.0001). However, KOR application did not alter PFC-BNST light-evoked EPSC transmission (paired t-test, baseline v. min 16-20, t4 = 0.6899, P > 0.1) (F) U69,593 reduces the fidelity of light-evoked BLA-BNST action potentials. When fitted with standard linear regression, the slope was significantly non-zero predrug application (F1,28 = 12.86, P < 0.01) but not post (F1,28 = 2.472, P > 0.05). The lines also showed significant different intercept points (F1,57 = 5.51, P < 0.05). Inset, representative traces of action potential fidelity at 20hz 5msec light stimulation pre (dark blue) and post (light blue) U69,593 application. Black boxes indicate light pulses.
KORs are expressed presynaptically, and modulate BLA-BNST anxiety-related behaviors
We next investigated the role of KOR in this pathway in vivo and ex vivo using a genetic approach. We injected AAV5-CaMKIIα-Cre-GFP (Cre) or AAV5-CaMKIIα-ChR2-eYFP (Control) into the BLA of conditional KOR knockout mice (Chefer et al., 2015) and assessed changes in anxiety-related behavior (Figure 3A-C). Viral deletion of KORs expressed on amygdala neurons resulted in an increase in EPM open arm time (Figure 3C) with no alteration in total distance traveled (Figure 3D). In these experiments, there was expression of the cre virus in both the CeA and adjacent cortex in subsets of animals; however, the expression in these regions did not correlate to any behavioral measures. In order to confirm the presynaptic locus of KOR modulation at BLA-BNST synapses in vitro, we injected a cocktail of CaMKIIα-ChR2-eYFP and CaMKIIα-Cre-GFP into the BLA of KOR KO mice. This approach deletes KORs expressed on amygdalar neurons, while simultaneously allowing us to optogenetically activate this specific circuit in slice electrophysiology experiments. KOR-mediated inhibition of the BLA input was absent when KORs were genetically deleted from the BLA (Figure 3E). Together, this confirms that KORs are expressed presynaptically on BLA neurons, and demonstrates that deletion of amygdala KORs results in an anxiolytic phenotype.
Figure 3. Deletion of KORs from amygdala neurons increases anxiolytic behavior in the EPM and blocks KOR agonist induced reduction of BLA to BNST inputs, while systemic activation of KORs occludes BLA-BNST driven anxiolytic behavior in the open field.

(A) Top, outline of experimental procedure. Floxed KOR mice were injected with either Control or Cre (behavior) or a cocktail of ChR2 and Cre (electrophysiology) bilaterally to the BLA. Following ample time for virus to express, experiments were conducted. Bottom, representative injection of Cre-GFP into the BLA of floxed KOR mice. (B) Representative heatmaps for Control (left) or Cre (right) mice. Enclosed arms are horizontal and open arms are vertical. Scale bar represents intensity of behavior. (C) Cre mice showed significantly greater open arm time compared to Control mice (unpaired t-test, Cre v. ChR2, t14 = 2.215, P < 0.05). (D) No differences in total distance traveled were seen (unpaired t-test, Cre- v. ChR2, t14 = 0.4226, P > 0.05). (E) Deletion of KORs from BLA neurons prevented the KOR-mediated inhibition of BLA-BNST synapses, confirming that KORs act pre-synaptically to inhibit evoked glutamate release. Mice were injected with ChR2 (AAV2- CaMKIIα-ChR2-eYFP) and Cre (AAV2-CaMKII-Cre-GFP) into the BLA, and light-evoked EPSCs were recorded in the BNST. U69,593 no longer inhibited light-evoked EPSCs (paired t-test, baseline v. min. 16-20, t4 = 1.474, P > 0.1). (F) Top, outline of experimental procedure. C57 mice were injected with either ChR2 or eYFP unilaterally to the BLA, and optical fibers were implanted above the BNST. Following ample time for virus to express, experiments were conducted. Bottom, representative injection of ChR2 into the BLA, and fiber placement in the BNST. Scale bar represents 100 μM. (G) A 2-way ANOVA revealed a significant effect of drug (F1,31=22.28, P < 0.001) as well as an interaction between drug and ChR2 (F1,31=5.210, P < 0.05) on number of entries into the center of the open field. Tukey's multiple comparisons showed a significant decrease of entries into the center in ChR2-U50 mice as compared to ChR2-Saline mice (P < 0.05). (H) A 2-way ANOVA revealed a trend towards a