

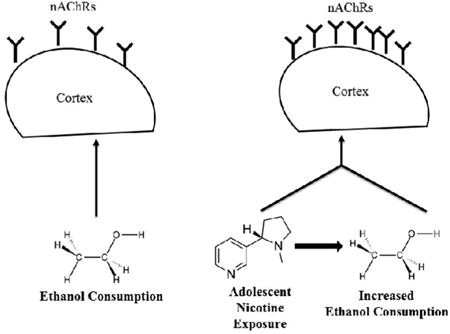
Graphical abstract
1. Introduction
Despite the deleterious effects resulting from cigarette use, 20% of the U.S. adult population continues to smoke and almost 80% of those adult smokers began smoking before 18 years of age [Center for Disease Control (CDC), 2011a; United States Department of Health and Human Services (USDHHS), 1994]. Individuals who begin smoking before the age of 18 are likely to increase tobacco use throughout adolescence and into adulthood (Hymowitz et al., 1997; Rose et al., 1996). Early initiation and high levels of cigarette smoking during adolescence are associated with reduced success in future quit attempts (Breslau & Peterson, 1996; Chassin et al., 2000; Laviola et al., 2003). Between the ages of 12 and 17 years, females (13.6%) have slightly higher rates of cigarette smoking than do their male counterparts (12.3%) (Pogun & Yararbas, 2009), and adult women have fewer successful quit attempts and long-term quit rates than men (Becker & Hu, 2008; Bjornson et al., 1995; Hatsukami et al., 1995). Because female adolescents have a higher rate of cigarette smoking than male counterparts and display a robust and continued use of cigarettes into adulthood, it is important to examine contributors to cigarette use in this vulnerable population.
Adolescent smokers often experiment with other abused drugs, specifically alcohol (Best et al., 2000; Lewinsohn et al., 1999; Torabi et al., 1993). Indeed, alcohol and tobacco products are frequently consumed together with binge alcohol consumption a particularly prevalent behavior among adolescents. Eighty-eight percent of adolescent ever-smokers report engaging in binge alcohol drinking (Best et al., 2000; Chen et al., 2002; Johnson et al., 2000) while only 5% of never smokers report engaging in binge alcohol drinking (Bobo & Husten, 2000). It is critical that we understand not only what drives nicotine intake in female adolescents, but how nicotine exposure might impact binge drinking of ethanol.
Use of animal models to investigate neurobiological alterations during adolescence helps to identify possible mechanisms that contribute to unique adolescent behaviors, e.g., drug abuse, risky behaviors, novelty seeking (Laviola et al., 2003; Spear, 2000b). Rodent models examining biological mechanisms that drive cigarette consumption often focus on nicotine, the primary addictive ingredient in tobacco (CDC, 2011b; USDHHS, 1988). Female rodents will readily consume and rapidly self-administer nicotine (Becker & Hu, 2008). Female rats also display rapid nicotine relapse following drug extinction (Donny et al., 2000; Isiegas et al., 2009). Moreover, age specific effects in rodents reveal that adolescent mice are more responsive to the positive rewarding properties of nicotine and less responsive to the negative properties of nicotine consumption than adults (Adriani et al., 2002;2003). Thus, a biological mechanism related to nicotine's activation of the reward pathway may influence increased nicotine use in adolescent females.
Exposure to drugs of abuse activate the mesolimbic dopamine system (Nestler, 2005). Indeed, both nicotine and ethanol activate the primary component of the reward pathway, the dopaminergic projections from the ventral tegmental area (VTA) to the nucleus accumbens (NAc), ultimately increasing synaptic dopamine (DA) levels (Corrigall et al., 1994; Larsson et al., 2005). A number of brain regions, (e.g., cortical regions, dorsal tegmental area) stimulate the core component of this reward pathway, and are active during rewarding experiences (Haber & Knutson, 2010; Omelchenko & Sesack, 2005;2006; Roesch & Olson, 2004).
Nicotinic acetylcholine receptors (nAChRs) are target receptors for endogenous acetylcholine and for pharmacologically administered nicotine (Picciotto et al., 2001). nAChRs are ligand gated ion channels comprised of five subunits, and are located in a number of regions within the brain reward pathway (Gotti et al., 2006; Picciotto et al., 2012). While nicotine directly activates nAChRs on DA neurons in the VTA, the way in which alcohol stimulates VTA activity is not yet fully known. It has been suggested that alcohol acts in part through nAChRs (Ericson et al., 2003; Hendrickson et al., 2010; Jones et al., 1999). One hypothesis proposes that alcohol may enhance stimulation of nAChRs through binding to specific sites on nAChRs that modulate excitability of the receptor (Borghese et al., 2003; Liu et al., 2013). Another hypothesis suggests that alcohol increases acetylcholine levels, in turn activating a greater number of nAChRs in regions of the reward pathway, and ultimately increasing activity of DA neurons in the VTA (Ericson et al., 2003; Liu et al., 2012; 2013). Thus, both nicotine and ethanol enhance reward pathway activation, in part through stimulation of cholinergic systems (Bito-Onon et al., 2011; Hendrickson et al., 2009; Imperato et al., 1986; Pidoplichko et al., 2004).
Adult rodents exposed to nicotine injections display high alcohol intake and preference in subsequent testing (Blomqvist et al., 1996; Lê et al., 2006; Olausson et al., 2001; Smith et al., 1999). Furthermore, chronic exposure to both nicotine and alcohol elevates nAChR levels for several days, and mecamylamine (an nAChR antagonist) pretreatment blocks alcohol-induced dopamine increases in the NAc and ethanol-induced conditioned place preference (Blomqvist et al., 1997; Dohrman & Reiter, 2003; Hendrickson et al., 2013; Zarrindast et al., 2010). Thus, experiments conducted in vivo also indicate that a neurobiological mechanism may drive nicotine-induced increases in alcohol use.
During adolescence, the brain undergoes changes through growth and development, particularly in cortical and limbic regions where remodeling of circuits is highly prevalent (Laviola & Marco, 2011). Thus, here the highly malleable adolescent brain is vulnerable to changes from exposure to environmental stimuli including exposure to drugs that may alter normal developmental trajectories (Laviola & Marco, 2011; Spear, 2000a; 2014). However, the behavioral and neurobiological effects of nicotine and alcohol exposure in adolescents are not currently known. Thus, we investigated how nicotine intake affects binge ethanol consumption and how exposure to nicotine with ethanol affects underlying neurobiological mechanisms that may modulate behavioral changes that differ from the mice in the ethanol only control group. We focused our examination on changes in nAChRs in several crucial brain reward regions (VTA, NAc, cortical regions, dorsal tegmental area).
Overall findings from this study reveal that nicotine consumption increases binge-like ethanol consumption. Moreover, adolescent nicotine and ethanol exposure was associated with increased α4β2*-nAChR density, as measured by epibatidine binding, compared to ethanol only exposed mice. It is possible that upregulation of nAChRs may contribute to the increase in ethanol consumption, but future work is needed to specifically address this hypothesis.
2. Materials
2.1 Animals
Twenty-six female C57BL/6J mice (The Jackson Laboratory, Bar Harbor, Maine) arrived on PND 27 at the Centralized Biological Laboratory, University Park, PA. Mice were individually housed on a 12-hour light/dark cycle [lights on 0200 hours] in a climate-controlled room with a temperature of 20.3 °C ± 0.8 °C and 62% relative humidity. Mice were housed in standard shoebox Plexiglas cages with 0.6 cm bedding (Bed-o'Cobs, The Andersons Agriservices, Inc. Maume, OH). Animals had continuous access to food (Lab Rodent Diet 5001, PMI Nutrition International, Inc., Brentwood, MO) throughout the experiment. All procedures were approved by the Pennsylvania State University Institutional Animal Care and Use Committee.
