

Abstract
Dietary potassium deficiency activates thiazide-sensitive sodium chloride cotransport along the distal nephron. This may explain, in part, the hypertension and cardiovascular mortality observed in individuals who consume a low potassium diet. Recent data suggest plasma potassium affects the distal nephron directly by influencing intracellular chloride, an inhibitor of the With no lysine kinase (WNK)-Ste20p-related proline-and alanine-rich kinase (SPAK) pathway. Since previous studies used extreme dietary manipulations, we sought to determine if the relationship between potassium and NCC is physiologically relevant and clarify the mechanisms involved. We report that modest changes in both dietary and plasma potassium affect the thiazide-sensitive sodium-chloride cotransporter, NCC, in vivo. Kinase assay studies showed that chloride inhibits WNK4 kinase activity at lower concentrations than it inhibits activity of WNK1 or WNK3. Also, chloride inhibited WNK4 within the range of distal cell chloride. Mutation of a previously identified WNK chloride-binding motif converted WNK4 effects on SPAK from inhibitory to stimulatory in mammalian cells. Disruption of this motif in WNKs 1, 3 and 4 had different effects on NCC, consistent with the three WNKs having different chloride sensitivities. Thus, potassium effects on NCC are graded within the physiological range, which explains how unique chloride-sensing properties of WNK4 enable kinase mediating effects of potassium on NCC in vivo.
Keywords: distal tubule, mineral metabolism, potassium channels, diuretics, cell signaling, potassium, distal convoluted tubule, thiazide-sensitive NaCl cotransporter, chloride
Introduction
The antihypertensive effect of dietary potassium (K+) supplementation has been recognized since the early part of the 20th century.1 Mechanistic explanations have been abundant,2 generally focusing on extra-renal systems as sites of primary action, with few authors attributing a primary role for the nephron in sensing and responding to K+ variation. Despite this, evidence for such a primary renal role is supported by a comprehensive model of long-term K+ regulation reported thirty years ago.3
Supporting this model, we recently reported on the molecular mechanism4 by which the kidney senses and responds to changes in K+ balance. Changes in plasma [K+] affect the membrane potential of renal epithelial cells in the distal convoluted tubule (DCT), thereby altering their intracellular chloride concentration ([Cl-]i). Such an effect might occur in multiple cell types in various organs, but two important facts cause these changes in the DCT to have great implications for K+ and blood pressure homeostasis.
First, DCT cells are the only cell type in the kidney to express the thiazide-sensitive Na-Cl cotransporter (NCC). NCC and its molecular regulators, Ste20p-related proline- and alanine-rich kinase (SPAK) and With no lysine (WNK) kinases, have been studied intensely since their dysregulation was implicated in the pathogenesis of Familial Hyperkalemic Hypertension (FHHt, also called pseudohypoaldosteronism type 2 or Gordon syndrome).5-7 Recent identification of a Cl--binding motif in WNK1 which, when bound, affects WNK1 autophosphorylation,8 helps explain how WNKs may mediate effects of altered [Cl-]i on NCC phosphorylation and, in turn, activity.9
Second, NCC-expressing DCT cells lie directly upstream of the connecting tubule (CNT) and cortical collecting duct (CCD). The CNT and CCD contain aldosterone-sensitive epithelial cells that are key sites of K+ regulation. Furthermore, their K+-secretory activity is modulated by sodium (Na+) delivery. Sodium reabsorption by the epithelial Na+ channel (ENaC) in these cells drives electrogenic K+ secretion via apical K+ channels.
Taken together, these observations imply that, when DCT cells become depolarized by high plasma [K+], [Cl-]i rises, inhibiting WNK kinase activity and, thus, NCC. This promotes a natriuresis, which will have antihypertensive effects and increase Na+ delivery to the CNT and CCD, driving K+ secretion. This scenario corresponds well with mathematical models of distal nephron function.4, 10
Although studies by our laboratory,4 and other groups,11-15 indicate that NCC phosphorylation and abundance are modulated by K+ balance, these studies have typically involved extreme manipulations of plasma [K+], using dietary and pharmacological approaches. Thus, these findings could have resulted from pathophysiologic perturbations without relevance to physiological balance. Given the narrow range in which plasma [K+] is regulated, data obtained at K+ concentrations less than 3mM and greater than 6mM should not be used to infer effects within a narrower range.