ChR2 effect (F1,38=3.606, P = 0.06) and interaction between ChR2 and drug (F1,38=3.097, P = 0.08) on amount of time spent in the center of the open field. Based on the previous findings, our planned comparisons revealed a significant increase amount of time spent in the center of the open field of ChR2-Saline mice as compared to eYFP-Saline mice (t17=3.034, P < 0.01) and ChR2-U50 mice as compared to ChR2-Saline mice (t18=2.178, P < 0.05), indicating that though ChR2 activation of BLA-BNST neurons produced an anxiolytic effect, this was blocked by the KOR agonist U50,488. In addition, changes in locomotor behavior were seen in ChR2-U50 and eYFP-U50 mice (data not shown). A 2-way ANOVA revealed a significant effect of U50,488 treatment on overall activity (F1,30=53.91, P < 0.0001). Tukey's post hoc tests showed that eYFP-U50 mice showed significantly decreased overall locomotor activity as compared to eYFP-saline mice (P < 0.05). Similarly, as expected, ChR2-U50 mice showed significant decreased overall locomotor as compared to ChR2-saline controls (P < 0.05). (I) Representative heatmaps for all groups. Scale bar indicates intensity of behavior.
In order to further understand the role of BNST KORs in anxiety-related behavior, we next probed this system using in vivo optogenetics (Figure 3F). Optogenetic activation of the BLA-BNST pathway produced a robust anxiolytic effect in the open field, replicating the effects found by others (Kim et al., 2013). Following administration of a sub-anxiogenic dose of the KOR agonist U50,488 (5mg/kg i.p.), activation of the BLA-BNST-induced anxiolytic phenotype, as well as locomotor behavior, was reduced as compared to control mice (Figure 3G-I). Together, these complementary behavioral approaches demonstrate that, globally, KORs are an important regulator of the BLA-BNST anxiety circuit, and that BLA KORs are critically involved in anxiogenic behavior.
Dynorphin is a heterosynaptic messenger in the BNST, and dynorphin neurons are preferentially modulated by KORs on BLA to BNST synapses
Finally, we assessed the capacity of dynorphin neurons in the BNST to release dynorphin following optogenetic stimulation. We stereotaxically injected ChR2 using a Cre-dependent vector (AAV5-EF1α-DIO-ChR2-EYFP) in the BNST of Preprodynorphin-IRES-Cre mice (Al-Hasani et al., 2015; Krashes et al., 2014). Dynorphin-positive (DYN+) neurons were located throughout the dorsal-lateral BNST (Figure 4A). Light activation of these cells reliably produced action potentials at up to 20 Hz and resulted in a monosynaptic IPSC (mean = 422.9, SEM = 149.0), but no EPSC (mean = -17.64, SEM = 6.414) (Figure 4B) onto neighboring, putatively non-dynorphin (DYN-) neurons, indicating BNST DYN+ neurons form robust local synapses that are exclusively GABAergic. We next probed whether optogenetic activation of these DYN+ neurons was capable of altering electrically-evoked glutamate release within the BNST. 5 hz activation of DYN+ neurons for 150 seconds produced a transient norBNI-sensitive (KOR-mediated) inhibition of eEPSCs (Figure 4C), however the EPSC amplitude returned to baseline within minutes. 20 hz stimulation for 150 seconds produced a robust and lasting inhibition of eEPSCs (Figure 4D); this inhibition was blocked by pre-application of the selective KOR antagonist norBNI. This effect was indistinguishable from that seen with KOR agonist application (Figure 1A-D). This effect was not seen when an identical approach was performed assessing a GABAergic dynorphin projection to the BNST from the paraventricular nucleus of the hypothalamus (PVN) (Figure 4E). 20 hz activation of PVN-BNST dynorphin neurons did produce an inhibition of eEPSCs, but this inhibition was not KOR-dependent, suggesting that another modulatory system may be playing a role in this distal inhibition by dynorphin neurons (Bodnar, 2013). Taken together, these experiments demonstrate that activation of local BNST dynorphin neurons can inhibit electrically-evoked glutamatergic transmission in the BNST. Additionally, these experiments suggest a possible timing-dependent component in the persistence of KOR plasticity, similar to what has been demonstrated with norepinephrine-induced LTD in the BNST (McElligott and Winder, 2008).