2.2 Compounds
(-)- Nicotine freebase, cytisine and other chemicals used to prepare buffer solutions were purchased from Sigma-Aldrich (St. Louis, MO). Koptec 190 proof ethanol was purchased from VWR (Radnor, PA), and diluted in tap water to produce a 20% v/v ethanol solution. The radioligands [125I] epibatidine (2200 Ci/mmol) and [125I] α -bungarotoxin (initial specific activity 237 mCi/mmol) were purchased from Perkin-Elmer (Waltham, MA). [125I] α-conotoxin MII (2200 Ci/mmol) was supplied by Dr. J. Michael McIntosh (University of Utah). Non-radioactive 6I-epibatidine was a gift of Dr. Kenneth Kellar (Georgetown University).
3. Methods
3.1. Procedure
3.1.1. Acclimation
Mice arrived on PND 27, were singly housed, and left undisturbed to acclimate to the light cycle. Light cycles were not fully reversed (lights on at 0200 hours instead of 0700) in order to allow for drinking-in-the-dark (DID) experiments to occur during daytime hours while also minimizing stress associated with a complete reversal of the light cycle and resultant change in diurnal rhythms. See Figure 1 for experimental timeline.
Figure 1. Experimental Timeline.

3.1.2 Baseline
During baseline (PND 32-34), mice had 24-hour access to tap water in a single drinking bottle. Experimenters entered the room five hours into the dark cycle (1700 hours) to obtain daily body weight, food and fluid consumption.
3.1.3. Nicotine Treatment
During the experiment mice were “periadolescent” (PND 35-44; Klein et al., 2003; 2004; Laviola et al., 2003; Spear, 2000a). During the first 7 days of the experiment, all mice were exposed to three glass drinking bottles filled with water or nicotine for 22 hours a day, and a single water bottle for two hours each day. For mice in the control group (N=12), all 3 bottles were filled with tap water, and for mice in the nicotine group (N=14) all 3 bottles were filled with 200 μg/ml (-)-nicotine freebase dissolved in tap water [the multiple bottle protocol was used to produce pronounced nicotine intake (Biondolillo & Pearce, 2007; Halder et al., 2013)]. This nicotine concentration was selected as adolescent mice voluntarily consume nicotine at this concentration without any signs of adverse side effects (Klein et al., 2003; 2004). Moreover, this concentration produces measurable cotinine levels in adolescent mice (Klein et al., 2003; 2004). A control cage with three bottles of water was housed in the same experimental room and bottles were manipulated in the same manner as those bottles on mouse cages. There was no appreciable loss of liquid on the control cages. Nicotine solutions were prepared fresh every 5 days based on published reports demonstrating no considerable nicotine loss in this time frame (Pekonen et al., 1993). In order to keep nicotine exposure consistent (22 hours/day) throughout the experiment the drinking bottles were removed from the cages 3 hours into the dark cycle and replaced with a single plastic water bottle for a 2 hour period for nicotine only days. Bottles from the 22 hour water or nicotine access period were then weighed and consumption values calculated for each mouse. Following the 2 hour limited water access period, the single bottle was removed and the appropriate treatment bottles for each mouse were placed back on the cage. The single water bottle was weighed before and after the 2 hour exposure period and consumption values were calculated for each mouse. Body and food weight were obtained following the 2 hour water period. This procedure continued for 6 days (PND 35-40) until mice were exposed to 2 hours of ethanol via the DID protocol for the last 4 days.
3.1.4 Drinking-in-the-Dark (DID) Treatment
During the four DID treatment days, nicotine exposure continued as above (22 hours/day). However, on the DID days, three hours into the dark cycle, the three (nicotine or water) bottles were removed and replaced with a single 25 ml plastic tube fitted with a rubber stopper and ball bearing sipper top filled with 20% v/v ethanol (Rhodes et al., 2005). Although several variations of the DID procedure have been developed, we chose the version that produces the highest ethanol consumption and BECs (see review by Thiele et al., 2014). Exposure to the 4 day DID protocol produces both blood ethanol concentrations (BEC; e.g., 80mg% - 199mg%) and behaviors (e.g., ataxia indexed by the rotorod) comparable to intoxicated humans in C57BL/6J mice (Rhodes et al., 2005; 2007). Mice had access to ethanol for two hours before the ethanol bottle was removed and the three nicotine or water bottles were replaced on the cages. This 2-hour limited access ethanol availability occurred for three days (PND 41-43). On the fourth and final day (PND 44) mice had access to ethanol for four hours. At the end of this four-hour period, the ethanol bottle was removed, weighed, and the mice were sacrificed via cervical dislocation. Blood was collected via cardiac puncture and whole brains were extracted and cut in half (sagittal). Each hemisphere was flash frozen in dry-ice chilled 2-methylbutane, and stored in a -80°C freezer until subsequent autoradiography procedures were performed.
3.2 Blood Ethanol Concentration (BEC) Assessment
Blood samples were immediately refrigerated for the subsequent BEC assay. Assays were conducted in the Biomarker Core Laboratory (http://bbh.hhd.psu.edu/) at Penn State University. Procedures for the BEC are outlined in Kamens et al., 2012. Briefly, blood samples mixed with perchloric acid were centrifuged at 1500×g for 10 minutes, and the supernatant was collected and neutralized with potassium hydroxide. This mixture was centrifuged at 1500×g for 10 minutes and the supernatant collected. Water blanks, standards, and samples were plated with 0.5M Tris/2.0mM NAD+ or 0.5MTris/2.0mM NAD+/alcohol dehydrogenase (Sigma, St. Louis, MO). After a 30 minute room temperature incubation samples were read at 340nm on a Synergy II plate reader (Biotek, Winooski, VT) and data were analyzed using Gen 5.0 software (Biotek, Winooski, VT).
3.3. Receptor Autoradiography
Ten brains from the control and nicotine treated groups were randomly selected for receptor autoradiography. Autoradiography procedures followed the protocol of Baddick & Marks, 2011 and Doura et al., 2008. Once removed from the -80 freezer, brains were mounted to a cryostat chuck using either Tissue Tek or M-1 Embedding Medium. Brains were sectioned at a 14 micron thickness using either an IEC or Leica cryostat and mounted on superfrost plus microscope slides (Fisher Scientific). Slides containing the sections were stored at -80°C until use. On the day of the experiment, the slides were warmed to room temperature (approximately 1 hour) under a vacuum in a sealed desiccator.
To examine the role of different types of nAChRs we utilized four binding conditions; total high affinity epibatidine binding to heteromeric nAChRs, epibatidine plus cytisine in which cytisine inhibits binding primarily to α4β2-nAChR sites retaining a mixed population of nAChR binding sites (including α3β2- α6β2-, and α3β4-nAChR sites), α-conotoxin MII which binds to nAChRs composed predominantly of α6β2, α3β2, α6β2β3, and α4α6β2β3 (i.e., primarily receptors that include α6 nAChR subunits), and α-bungartoxin which binds with high affinity to α7 nAChRs (Baddick & Marks, 2011; Grady et al., 2007). Adjacent sets of slices were exposed to one of four different binding conditions: epibatidine, epibatidine + cytisine, α-bungarotoxin, or α-conotoxin MII. For the epibatidine and epibatidine + cytisine conditions, samples were incubated in 1× KRH solution (NaCl, 140 mM; KCl, 1.5 mM; CaCl2, 2 mM; MgSO4, 1 mM; HEPES 25 mM; pH = 7.5). For total epibatidine binding, slides were incubated at room temperature with 200 pM [125I] epibatidine with a specific activity of 110 Ci/mmol (attained by diluting a commercial sample of 2200 Ci/mmol with unlabeled 6I-epibatidine for 2 hours. The 2nd condition contained 200 pM [125I] epibatidine and non-radioactive 50 nM cytisine and these samples were incubated for 2 hours at room temperature. Both [125I] epibatidine incubations were done on the same day. In the 3rd condition, samples were treated with 1× KRH containing 1mM PMSF for 10 minutes before an incubation with 1nM [125I] α-bungarotoxin with a specific activity of 71 Ci/mmol in 1×KRH containing 0.1% BSA for 4 hours at room temperature. For the 4th binding condition samples were incubated with 1× KRH containing 1mM PMSF, for 10 minutes followed by incubation with 0.3 nM α-conotoxin MII (2200 Ci/mmol) in 1×KRH containing 0.1% BSA, 5 mM EDTA, 5 mM EGTA and protease inhibitors (aprotinin, leupetin and pepstatin -10 μg/ml each)for 3 hours at room temperature. In order to determine non-specific binding, an independent set of slides in each experimental condition incubated with 10 μM of nicotine tartrate for the epibatdine, 1 mM of nicotine tartrate for the α-bungarotoxin, and 100 μM of nicotine tartrate for the α-conotoxin condition.