Figure 4. Optogenetic activation of BNST Dynorphin neurons inhibits eEPSCs in nonChR2 expressing neurons, and KORs preferentially inhibit glutamate release onto DYN+ neurons.
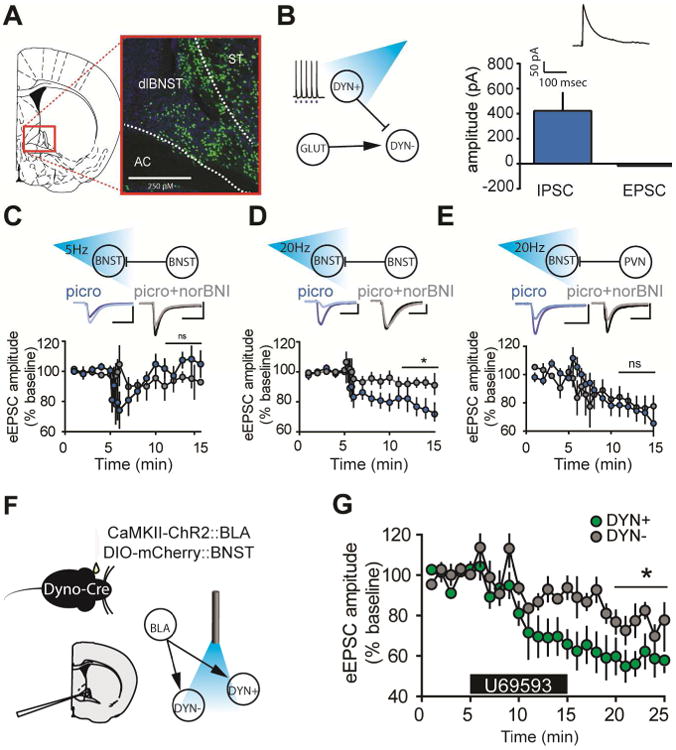
(A) Localization of DYN+ neurons in the BNST. DYN+ cells were found throughout the dorsal BNST in a Dyn-L10-EGFP reporter mouse. (B) Left, local dynorphin cells showed reliable light evoked action potentials at 20 hz. These cells synapsed locally, producing a light-evoked IPSC of approximately 423 pA, but no light-evoked EPSC. (C) 5 hz stimulation of DYN+ neurons produced a significant transient change in eEPSC amplitude (paired t-test, baseline v. min. 5-8, t4 = 6.033, P < 0.05), which returned to baseline (paired t-test, baseline v. min 11-15, t4 = 2.474, P > 0.05). (D) 20 hz stimulation of local DYN+ neurons produces a significant and lasting inhibition of eEPSC (paired t-test, baseline v. min 11-15, t4 = 10.42, P < 0.001). This effect is partially blocked by norBNI (unpaired t-test acsf v. norBNI block, min 16-20, t8 = 10.59, P < 0.0001). (E) 20 hz optogenetic stimulation of PVN to BNST DYN+ neurons produces a significant inhibition of eEPSC (paired t-test, baseline v. min 11-15, t4 = 8.566, P < 0.001) but this was not significantly different from the inhibition seen when in the presence of the KOR antagonist norBNI (unpaired t-test, acsf+picrotoxin v. acsf+picrotoxin+norBNI, t8 = 1.191, P > 0.1). (F) Mice were injected with a cre-inducible mCherry (AAV5-EF1α-DIO-mCherry) to the BNST, and ChR2 (AAV2-CaMKIIα-ChR2-EYFP) to the BLA. Optogenetic activation of BLA-BNST DYN+ and DYN- neurons was then assessed. (G) Application of the KOR agonist U69,593 significantly reduced the BLA-BNST eEPSC amplitude onto both DYN+ (paired t-test, baseline v. min 21-25, t4 = 12.14, P < 0.001) and DYN- neurons (paired t-test, baseline v. min 21-25, t4 = 9.039, P < 0.001). However, the effect of KOR-mediated inhibition was significantly larger on DYN+ neurons as compared to DYN- neurons (unpaired t-test, DYN+ min 21-25 v. DYN- 21-25, t8 = 7.071, P < 0.001), indicating KOR activation in the BNST may preferentially inhibit BLA-DYN+ neurons. See also Supplemental Figures 1 and 2.
We next assessed the relationship between BLA inputs to the BNST and BNST dynorphin neurons. In order to assess the BLA inputs onto DYN+ and DYN- neurons separately, ChR2 (AAV2-CaMKIIα-ChR2-eYFP) was injected into the BLA and a cre-dependent fluorophore (AAV5-EF1α-DIO-mCherry) was injected into the BNST of Preprodynorphin-IRES-Cre mice (Figure 4F). DYN+ neurons had smaller membrane resistance than their DYN- counterparts, and a trend towards significant differences in cell capacitance (Supplementary Figure 1A-B), consistent with known classifications of BNST neuron types (Hammack et al., 2007). DYN- neurons had more action potentials per current injection when held at RMP, indicative of greater intrinsic excitability (Supplementary Figure 1C-K). Both DYN+ and DYN- neurons received a glutamate projection from the BLA, as well as a polysynaptic GABAergic projection, but no basal differences were seen with BLA innervation of these cells as indicated by similar paired pulse ratio and amplitude of evoked responses (Supplementary Figure 2A-D). We next examined KOR modulation of BLA inputs to these two discrete cell types. We found that KOR activation more robustly inhibited BLA-induced eEPSCs in DYN+ neurons as compared to DYN- neurons (Figure 2G). Taken together, these data suggest that there is both pathway- and cell- type dependent modulation of glutamate function in the BNST by KOR, allowing for important gating of glutamate transmission at DYN+ and DYN- neurons.
Discussion
The BNST has been shown to orchestrate both rewarding and aversive behaviors (Jennings et al., 2013; Kim et al., 2013). KORs have historically been thought to modulate aversive (or negatively regulate rewarding) systems (Chefer et al., 2005) or anxiety related behaviors, though recent literature has shown that that the neuroanatomical location of KORs may be important in behavioral effects seen with their activation. Castro and Berridge (2014) demonstrated that activation of KORs in the nucleus accumbens can mediate both conditioned place preference and conditioned place aversion. This work was expanded recently expanded upon when Al-Hasani et al. (2015) demonstrated that activation of DYN+ neurons themselves in distinct subpopulations of the nucleus accumbens is responsible for rewarding or aversive behavioral phenotypes, and found that photo-activation of the aversive subpopulation of accumbens dynorphin neurons did not results in anxiogenic behavior. The diversity in effects seen with KOR activation, coupled with the multitude of behavioral outcomes seen with the BNST activation in this study, places KORs in the BNST at the interesting and likely position of being able to modulate anxiety-related behaviors. Furthermore, these results suggest that KORs are poised to regulate negative affective behavioral states (aversion) and anxiety via discrete brain subnuclei.
We demonstrate that KOR activation in the BNST inhibits glutamatergic inputs by a presynaptic, p38-, and calcium- dependent mechanism. Previous work has shown p38-dependent effects of KOR modulation in other brain regions, such as the dorsal raphe nucleus (Bruchas et al., 2011; Land et al., 2008; Lemos et al., 2012), and others have postulated that this intracellular signaling pathway may be critical for the dysphoric effects of KOR agonists (Bruchas et al., 2007). In addition, previous work by our lab demonstrated that KORs inhibit BNST GABAergic transmission via MEK/ERK signaling (Li et al., 2012a). We therefore show that KORs within the BNST are capable of altering neurotransmitter transmission via pharmacologically distinct mechanisms at different synapses. Differential use of signaling pathways by KORs has been demonstrated in the nucleus accumbens (Hjelmstad and Fields, 2003), and these pharmacologically distinct signaling mechanisms may play a key role in the development of functionally biased compounds. Development or identification of biased ligands, capable of targeting individual signaling pathways, may be useful in developing therapeutic treatments without the unpleasant side-effects seen with KOR activation (White et al., 2014).