Following incubation, samples were washed according to conditions listed in Baddick & Marks, 2011. Slides were dried with a gentle stream of air and desiccated under vacuum overnight. Slides were then placed in cassettes and exposed to Packard Cyclone Super Resolution Screens to develop for the following amount of time: Epibatidine - 6 days, Epibatidine + Cytisine - 6 days, α- bungarotoxin – 14 days, α-conotoxin MII – 5 days. A subset of slides from each group was removed and exposed to Kodak MR (Kodak, Rochester, NY) film for 5 days to obtain representative, higher resolution images.
3.4 Image Analysis
Screens were imaged using a Cyclone Phosphoimager (Perkin Elmer). Images were opened in Optiquant (Perkin Elmer) and brain regions of interest for each ligand were selected and outlined. Our regions of interest included the VTA, NAc, cortical regions, and dorsal tegmental area as the presence of nAChRs have been found in all these regions and they are also crucial in the activation of the reward pathway. The density of nAChR sites labeled by each ligand was calculated by converting pixels/mm2 to Fmol/mg wet weight using a standard curve constructed with tissue standards with known amounts of 125I.
3.5 Statistical Analyses
Statistical Program for Social Science [SPSS (SPSS, Chicago, IL)] was used for statistical analyses. Mixed factorial repeated measures ANOVAs were run including group (water or nicotine) and experimental day (1-10) as factors with the following dependent variables: body weight (g), food intake (g), water or nicotine intake (ml). To examine ethanol consumption (g/kg), a repeated measures ANOVA was run on the final 4 days of the experiment including group and day as factors. Significant main effects and interactions were followed by appropriate post-hoc comparisons. To examine differences in nAChR density levels, one-way ANOVAs were run on each brain region of interest (cortical regions, ventral tegmental area, nucleus accumbens shell and dorsal tegmental area). All analyses were two-tailed with the alpha level set at 0.05. Results are reported as mean (± S.E.M.) in text and figures.
4. Results
4.1 Baseline
Average body weight, food consumption, and liquid intake for the 2 baseline days are listed in Table 1. While there were no significant effects of group on body weight or liquid intake during baseline days, there was a significant main effect of group on food consumption [F(1,25)=59.65, p <0.05]. Mice in the nicotine group consumed more food during baseline than did mice in the control group (Table 1).
Table 1. Values for Average Baseline Body Weight (g), Food Consumption (g), and Liquid Intake (mL).
| Body Weight (g) | Food Consumption (g) | Liquid Intake (mL) | |
|---|---|---|---|
| Control | 14.93 ± 0.44 | 3.87 ± 0.18* | 6.33 ± 0.30 |
| Nicotine | 15.07 ± 0.20 | 5.35 ± 0.09 | 6.57 ± 0.23 |
group differences p< 0.05
4.2 All Experimental Days (1-10)
In adolescent mice, body weight generally increased over the course of the experimental days as illustrated by the significant main effect of day on body weight with body weight consistently increasing during the first 7 days of the experiment, then remaining fairly stable until the end of the experiment [F(9,216) = 18.48, p< 0.05] (Figure 2a). While there was no significant main effect of group on body weight over the course of the ten experimental days, there was a significant day × group interaction on body weight [F(9,216) = 1.97, p <0.05] over the course of the experimental days (See Figure 2a). However, post hoc analyses on each day did not support significant group differences on any experimental day.
Figure 2.

a. Body weight (g) for control and nicotine-exposed mice across experiment days. No significant group differences were observed and both groups appear to have a general increase in weight over time consistent with normal development. b. Food Consumption (g) for control and nicotine-exposed mice across experiment days. * Significant difference in food consumption between nicotine-exposed and control mice.
In adolescent mice, there were subtle differences in food consumption during the experiment (Figure 2b). Similar to body weight, there was no significant main effect of group on food consumption during the ten experimental days. There was a significant main effect of day [F(9,216) = 27.47, p <0.05] as food consumption varied over the course of the experiment and was highest on days 2-5, 9, and 10 and lowest on days 6-8 (Figure 2b). There was also significant day × group interaction effect on food consumption over the course of the experiment [F(9,216) = 21.10, p < 0.05] (Figure 2b). Investigation of group differences in food consumption revealed that nicotine-exposed mice ate significantly less food than did control mice during the first 2 days of nicotine exposure and on the 5th day [Fs(1,24) > 12.01, p <0.05], but were not statistically different than control mice for the other nicotine only days. In contrast, during days 7, 8, and 9 (DID days) nicotine-exposed mice consumed significantly more food than mice in the control group [Fs(1, 24) > 28.17, p < 0.05] (Figure 2b).
Analysis of nicotine consumption (mg/kg) revealed a significant main effect of day [F(9,117) =20.81, p <0.05] (Figure 3). Nicotine consumption was elevated during the initial 2 days of exposure, a finding that is consistent with initial drug exposure (Klein et al., 2004). Consumption levels decreased and remained stable during days 3-7. After DID exposure began (day 8), mice significantly increased nicotine consumption (Figure 3).
Figure 3.

Nicotine Consumption (mg/kg) across all 10 experimental days. * denotes nicotine consumption values higher than other days
Control adolescent mice drank more liquid than nicotine exposed mice during the first 7 days of the experiment, but not during DID days (Figure 4a). The main effect of group revealed that control mice consumed significantly more liquid during the 22 hour liquid intake period than nicotine-exposed mice [F(1,25) = 85.25 p <0.05] (Figure 4a). These liquid intake results are not surprising as mice exposed to high concentrations of nicotine often consume less liquid than mice exposed to low nicotine concentrations and/or control mice (Adriani et al., 2002; Halder et al., 2013; Klein et al., 2004). An overall significant main effect of day [F(9,216) = 15.75, p<0.05] revealed that intake was high on the first few days and last few days, but was significantly lower during the middle of the experiment (days 4,6,7) (Figure 4a). There was a significant day × group effect on 22 hour liquid intake over the course of the experiment [F(9,216)= 14.41, p < 0.05) (Figure 4a). Control mice consumed more fluid than nicotine-exposed mice on experimental days 1-7 (during nicotine treatment) [Fs(1,25) > 43.31, p<0.05], but there were no significant group differences days 8-10 (during DID days; see Figure 4a). While there were no main effects of group or day on limited access water intake, there was a significant day × group interaction effect [F(1, 24) = 5.84, p <0.05]. Nicotine exposed mice drank significantly more water during the two hour limited access water period on days 1 and 3 than control mice (See Figure 4b). There were no significant group differences in 2 hour limited access water intake on the remainder of the experimental days.
Figure 4.

a. 22 hour liquid (water or nicotine) intake (mL) for control and nicotine-exposed mice across experimental days. b. 2 hour limited access water intake (mL) over the first 6 experiment days (prior to DID protocol) * significant difference between control and nicotine-exposed mice.
4.3 Drinking-in-the-Dark (DID)
Adolescent mice exposed to nicotine consumed more ethanol and had higher BECs than control mice on average (Figure 5a). While there was no significant main effect of group over the course of the 4 DID days, there was a significant main effect of day [F(1,24) = 73.15, p<0.05]. Not surprisingly, ethanol consumption on DID day 4 was significantly higher than the other 3 DID days (Figure 5a). This is likely due to the increased length of ethanol exposure on the last day compared to the first 3 DID days (4 hours vs. 2 hours, respectively). There was also a significant day × group interaction across DID days [F(3,72) = 4.00, p <0.05] as significant group differences appeared on the DID day 4. When ethanol consumption on day 4 was examined, mice in the nicotine group drank significantly more ethanol than did mice in the control group [F(1,24) = 4.21, p< 0.051] (Figure 5a). Mice in the nicotine group also had significantly greater BECs than mice in the control group [F(1,23) = 6.78, p<0.05] (Figure 5b). Two BEC values from control mice returned 0 mg% despite the fact that these animals had measurable alcohol consumption. Data from these animals were excluded from the BEC analysis, but these animals were included in remaining analyses (e.g., body weight, food consumption, liquid intake) for which these mice had data. These findings are consistent with prior DID studies, that report the greatest ethanol consumption and BEC differences on DID day 4 (Rhodes et al., 2007).
Figure 5.

Ethanol consumption on DID Days. DID ethanol consumption (g/kg) (4 days) (a) Blood ethanol concentration (BEC; mg%) following DID day 4 (4 hours) (b) in control and nicotine-exposed mice following DID protocol. *Significant difference between control and nicotine-exposed mice
4.4 Autoradiography
4.4.1. [125I] Epibatidine binding
Mice in the nicotine group displayed higher mean nAChR density than mice in the control group (Figure 6) in the: frontal cortex [F(1,19) = 5.48, p< 0.05], orbitofrontal cortex [F(1,19) = 4.92. p <0.05], outer cortex [F(1,19) = 4.34, p < 0.05], and inner cortex [F(1,19) =7.67, p< 0.05]. Mean binding values for brain regions of interest can be found in table 2.
Figure 6.

Total epibatidine binding (Fmol/mg) in the frontal cortex (a), orbitofrontal cortex (b), outer cortex (c), inner cortex (d) in control and nicotine-exposed mice, following ethanol consumption in the DID protocol. Autoradiographic images of anterior-coronal mouse hemisections between (e) 1.70 mm Bregma and -1.58 mm Bregma, (f) -1.94 mm Bregma and -4.16 mm Bregma.
Table 2.
Mean Ligand Binding Values (Fmol/mg) in Brain Regions of Interest for All Four Binding Conditions.
| Frontal Cortex | Orbitofrontal Cortex | Outer Cortex | Inner Cortex | Cingulate Cortex | Dorsal Tegment al Area | Ventral Tegmental Area | Nucleus Accumbens | |
|---|---|---|---|---|---|---|---|---|
| Control | ||||||||
| Epi | 6.08±0.48 | 11.50±0.87 | 6.05±0.58 | 10.39±076 | 8.17±1.20 | 20.05±1.61 | 24.46±1.90 | 9.51±0.83 |
| Cytres | -- | -- | -- | -- | -- | -- | -- | 0.76±0.07 |
| α-CTX MII | -- | -- | -- | -- | -- | -- | -- | 5.27±0.38 |
| α –BTX | -- | -- | 2.28±0.21 | 2.66±0.28 | -- | 11.54±1.74 | -- | -- |
| Nicotine | ||||||||
| Epi | 7.78±0.55* | 14.58±1.08 | 7.63±0.49* | 13.44±0.80* | 10.31±0.90 | 29.63±5.00 | 25.13±1.84 | 10.53±0.72 |
| Cytres | -- | -- | -- | -- | -- | -- | -- | 075±0.06 |
| α-CTX MII | -- | -- | -- | -- | -- | -- | -- | 5.06±0.24 |
| α –BTX | -- | -- | 2.68±0.26 | 2.96±0.27 | -- | 16.29±3.41 | -- | -- |
indicates significant group differences in mean binding values
4.4.2. [125I] Epibatidine plus 50 nM cytisine binding
There were no significant differences in nAChR density between nicotine-treated and control mice for cytisine-resistant epibatidine binding sites in any of our brain region of interest (table 2)
4.4.3. [125I] α-bungarotoxin binding
There were no significant differences in the density of bungarotoxin binding sites between nicotine-treated and control mice for the three brain regions of interest (table 2).
4.4.4 [125I] α-conotoxin MII binding
There were no significant differences in α-conotoxin MII binding between nicotine-exposed and control mice in the brain regions of interest (table 2).
5. Discussion
The results from this experiment revealed that nicotine intake increased both binge ethanol consumption (g/kg) and resulting BEC (mg%) in adolescent female C57BL/6J mice. Further, investigation of the neurobiological mechanisms underlying this effect revealed increased nAChR density in the cortex. Exposure to nicotine prior to ethanol increased epibatidine binding in the frontal, orbitofrontal, inner, and outer cortex more than ethanol exposure alone. These findings are similar to past adult rodent studies that report upregulation of nAChRs, composed of α4 and β2 subunits, in the cortex following exposure to chronic nicotine (for example, Marks et al., 2004; Marks et al., 2011; Marks et al., 1992; Pauly et al., 1996; Picciotto et al., 2008; Schwartz & Kellar, 1985). Because cytisine-sensitive epibatidine binding measures predominantly α4β2*-nAChRs (Marks et al., 1992; Baddick & Marks, 2011; Doura et al., 2008) our results suggest that that the upregulation of α4β2*nAChRs may be driven by biological mechanisms activated by either nicotine alone, or nicotine and ethanol exposure. It is possible that upregulation of this receptor underlies the increased binge ethanol consumption observed, however further work is necessary to address this hypothesis.
Our findings are consistent with prior studies in adult rodent models that have shown nicotine consumption increases responding for ethanol and ethanol consumption (Clark et al., 2001; Doyon et al., 2013). In adolescent mouse studies, one experiment revealed increases in ethanol consumption and BEC in mice exposed to cigarette smoke compared to control counterparts (Burns & Proctor, 2013). To our knowledge, no experiments have directly investigated how exposure to nicotine affects subsequent ethanol consumption in adolescent mice. While cigarette smoke has many components besides nicotine, findings from the current study and studies where nicotine exposure was given prior to ethanol suggest that nicotine is one of the primary compounds in cigarettes that may increase ethanol consumption (Burns & Proctor, 2013; Laviola et al., 2003). It is important to note that in the current study, on average, we found few group differences in 2 hour water intake between groups during the nicotine treatment period. Any significant group differences were observed during the initial days, but did not persist as no group differences were seen during the last 3 days of limited access water exposure. This finding indicates that the increase in intake of alcohol is not due to a mere increase in thirst. Furthermore, during the first 3 days of ethanol exposure (2 hour exposure) no significant group differences in ethanol consumption were observed. Thus, results from the current study are in line with past behavioral findings, and support the idea that biological mechanisms, aside from thirst, may drive nicotine-induced increases in ethanol consumption in adolescents (Laviola et al., 1999; 2003).