Beyond a pharmacologically distinct mechanism of KOR inhibition, we also demonstrate a pathway-specific inhibition of glutamatergic inputs to the BNST. KORs inhibit BLA but not PFC inputs to the BNST; this mechanism may provide for gating of information flow both to and from the BNST. Future work should address other inputs to the BNST, including the multitude of inputs recently identified in the human brain (Avery et al., 2014). We also demonstrate a mechanism by which KORs are activated: GABAergic/dynorphin co-expressing neurons in the BNST release dynorphin to presynaptically inhibit glutamate inputs to the BNST. This signaling mechanism through which dynorphin regulates transmission may be a common motif throughout the brain: it was originally postulated to be the mechanism of inhibition in the hippocampus (Drake et al., 1994), and, more recently, was shown to be the mechanism of inhibition in the PVN (Iremonger and Bains, 2009). We not only show similar findings in the BNST, but we expand upon this literature by demonstrating an optogenetic mechanism for activation of such release. This allows for peptidergic and optogenetic coupling, greatly expanding upon the ability to assess peptide release both in vitro and in vivo.
In addition, we used complementary approaches to identify behavioral changes in anxiety-related behaviors following manipulations of the KOR system. When KORs were genetically deleted from BLA neurons using a viral cre approach coupled with the floxed KOR mouse, we saw a decrease in anxiety-related behavior in the EPM. Though other projections such as those to the CeA and hippocampus are likely also important pathways in this phenotype, the BLA-BNST has been previously demonstrated to be involved in anxiety-like behavior and the EPM (Kim et al., 2013). Therefore, KORs are likely to be a key regulator of this pathway and phenotype. In a complementary series of experiments, we demonstrated in vivo activation of BLA-BNST neurons produced a robust anxiolytic phenotype (replicating work by Kim et al.). Importantly, systemic activation of KORs occludes the anxiolytic effect seen with BLA-BNST activation; though optogenetic activation of the BLA-BNST pathway can produce a robust anxiolytic phenotype, this pathway is gated by the dynorphin/KOR system.
Importantly, we found this KOR inhibition exhibited both cell-type and pathway-dependent differences in function. Though BLA inputs to the BNST show similar properties onto both DYN+ and DYN- neurons, there is a greater KOR-mediated inhibition of BLA-DYN+ synapses versus BLA-DYN- synapses. These results suggest a model in which activation of the BNST by the BLA excites DYN+ neurons, and this activation leads to a local retrograde release of dynorphin. This dynorphin release then gates inputs to the BNST, potentially providing a homeostatic balance within the system during times of heightened activity, suggesting a mechanism by which KOR antagonists exhibit anxiolytic actions. In addition, this further illuminates the micro-circuitry of the BNST and how it may coordinate such a broad range of behavioral states, such as those involved in stress and addiction. Further, this raises a strategy for development of anxiolytic compounds—blocking endogenous inhibitors of defined circuits that reduce anxiety.
Further work is necessary to elucidate the downstream effects of BNST dynorphin neurons. Specifically, an understanding is needed of their projection and activation patterns, this is especially important in light of several recent findings detailing various sub-domains in the BNST that are linked with specific behaviors. This work provides an important framework to begin that research: though the circuitry of DYN+ and DYN-neurons may be equal, their activation and inhibition may not be. Taken together, this work demonstrates a pathway specific, p38- and calcium- dependent form of KOR inhibition of glutamate. It also demonstrates a mechanism by which local dynorphin release may mediate this presynaptic effect. Finally, this work shows that KORs may preferentially inhibit glutamatergic inputs onto DYN+ neurons, versus DYN- neurons, allowing for specific gating of information flow. This circuit-based approach has numerous advantages over existing approaches, as it allows higher precision in defining the substrate and mechanism of action. Given the lack of efficacious and well-tolerated anxiolytic medications currently available, this work introduces a site-specific manipulation of the mammalian anxiety system.