One unexpected, but particularly interesting finding, in this current study was the increase in nicotine consumption during the last 3 experiment days compared to first 7 experiment days (i.e. following initiation of the 2 h ethanol access period, see Figure 4a). While the purpose of this study was not to investigate how exposure to ethanol affects nicotine consumption, the increased nicotine intake following the availability of ethanol suggests that exposure to either drug alone may increase the use of the other. Consistent with our results, prior studies have reported that exposure to either nicotine or ethanol promotes subsequent use of ethanol or nicotine, respectively, in adult rodents (Lê et al., 2006; Olausson et al., 2001). Furthermore, the use of these two drugs together is very popular in human adolescent populations (Bobo & Husten, 2000; Chen et al., 2002). Specifically, adolescents that report binge drinking in the last 30 days, are 5 times more likely to smoke than adolescents who do not binge drink. Additionally, adolescent smokers have higher rates of alcohol use disorders than do their non-smoking counterparts (Bobo & Husten, 2000; Grucza & Bierut, 2006). Our current findings combined with prior studies seem to suggest that not only does exposure to one of these drugs promote use of the other drug, but exposure to one of these drugs promotes increased or excessive consumption of the other drug. Thus, findings from this study and past adult rodent studies seem to suggest a biological mechanisms may drive the excessive consumption of these two drugs together.
During adolescence the brain undergoes a number of structural and functional changes (Spear, 2000a;b; 2013). Therefore, certain brain regions have a high level of plasticity during adolescence and are particularly vulnerable to alterations induced by environmental agents (Gogtay et al., 2004; Laviola & Marco, 2011; Spear, 2013). One particularly malleable region is the prefrontal cortex, as it shows increased cholinergic input throughout adolescent development (Crews et al., 2000; Gould et al., 1991), a neurodevelopmental change thought to be responsible for many of the behaviors that characterize adolescence (e.g., risk taking, reward seeking, drug use) (Spear, 2000a,b). Our findings that nicotine and ethanol-exposed mice had increased epibatidine binding in frontal cortical regions compared to control mice that were exposed to ethanol only suggests that increases in α4β2* nAChRs may contribute to subsequent changes in behavior, such as increased ethanol consumption (see also Adriani et al., 2003 for increased vulnerability to subsequent nicotine self-administration). More research is with additional control groups is required before such conclusions can be made.
We observed no group differences in nAChR levels (per lack of group differences in binding of radioactive ligand) in primary regions of the reward pathway such as the VTA or NAc. These results are not completely unexpected as past studies have shown more robust effects of nicotine on nAChR levels in the cerebral cortex than in other regions in the reward pathway (Marks et al., 1986; Marks et al., 2004). In this study, the comparative increase in α4β2* nAChR density in cortical regions following nicotine and ethanol exposure are similar to findings from past studies with only nicotine administration, and suggest a complex activation of the reward pathway by nicotine. The current findings may suggest that nicotine exposure is influential in the stimulation of cortical regions of the reward pathway that subsequently induce increases in binge ethanol consumption. Thus, it is possible that nicotine may modulate alterations in the activity level of brain regions (i.e., the frontal cortices) associated with regulation of executive functions and decision-making, and in turn these regions may affect stimulation of the core of the reward pathway (e.g., dorsal tegmental area). In light of our findings, it is tempting to conclude that nicotine-induced increases in activation of cortical regions could contribute to alterations in decisions to engage in rewarding behaviors (e.g., enhanced consumption of alcohol or nicotine), however more research is required before such conclusions can be made.
One limitation of the current study was that we did not have a nicotine-only or a water-only condition. Therefore, our ability to interpret these findings is somewhat hindered as mice exposed to nicotine were also exposed to ethanol. While our results show that nicotine and ethanol exposed mice have higher levels of α4β2 nAChRs in the cortex than mice exposed to ethanol alone, we cannot determine from this study whether nicotine alone would produce similar results. Previous studies with adult female mice have reported that oral nicotine exposure produces upregulation of α4β2 nAChRs in several regions of the cortex (e.g., entorhinal, frontal, parietal, retrosplenial) (Sparks & Pauly, 1999). Additionally, cultured neurons from adult mice exposed to chronic nicotine had increased binding of epibatidine and upregulation of α4β2 nAChRs in multiple brain regions, with regions of the cortex demonstrating some of the largest upregulation (Lomazzo et al., 2011; Marks et al., 2004; 2011; Zambrano et al., 2012). Our results in conjunction with past studies, suggesting that the chronic exposure to nicotine may lead to increases in α4β2 nAChR in cortical regions. However, other adolescent mouse studies have shown that administration of oral nicotine and ethanol injections within a short time window upregulate nAChR levels in the cortex and midbrain to levels higher than either drug alone (Trauth et al., 1999). In this way, the interaction effect of both nicotine and ethanol administration on nAChR levels in adolescents appears complex. Future research should directly examine if oral nicotine or oral ethanol alone increases nAChRs, or if the combination of nicotine and ethanol is necessary to observe these neurobiological changes.
In humans, alcohol and nicotine are commonly consumed at the same time. While the current study did not examine co-use per se, as nicotine and ethanol were never administered simultaneously, mice were exposed to both drugs within the same day, and there were no times during the DID treatment period when mice in the nicotine group were not exposed to a drug. Findings from this study reveal that nicotine-exposed mice consumed significantly more ethanol than controls. Additionally, these mice consumed more nicotine during the DID treatment period than they did during the nicotine only treatment period. These results are in line with past studies that seem to suggest that there is a biological mechanism that drives use of nicotine and ethanol within a short time period of each other in adolescence (Laviola et al., 1999). Future studies should examine quantity of co-use or different combinations of use of these two drugs and relationship with changes in cortical nAChRs.
Adolescence is a period of sensation and novelty seeking, including experimentation with drugs of abuse, and results from this study are some of the first to reveal that nicotine exposure makes adolescents more susceptible to ethanol binge-like consumption in part through alterations in nAChRs levels in certain brain regions (e.g., cortex). Results from the current study revealed that food consumption was fairly similar between nicotine-exposed mice and control mice during the first 7 days of the experiment, but during DID days nicotine-exposed mice had food consumption levels that were greater than those of controls. It is interesting that food consumption increases during the ethanol exposure period for nicotine exposed-mice. While food consumption changes could be a results of nicotine's effects on appetite in these mice, it is also possible that exposure to nicotine and/or ethanol affects the way the reward pathway responds to other rewarding stimuli (e.g., food). Thus, future research should investigate whether nicotine-induced enhancement of ethanol consumption is unique to this combination of drugs, or if nicotine and/or ethanol exposure in adolescents primes the brain reward pathway to engage in other types of reward behavior (e.g., food and sucrose consumption, novelty seeking) or consumption of other drugs of abuse (e.g., psychostimulants, opiates).
Overall, results from the current study suggest that nicotine exposure in female adolescent mice is a complex process that activates nAChRs in several crucial reward pathway regions, including regions involved in higher level cognitive function (e.g., cortical regions). In this way, nicotine exposure may affect the overall modulation of the reward pathway beyond activation of the primitive dopaminergic projections from the VTA to NAc alone. Nicotine's effects on overall reward pathway activation may influence the way that adolescents engage in other rewarding behaviors, in particular use of ethanol. Because adolescence is a period of novelty seeking, these findings raise concerns as they suggest that there is a biological mechanism that influences the use of nicotine and alcohol, rather than just societal or peer pressures. Moreover, they suggest that experimentation with nicotine may contribute to ethanol consumption in adolescence, and that ethanol consumption may in-turn increase nicotine intake. Further understanding of the neurobiological mechanism that drives this enhanced drug use may help in the development of pharmacological treatments or prevention techniques.
Highlights.
Adolescent female mice were exposed to nicotine and then a binge ethanol paradigm
Nicotine-exposed mice had higher ethanol consumption and BEC than controls
nAChR density in the reward pathway was measured after nicotine and ethanol intake
Nicotine-exposed mice had higher epibatidine binding in cortex than controls
Acknowledgments
This work has been supported by the Penn State Social Science Research Institute through a SSRI/CBBC Level 1 Funding grant (Klein), K01 AA019447 (Kamens), and R01 DA003194 (Marks). Thank you to Dr. Allen Phillips, Dr. Will Horton, Dr. Melissa Mercincavage, Caitlin Short, and Dr. Kim Walter for their technical assistance and aid with this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adriani W, Macrì S, Pacifici R, Laviola G. Peculiar vulnerability to nicotine oral self-administration in mice during early adolescence. Neuropsychopharmacology. 2002;27:212–224. doi: 10.1016/S0893-133X(02)00295-6. [DOI] [PubMed] [Google Scholar]
- Adriani W, Granstrem O, Macrì S, Izykenova G, Dambinova S, Laviola G. Behavioral and neurochemical vulnerability during adolescence in mice: studies with nicotine. Neuropsychopharmacology. 2003;29:869–878. doi: 10.1038/sj.npp.1300366. [DOI] [PubMed] [Google Scholar]
- Baddick CG, Marks MJ. An autoradiographic survey of mouse brain nicotinic acetylcholine receptors defined by null mutants. Biochem Pharmacol. 2011;82:828–841. doi: 10.1016/j.bcp.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker JB, Hu M. Sex differences in drug abuse. Front Neuroendocrin. 2008;29:36–47. doi: 10.1016/j.yfrne.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best D, Rawaf S, Rowley J, Floyd K, Manning V, Strang J. Drinking and smoking as concurrent predictors of illicit drug use and positive drug attitudes in adolescents. Drug Alcohol Depen. 2000;60:319–321. doi: 10.1016/s0376-8716(00)00113-7. [DOI] [PubMed] [Google Scholar]
- Biondolillo KD, Pearce AR. Availability influences initial and continued ingestion of nicotine by adolescent female rats. Neuropsychobiology. 2007;55:73–80. doi: 10.1159/000103905. [DOI] [PubMed] [Google Scholar]
- Bito-Onon JJ, Simms JA, Chatterjee S, Holgate J, Bartlett SE. Varenicline, a partial agonist at neuronal nicotinic acetylcholine receptors, reduces nicotine-induced increases in 20% ethanol operant self-administration in Sprague-Dawley rats. Addict Biol. 2011;16:440–449. doi: 10.1111/j.1369-1600.2010.00309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornson W, Rand C, Connett JE, Lindgren P, Nides M, Pope F, Buist A, Hoppe-Ryan C, O'Hara P. Gender differences in smoking cessation after 3 years in the lung health study. Am J Public Health. 1995;85:223–230. doi: 10.2105/ajph.85.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomqvist O, Ericson M, Engel JA, Söderpalm B. Accumbal dopamine overflow after ethanol: localization of the antagonizing effect of mecamylamine. Eur J Pharmacol. 1997;334:149–156. doi: 10.1016/s0014-2999(97)01220-x. [DOI] [PubMed] [Google Scholar]
- Blomqvist O, Ericson M, Johnson DH, Engel JA, Söderpalm B. Voluntary ethanol intake in the rat: effects of nicotinic acetylcholine receptor blockade or subchronic nicotine treatment. Eur J Pharmacol. 1996;314:257–267. doi: 10.1016/s0014-2999(96)00583-3. [DOI] [PubMed] [Google Scholar]
- Bobo JK, Husten C. Sociocultural influences on smoking and drinking. Alcohol Research & Health: The Journal of the National Institute on Alcohol Abuse and Alcoholism. 2000;24:225–232. [PMC free article] [PubMed] [Google Scholar]
- Borghese CM, Henderson LA, Bleck V, Trudell JR, Harris RA. Sites of Excitatory and inhibitory actions of alcohols on neuronal α2β4 nicotinic acetylcholine receptors. J Pharmacol Exp Ther. 2003;307:42–52. doi: 10.1124/jpet.102.053710. [DOI] [PubMed] [Google Scholar]
- Breslau N, Peterson EL. Smoking cessation in young adults: age at initiation of cigarette smoking and other suspected influences. Am J of Public Health. 1996;86:214–220. doi: 10.2105/ajph.86.2.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns BE, Proctor WR. Cigarette smoke exposure greatly increases alcohol consumption in adolescent C57BL/6 mice. Alcohol Clin Exp Res. 2013;37:E364–372. doi: 10.1111/j.1530-0277.2012.01911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control. Current cigarette smoking among adults — United States, 2011. Morbidity and Mortality Weekly Report. 2011a;61:889–894. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6144a2.htm. [PubMed] [Google Scholar]
- Centers for Disease Control. Smoking and tobacco use: Chemicals in tobacco smoke. 2011b Available at: http://www.cdc.gov/tobacco/data_statistics/sgr/2010/consumer_booklet/chemicals_smoke/
- Chassin L, Presson CC, Pitts SC, Sherman SJ. The natural history of cigarette smoking from adolescence to adulthood in a midwestern community sample: multiple trajectories and their psychosocial correlates. Health Psychol. 2000;19:223–231. [PubMed] [Google Scholar]
- Chen X, Unger JB, Palmer P, Weiner MD, Johnson CA, Wong MM, Austin G. Prior cigarette smoking initiation predicting current alcohol use: Evidence for a gateway drug effect among California adolescents from eleven ethnic groups. Addict Behav. 2002;27:799–817. doi: 10.1016/s0306-4603(01)00211-8. [DOI] [PubMed] [Google Scholar]
- Clark A, Lindgren S, Brooks SP, Watson WP, Little HJ. Chronic infusion of nicotine can increase operant self-administration of alcohol. Neuropharmacology. 2001;41:108–117. doi: 10.1016/s0028-3908(01)00037-5. [DOI] [PubMed] [Google Scholar]
- Corrigall WA, Coen KM, Adamson KL. Self-administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res. 1994;653:278–284. doi: 10.1016/0006-8993(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Crews FT, Braun CJ, Hoplight B, Switzer RC, Knapp DJ. Binge ethanol consumption causes differential brain damage in young adolescent tats compared with adult rats. Alcohol Clin Exp Res. 2000;24:1712–1723. [PubMed] [Google Scholar]
- Dohrman DP, Reiter CK. Chronic ethanol reduces nicotine-induced dopamine release in PC12 cells. Alcohol Clin Exp Res. 2003;27:1846–1851. doi: 10.1097/01.ALC.0000095923.41707.C8. [DOI] [PubMed] [Google Scholar]
- Donny EC, Caggiula AR, Rose C, Jacobs KS, Mielke MM, Sved AF. Differential effects of response-contingent and response-independent nicotine in rats. Eur J Pharmacol. 2000;402:231–240. doi: 10.1016/s0014-2999(00)00532-x. [DOI] [PubMed] [Google Scholar]
- Doura MB, Gold AB, Keller AB, Perry DC. Adult and periadolescent rats differ in expression of nicotinic cholinergic receptor subtypes and in the response of these subtypes to chronic nicotine exposure. Brain Res. 2008;1215:40–52. doi: 10.1016/j.brainres.2008.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon WM, Dong Y, Ostroumov A, Thomas AM, Zhang TA, Dani JA. Nicotine decreases ethanol-induced dopamine signaling and increases self-administration via stress hormones. Neuron. 2013;79:530–540. doi: 10.1016/j.neuron.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericson M, Molander A, Löf E, Engel JA, Söderpalm B. Ethanol elevates accumbal dopamine levels via indirect activation of ventral tegmental nicotinic acetylcholine receptors. Eur J Pharmacol. 2003;467:85–93. doi: 10.1016/s0014-2999(03)01564-4. [DOI] [PubMed] [Google Scholar]