Supplementary Material
Highlights.
KORs presynaptically inhibit BLA glutamate transmission in the BNST
Optogenentic activation of local dynorphin neurons produces inhibition of glutamate
Deletion of amygdala KORs produces an anxiolytic phenotype in the elevated plus maze
Acknowledgments
This work was supported by the following grants: R01AA019454, R00AA017668, NARSAD Young Investigator Award, and U01AA020911 (TLK); F31AA02228001 (NAC); R01DK096010, R01DK089044, R01DK071051, R01DK075632, R37DK053477, BNORC Transgenic Core P30 DK046200, and BADERC Transgenic Core P30 DK57521 (BBL); F32 DK089710 (MJK); R01DA033396, and McDonnell Center for Systems Neuroscience (MRB).
Footnotes
Author contributions: NAC, DWB, JGM, RA, MRB, and TLK designed the experiments; NAC, DWB, JAH, AK, JGM, RA, NMM, WY, and ZLS conducted the experiments; MJK, BBL, and JLW provided the transgenic mice; NAC and TLK wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Hasani R, McCall JG, Shin G, Gomez AM, Schmitz GP, Bernardi JM, Pyo CO, Park SI, Marcinkiewcz CM, Crowley NA, et al. Distinct Subpopulations of Nucleus Accumbens Dynorphin Neurons Drive Aversion and Reward. Neuron. 2015;87:1063–1077. doi: 10.1016/j.neuron.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Kupferschmidt DA, Lovinger DM. Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nature neuroscience. 2014;17:540–548. doi: 10.1038/nn.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery SN, Clauss JA, Winder DG, Woodward N, Heckers S, Blackford JU. BNST neurocircuitry in humans. NeuroImage. 2014;91:311–323. doi: 10.1016/j.neuroimage.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter AJ, Scott KM, Vos T, Whiteford HA. Global prevalence of anxiety disorders: a systematic review and meta-regression. Psychol Med. 2013;43:897–910. doi: 10.1017/S003329171200147X. [DOI] [PubMed] [Google Scholar]
- Bodnar RJ. Endogenous opiates and behavior: 2012. Peptides. 2013;50:55–95. doi: 10.1016/j.peptides.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C. Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:11614–11623. doi: 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain research. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Lemos JC, Chavkin C. CRF1-R activation of the dynorphin/kappa opioid system in the mouse basolateral amygdala mediates anxiety-like behavior. PloS one. 2009;4:e8528. doi: 10.1371/journal.pone.0008528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, Lemos JC, Hagan CE, Neumaier JF, Quintana A, et al. Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron. 2011;71:498–511. doi: 10.1016/j.neuron.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro DC, Berridge KC. Opioid hedonic hotspot in nucleus accumbens shell: mu, delta, and kappa maps for enhancement of sweetness “liking” and “wanting”. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:4239–4250. doi: 10.1523/JNEUROSCI.4458-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- Chefer VI, Backman CM, Gigante ED, Shippenberg TS. Kappa opioid receptors on dopaminergic neurons are necessary for kappa-mediated place aversion. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2013;38:2623–2631. doi: 10.1038/npp.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chefer VI, Czyzyk T, Bolan EA, Moron J, Pintar JE, Shippenberg TS. Endogenous kappa-opioid receptor systems regulate mesoaccumbal dopamine dynamics and vulnerability to cocaine. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:5029–5037. doi: 10.1523/JNEUROSCI.0854-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley NA, Kash TL. Kappa opioid receptor signaling in the brain: Circuitry and implications for treatment. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2015 doi: 10.1016/j.pnpbp.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Walker DL, Miles L, Grillon C. Phasic vs sustained fear in rats and humans: role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2010;35:105–135. doi: 10.1038/npp.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K. Circuit dynamics of adaptive and maladaptive behaviour. Nature. 2014;505:309–317. doi: 10.1038/nature12982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong HW, Petrovich GD, Swanson LW. Topography of projections from amygdala to bed nuclei of the stria terminalis. Brain research Brain research reviews. 2001;38:192–246. doi: 10.1016/s0165-0173(01)00079-0. [DOI] [PubMed] [Google Scholar]