- Gogtay N, Giedd JN, Lusk L, Hayashi KM, Greenstein D, Vaituzis AC, Nugen TF, Herman DH, Clasen LS, Toga AW, Rapoport JL, Thompson PM. Dynamic mapping of human cortical development during childhood through early adulthood. Proc Natl Acad Sci USA. 2004;101:8174–8179. doi: 10.1073/pnas.0402680101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. 2006;27:482–491. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Gould E, Woolf NJ, Butcher LL. Postnatal development of cholinergic neurons in the rat: I. Forebrain. Brain Res Bull. 1991;27:767–789. doi: 10.1016/0361-9230(91)90209-3. [DOI] [PubMed] [Google Scholar]
- Grady SR, Salminen O, Laverty DC, Whiteaker P, McIntosh JM, Collins AC, Marks MJ. The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem Pharmacol. 2007;74:1235–1246. doi: 10.1016/j.bcp.2007.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grucza RA, Bierut LJ. Cigarette smoking and the risk for alcohol use disorders among adolescent drinkers. Alcoholism, Clinical and Experimental Research. 2006;30(12):2046–2054. doi: 10.1111/j.1530-0277.2006.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Knutson B. The reward circuit: linking primate anatomy and human imaging. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacol. 2010;35:4–26. doi: 10.1038/npp.2009.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder S, Lynch JM, Pearce AR. The multiple bottle effect is overridden in male and female rats by simultaneous presentation of two oral nicotine solutions. Am J Drug Alcohol Abuse. 2013;39:161–167. doi: 10.3109/00952990.2013.776065. [DOI] [PubMed] [Google Scholar]
- Hatsukami D, Skoog K, Allen S, Bliss R. Gender and the effects of different doses of nicotine gum on tobacco withdrawal symptoms. Exp Clin Psychopharmacol. 1995;3:163–173. [Google Scholar]
- Hendrickson L, Guildford M, Tapper A. Neuronal nicotinic acetylcholine receptors: common molecular substrates of nicotine and alcohol dependence. Front Psychiatry. 2013;429:1–16. doi: 10.3389/fpsyt.2013.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson LM, Zhao-Shea R, Pang X, Gardner PD, Tapper AR. Activation of alpha4 nAChRs is necessary and sufficient for varenicline-induced reduction of alcohol consumption. J Neurosci. 2010;30:10169–10176. doi: 10.1523/JNEUROSCI.2601-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson LM, Zhao-Shea R, Tapper AR. Modulation of ethanol drinking-in-the-dark by mecamylamine and nicotinic acetylcholine receptor agonists in C57BL/6J mice. Psychopharmacology. 2009;204:563–572. doi: 10.1007/s00213-009-1488-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hymowitz N, Cummings KM, Hyland A, Lynn WR, Pechacek TF, Hartwell TD. Predictors of smoking cessation in a cohort of adult smokers followed for five years. Tob Control. 1997;6:S57–S62. doi: 10.1136/tc.6.suppl_2.s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperato A, Mulas A, Di Chiara G. Nicotine preferentially stimulates dopamine release in the limbic system of freely moving rats. Eur J Pharmacology. 1986;132:337–338. doi: 10.1016/0014-2999(86)90629-1. [DOI] [PubMed] [Google Scholar]
- Isiegas C, Mague SD, Blendy JA. Sex differences in response to nicotine in C57Bl/6:129SvEv mice. Nicotine Tob Res. 2009;11:851–858. doi: 10.1093/ntr/ntp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PB, Boles SM, Vaughan R, Kleber HD. The co-occurrence of smoking and binge drinking in adolescence. Addict Behav. 2000;25:779–783. doi: 10.1016/s0306-4603(99)00066-0. [DOI] [PubMed] [Google Scholar]
- Jones S, Sudweeks S, Yakel JL. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22:555–561. doi: 10.1016/s0166-2236(99)01471-x. [DOI] [PubMed] [Google Scholar]
- Kamens HM, Hoft NR, Cox RJ, Miyamoto JH, Ehringer MA. The α6 nicotinic acetylcholine receptor subunit influences ethanol-induced sedation. Alcohol. 2012;46:463–471. doi: 10.1016/j.alcohol.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein LC, Stine MM, Pfaff DW, Vandenbergh DJ. Maternal nicotine exposure increases nicotine preference in periadolescent male but not female C57Bl/6J mice. Nicotine Tob Res. 2003;5:117–124. doi: 10.1080/14622200307257. [DOI] [PubMed] [Google Scholar]
- Klein LC, Stine MM, Vandenbergh DJ, Whetzel CA, Kamens HM. Sex differences in voluntary oral nicotine consumption by adolescent mice: a dose-response experiment. Pharmacol Biochem Behav. 2004;78:13–25. doi: 10.1016/j.pbb.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Larsson A, Edström L, Svensson L, Söderpalm B, Engel JA. Voluntary ethanol intake increases extracellular acetylcholine levels in the ventral tegmental area in the rat. Alcohol Alcoholism. 2005;40:349–358. doi: 10.1093/alcalc/agh180. [DOI] [PubMed] [Google Scholar]
- Laviola G, Marco EM. Passing the knife edge in adolescence: Brain pruning and specification of individual lines of development. Neurosci Biobehav Rev. 2011;35:1631–1633. doi: 10.1016/j.neubiorev.2011.05.011. [DOI] [PubMed] [Google Scholar]
- Laviola G, Adriani W, Terranova ML, Gerra G. Psychobiological risk factors for vulnerability to psychostimulants in human adolescents and animal models. Neurosci Biobehav Rev. 1999;23(7):993–1010. doi: 10.1016/s0149-7634(99)00032-9. [DOI] [PubMed] [Google Scholar]
- Laviola G, Macrì S, Morley-Flectcher S, Adriani W. Risk-taking behavior in adolescent mice: psychobiological determinants and early epigenetic influence. Neurosci Biobehav Rev. 2003;27:19–31. doi: 10.1016/s0149-7634(03)00006-x. [DOI] [PubMed] [Google Scholar]
- Lê AD, Li Z, Funk D, Shram M, Li TK, Shaham Y. Increased vulnerability to nicotine self-administration and relapse in alcohol-naive offspring of rats selectively bred for high alcohol intake. J Neurosci. 2006;26:1872–1879. doi: 10.1523/JNEUROSCI.4895-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewinsohn PM, Rohde P, Brown RA. Level of current and past adolescent cigarette smoking as predictors of future substance use disorders in young adulthood. Addiction. 1999;94:913–921. doi: 10.1046/j.1360-0443.1999.94691313.x. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhao-Shea R, McIntosh JM, Gardner PD, Tapper AR. Nicotine persistently activates ventral tegmental area dopaminergic neurons via nicotinic acetylcholine receptors containing α4 and α6 subunits. Mol Pharmacol. 2012;81:541–548. doi: 10.1124/mol.111.076661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhao-Shea R, McIntosh JM, Tapper AR. Nicotinic acetylcholine receptors containing the α6 subunit contribute to ethanol activation of ventral tegmental area dopaminergic neurons. Biochem Pharmacol. 2013;86:1194–1200. doi: 10.1016/j.bcp.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomazzo E, Hussmann GP, Wolfe BB, Yasuda RP, Perry DC, Kellar KJ. Effects of chronic nicotine on heteromeric neuronal nicotinic receptors in rat primary cultured neurons. J Neurochem. 2011;119:153–164. doi: 10.1111/j.1471-4159.2011.07408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, McClure-Begley TD, Whiteaker P, Salminen O, Brown RWB, Cooper J, et al. Lindstrom JM. Increased nicotinic acetylcholine receptor protein underlies chronic nicotine-induced up-regulation of nicotinic agonist binding sites in mouse brain. J Pharmacol Exp Ther. 2011;337:187–200. doi: 10.1124/jpet.110.178236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1992;12:2765–2784. doi: 10.1523/JNEUROSCI.12-07-02765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Rowell PP, Cao JZ, Grady SR, McCallum SE, Collins AC. Subsets of acetylcholine-stimulated 86Rb+ efflux and [125I]-epibatidine binding sites in C57BL/6 mouse brain are differentially affected by chronic nicotine treatment. Neuropharmacology. 2004;46:1141–1157. doi: 10.1016/j.neuropharm.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Stitzel JA, Romm E, Wehner JM, Collins AC. Nicotinic binding sites in rat and mouse brain: comparison of acetylcholine, nicotine, and alpha-bungarotoxin. Molecular Pharmacology. 1986;30:427–436. [PubMed] [Google Scholar]
- Nestler EJ. Is there a common molecular pathway for addiction? Nat Neurosci. 2005;8:1445–1449. doi: 10.1038/nn1578. [DOI] [PubMed] [Google Scholar]
- Olausson P, Ericson M, Löf E, Engel JA, Söderpalm B. Nicotine-induced behavioral disinhibition and ethanol preference correlate after repeated nicotine treatment. Eur J Pharmacol. 2001;417:117–123. doi: 10.1016/s0014-2999(01)00903-7. [DOI] [PubMed] [Google Scholar]
- Omelchenko N, Sesack SR. Laterodorsal tegmental projections to identified cell populations in the rat ventral tegmental area. J Comp Neurol. 2005;483:217–235. doi: 10.1002/cne.20417. [DOI] [PubMed] [Google Scholar]
- Omelchenko N, Sesack SR. Cholinergic axons in the rat ventral tegmental area synapse preferentially onto mesoaccumbens dopamine neurons. J Compar Neurol. 2006;494:863–875. doi: 10.1002/cne.20852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly JR, Marks MJ, Robinson SF, van de Kamp JL, Collins AC. Chronic nicotine and mecamylamine treatment increase brain nicotinic receptor binding without changing alpha 4 or beta 2 mRNA levels. J Pharmacol Exp Ther. 1996;278:361–369. [PubMed] [Google Scholar]
- Pekonen K, Karlsson C, Laakso I, Ahtee L. Plasma nicotine and cotinine concentrations in mice after chronic oral nicotine administration and challenge doses. Eur J Pharm Sci. 1993;1:13–18. [Google Scholar]
- Picciotto MR, Addy NA, Mineur YS, Brunzell DH. It is not “either/or”: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol. 2008;84:329–342. doi: 10.1016/j.pneurobio.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Caldarone BJ, Brunzell DH, Zachariou V, Stevens TR, King SL. Neuronal nicotinic acetylcholine receptor subunit knockout mice: physiological and behavioral phenotypes and possible clinical implications. Pharmacology & Therapeutics. 2001;92:89–108. doi: 10.1016/s0163-7258(01)00161-9. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. 2012;76:116–129. doi: 10.1016/j.neuron.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidoplichko VI, Noguchi J, Areola OO, Liang Y, Peterson J, Zhang T, Dani JA. Nicotinic cholinergic synaptic mechanisms in the ventral tegmental area contribute to nicotine addiction. Learn Memory. 2004;11:60–69. doi: 10.1101/lm.70004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogun S, Yararbas G. Sex differences in nicotine action. Handb Exp Pharmacol. 2009;192:261–291. doi: 10.1007/978-3-540-69248-5_10. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Ford MM, Yu CH, Brown LL, Finn DA, Garland T, Crabbe JC. Mouse inbred strain differences in ethanol drinking to intoxication. Genes Brain Behav. 2007;6:1–18. doi: 10.1111/j.1601-183X.2006.00210.x. [DOI] [PubMed] [Google Scholar]
- Roesch MR, Olson CR. Neuronal activity related to reward value and motivation in primate frontal cortex. Science. 2004;304:307–310. doi: 10.1126/science.1093223. [DOI] [PubMed] [Google Scholar]
- Rose JS, Chassin L, Presson CC, Sherman SJ. Prospective predictors of quit attempts and smoking cessation in young adults. Health Psychol. 1996;15:261–268. doi: 10.1037//0278-6133.15.4.261. [DOI] [PubMed] [Google Scholar]
- Schwartz RD, Kellar KJ. In Vivo Regulation of [3H]Acetylcholine Recognition Sites in Brain by Nicotinic Cholinergic Drugs. J Neurochem. 1985;45:427–433. doi: 10.1111/j.1471-4159.1985.tb04005.x. [DOI] [PubMed] [Google Scholar]
- Smith BR, Horan JT, Gaskin S, Amit Z. Exposure to nicotine enhances acquisition of ethanol drinking by laboratory rats in a limited access paradigm. Psychopharmacology. 1999;142:408–412. doi: 10.1007/s002130050906. [DOI] [PubMed] [Google Scholar]
- Sparks JA, Pauly JR. Effects of continuous oral nicotine administration on brain nicotinic receptors and responsiveness to nicotine in C57Bl/6 mice. Psychopharmcology. 1999;141:145–153. doi: 10.1007/s002130050818. [DOI] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000a;24:417–463. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Spear LP. Neurobehavioral changes in adolescence. Curr Dir Psychol Sci. 2000b;9:111–114. [Google Scholar]
- Spear LP. Adolescent neurodevelopment. J Adolescent Health. 2013;52:S7–S13. doi: 10.1016/j.jadohealth.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP. Adolescents and alcohol: acute sensitivities, enhanced intake, and later consequences. Neurotoxicol Teratol. 2014;41:51–59. doi: 10.1016/j.ntt.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele TE, Navarro M. “Drinking in the dark” (DID) procedures: A model of binge-like ethanol drinking in non-dependent mice. Alcohol. 2014;48:235–41. doi: 10.1016/j.alcohol.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torabi MR, Bailey WJ, Majd-Jabbari M. Cigarette smoking as a predictor of alcohol and other drug use by children and adolescents: Evidence of the “gateway drug effect”. J School Health. 1993;63:302–306. doi: 10.1111/j.1746-1561.1993.tb06150.x. [DOI] [PubMed] [Google Scholar]
- Trauth JA, Seidler FJ, McCook EC, Slotkin TA. Adolescent nicotine exposure causes persistent upregulation of nicotinic cholinergic receptors in rat brain regions. Brain Res. 1999;851:9–19. doi: 10.1016/s0006-8993(99)01994-0. [DOI] [PubMed] [Google Scholar]
- United States Department of Health and Human Services. Preventing tobacco use among young people: A report of the surgeon general. Atlanta, Georgia: U.S Department of Health and Human Services, CDC, National Center for Chronic Disease Prevention and Health Promotion Office on Smoking and Health; 1994. [Google Scholar]
- United States Department Health and Human Services. A report of the Surgeon General. Washington, DC: U.S Government Printing Office; 1988. The health consequences of smoking: nicotine addiction. [Google Scholar]
- Zambrano CA, Salamander RM, Collins AC, Grady SR, Marks MJ. Regulation of the distribution and function of [125I]epibatidine binding sites by chronic nicotine in mouse embryonic neuronal cultures. The Journal of Pharmacology and Experimental Therapeutics. 2012;342(2):245–254. doi: 10.1124/jpet.112.192542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrindast MR, Meshkani J, Rezayof A, Beigzadeh R, Rostami P. Nicotinic acetylcholine receptors of the dorsal hippocampus and the basolateral amygdala are involved in ethanol-induced conditioned place preference. Neuroscience. 2010;168:505–513. doi: 10.1016/j.neuroscience.2010.03.019. [DOI] [PubMed] [Google Scholar]