- Drake CT, Terman GW, Simmons ML, Milner TA, Kunkel DD, Schwartzkroin PA, Chavkin C. Dynorphin opioids present in dentate granule cells may function as retrograde inhibitory neurotransmitters. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1994;14:3736–3750. doi: 10.1523/JNEUROSCI.14-06-03736.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix-Ortiz AC, Beyeler A, Seo C, Leppla CA, Wildes CP, Tye KM. BLA to vHPC inputs modulate anxiety-related behaviors. Neuron. 2013;79:658–664. doi: 10.1016/j.neuron.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammack SE, Mania I, Rainnie DG. Differential expression of intrinsic membrane currents in defined cell types of the anterolateral bed nucleus of the stria terminalis. Journal of neurophysiology. 2007;98:638–656. doi: 10.1152/jn.00382.2007. [DOI] [PubMed] [Google Scholar]
- Hjelmstad GO, Fields HL. Kappa opioid receptor activation in the nucleus accumbens inhibits glutamate and GABA release through different mechanisms. Journal of neurophysiology. 2003;89:2389–2395. doi: 10.1152/jn.01115.2002. [DOI] [PubMed] [Google Scholar]
- Holden C. Excited by Glutamate. Science. 2003;300:1866–1868. doi: 10.1126/science.300.5627.1866. [DOI] [PubMed] [Google Scholar]
- Holmes A, Fitzgerald PJ, MacPherson KP, DeBrouse L, Colacicco G, Flynn SM, Masneuf S, Pleil KE, Li C, Marcinkiewcz CA, et al. Chronic alcohol remodels prefrontal neurons and disrupts NMDAR-mediated fear extinction encoding. Nature neuroscience. 2012;15:1359–1361. doi: 10.1038/nn.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert GW, Muly EC. Distribution of AMPA receptor subunit glur1 in the bed nucleus of the stria terminalis and effect of stress. Synapse (New York, NY) 2014;68:194–201. doi: 10.1002/syn.21729. [DOI] [PubMed] [Google Scholar]
- Iremonger KJ, Bains JS. Retrograde opioid signaling regulates glutamatergic transmission in the hypothalamus. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:7349–7358. doi: 10.1523/JNEUROSCI.0381-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings JH, Sparta DR, Stamatakis AM, Ung RL, Pleil KE, Kash TL, Stuber GD. Distinct extended amygdala circuits for divergent motivational states. Nature. 2013;496:224–228. doi: 10.1038/nature12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP. Neuroscience: anxiety is the sum of its parts. Nature. 2013;496:174–175. doi: 10.1038/nature12087. [DOI] [PubMed] [Google Scholar]
- Kash TL. The role of biogenic amine signaling in the bed nucleus of the stria terminals in alcohol abuse. Alcohol (Fayetteville, NY) 2012;46:303–308. doi: 10.1016/j.alcohol.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Adhikari A, Lee SY, Marshel JH, Kim CK, Mallory CS, Lo M, Pak S, Mattis J, Lim BK, et al. Diverging neural pathways assemble a behavioural state from separable features in anxiety. Nature. 2013;496:219–223. doi: 10.1038/nature12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AT, Meloni EG, Thomas JB, Carroll FI, Carlezon WA., Jr Anxiolytic-like effects of kappa-opioid receptor antagonists in models of unlearned and learned fear in rats. The Journal of pharmacology and experimental therapeutics. 2007;323:838–845. doi: 10.1124/jpet.107.127415. [DOI] [PubMed] [Google Scholar]
- Knoll AT, Muschamp JW, Sillivan SE, Ferguson D, Dietz DM, Meloni EG, Carroll FI, Nestler EJ, Konradi C, Carlezon WA., Jr Kappa opioid receptor signaling in the basolateral amygdala regulates conditioned fear and anxiety in rats. Biological psychiatry. 2011;70:425–433. doi: 10.1016/j.biopsych.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Shah BP, Madara JC, Olson DP, Strochlic DE, Garfield AS, Vong L, Pei H, Watabe-Uchida M, Uchida N, et al. An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature. 2014 doi: 10.1038/nature12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, Hnasko TS, Palmiter RD, Chavkin C. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19168–19173. doi: 10.1073/pnas.0910705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JC, Roth CA, Messinger DI, Gill HK, Phillips PE, Chavkin C. Repeated stress dysregulates kappa-opioid receptor signaling in the dorsal raphe through a p38alpha MAPK-dependent mechanism. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:12325–12336. doi: 10.1523/JNEUROSCI.2053-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepine JP. The epidemiology of anxiety disorders: prevalence and societal costs. J Clin Psychiatry. 2002;63:4–8. [PubMed] [Google Scholar]
- Li C, Pleil KE, Stamatakis AM, Busan S, Vong L, Lowell BB, Stuber GD, Kash TL. Presynaptic inhibition of gamma-aminobutyric acid release in the bed nucleus of the stria terminalis by kappa opioid receptor signaling. Biological psychiatry. 2012a;71:725–732. doi: 10.1016/j.biopsych.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Pleil KE, Stamatakis AM, Busan S, Vong L, Lowell BB, Stuber GD, Kash TL. Presynaptic inhibition of gamma-aminobutyric acid release in the bed nucleus of the stria terminalis by kappa opioid receptor signaling. Biol Psychiatry. 2012b;71:725–732. doi: 10.1016/j.biopsych.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElligott ZA, Winder DG. Alpha1-adrenergic receptor-induced heterosynaptic long-term depression in the bed nucleus of the stria terminalis is disrupted in mouse models of affective disorders. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2008;33:2313–2323. doi: 10.1038/sj.npp.1301635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleil KE, Rinker JA, Lowery-Gionta EG, Mazzone CM, McCall NM, Kendra AM, Olson DP, Lowell BB, Grant KA, Thiele TE, et al. NPY signaling inhibits extended amygdala CRF neurons to suppress binge alcohol drinking. Nature neuroscience. 2015;18:545–552. doi: 10.1038/nn.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin JF, Arbour D, Laforest S, Drolet G. Neuroanatomical characterization of endogenous opioids in the bed nucleus of the stria terminalis. Progress in neuro-psychopharmacology & biological psychiatry. 2009;33:1356–1365. doi: 10.1016/j.pnpbp.2009.06.021. [DOI] [PubMed] [Google Scholar]
- Ravindran SM. The pharmacologic treatment of anxiety disorders: a review of progress. J Clin Psychiatry. 2010;71:839–854. doi: 10.4088/JCP.10r06218blu. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:2046–2050. doi: 10.1073/pnas.89.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparta DR, Stamatakis AM, Phillips JL, Hovelso N, van Zessen R, Stuber GD. Construction of implantable optical fibers for long-term optogenetic manipulation of neural circuits. Nat Protoc. 2012;7:12–23. doi: 10.1038/nprot.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svingos AL, Colago EE. Kappa-Opioid and NMDA glutamate receptors are differentially targeted within rat medial prefrontal cortex. Brain research. 2002;946:262–271. doi: 10.1016/s0006-8993(02)02894-9. [DOI] [PubMed] [Google Scholar]
- Tejeda HA, Counotte DS, Oh E, Ramamoorthy S, Schultz-Kuszak KN, Backman CM, Chefer V, O'Donnell P, Shippenberg TS. Prefrontal cortical kappa-opioid receptor modulation of local neurotransmission and conditioned place aversion. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2013;38:1770–1779. doi: 10.1038/npp.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, Deisseroth K. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–362. doi: 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez GR, Harshberger E. kappa opioid regulation of anxiety-like behavior during acute ethanol withdrawal. Pharmacology, biochemistry, and behavior. 2012;102:44–47. doi: 10.1016/j.pbb.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology. 2010;210:121–135. doi: 10.1007/s00213-010-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KL, Scopton AP, Rives ML, Bikbulatov RV, Polepally PR, Brown PJ, Kenakin T, Javitch JA, Zjawiony JK, Roth BL. Identification of novel functionally selective kappa-opioid receptor scaffolds. Molecular pharmacology. 2014;85:83–90. doi: 10.1124/mol.113.089649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.