If variations in plasma [K+] within the physiological range do influence NCC, this raises a question concerning the signaling pathway involved. WNKs 1,5 3,16 and 4,5 are all expressed in the DCT, but we proposed that WNK4 plays a dominant role in the response to plasma [K+]. While there has been a consensus that WNKs 17 and 316 stimulate NCC activity, the effects of WNK4 on NCC activity have been more controversial.17 Studies employing heterologous expression of WNK4 in Xenopus oocytes6, 7 and mammalian cell systems18, 19 typically report that WNK4 inhibits NCC. In contrast, data from WNK4-/- mice suggest that WNK4 stimulates NCC.20, 21 Recently, Bazúa-Valenti et al. reported that WNK4 activity is modulated in Xenopus oocytes by exposure to low chloride hypotonic solution. This provides a potential explanation for the discordant WNK4 data,22 as the [Cl-]i in oocytes is near 50 mM.22 Here, we test the hypotheses that physiologically relevant differences in dietary K+ intake modulate both plasma [K+] and NCC, and that unique Cl--sensing properties of WNK4, distinct from those of WNK1 and WNK3, make it able to transduce these physiological K+ changes to NCC activity.
Results
Phosphorylated NCC and plasma [K+] are correlated across a range of plasma [K+] values
To determine if the inverse relationship between NCC and plasma [K+] is continuous across a range of plasma [K+], we examined pooled data from multiple mouse experiments in which dietary Na+ and K+ manipulation, aldosterone infusion, and combinations of these (described in Materials and Methods) were used (Fig 1). Taken together, the results suggest that pNCC abundance is a linear function of plasma [K+] within the investigated range. According to the model, from 4.5 mM to 3.0 mM, every 0.1 mM decrease in plasma [K+] is associated with a 15% increase (relative to baseline) in normalized pNCC abundance.
Figure 1. The relationship between pNCC and plasma [K+] is linear across physiological levels of [K+].
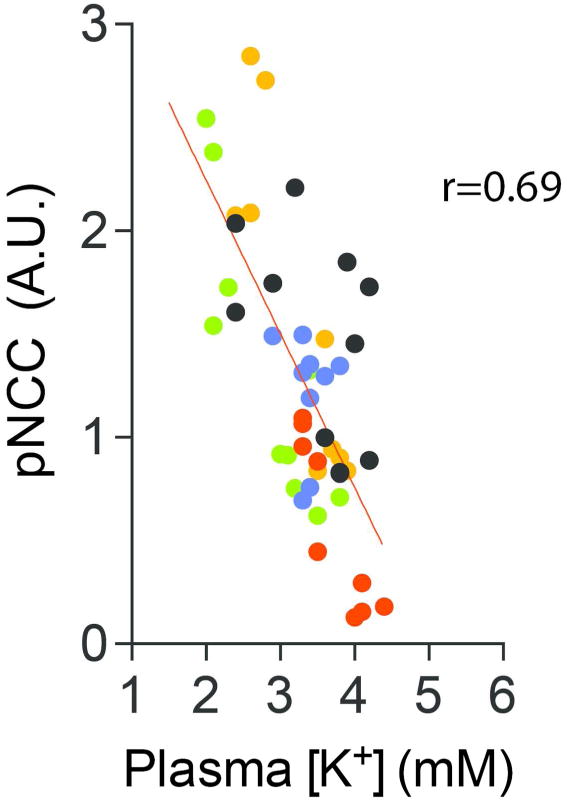
Pooled data from multiple animal experiments (see Methods for details) were correlated to determine the relationship between pNCC and plasma [K+]. Phospho-NCC-T53 abundance was normalized to β-actin and each experiment had an internal control group (vehicle treatment or normal diet) whose pNCC average was set to 1. For each experiment, treatment groups were compared to their internal control group on normal diet. p<0.0001 for correlation. All mice used were wild-type. Colors indicate different experiments. Red: C57Bl/6 mice on normal (1%) or high K+ (5%) diet, Blue: C57Bl/6 mice on normal (0.49%) or low NaCl (0.01-0.02%) diet, Black: C57Bl/6 mice that were either sham-treated or aldosterone-infused, Orange: Same as black, but BALB/c mice, Green: C57Bl/6 mice that were either sham-treated (on normal K+ diet), aldosterone-infused (on normal K+ diet), or aldosterone-infused (on high K+ diet).
Modest changes in dietary K+ content affect plasma [K+] and NCC in a graded fashion
Most studies investigating the relation between NCC abundance and K+, including ours, have employed severe dietary K+ manipulations with 0 to 0.03% K+ used for a low K+ diet and 5% K+ used for a high K+ diet.4, 11, 12, 14 As these diets are unphysiological, we tested effects of more modest dietary K+ changes on plasma [K+] and NCC. A dietary K+ titration was performed and effects on pNCC abundance were determined. The K+ titration produced a linear increase in plasma [K+] ranging from 2.90 to 3.85 mM (Fig 2a). The slope of the relation between dietary K+ and plasma [K+] was linear and positive. The linear increase in plasma [K+] was correlated with a linear decrease in NCC and pNCC abundance (Fig 2b, c, d), suggesting that the inverse relationship persists throughout the normal range of dietary K+ consumption and plasma [K+].
Figure 2. Effects of increasing dietary K+ content on plasma [K+] and NCC.
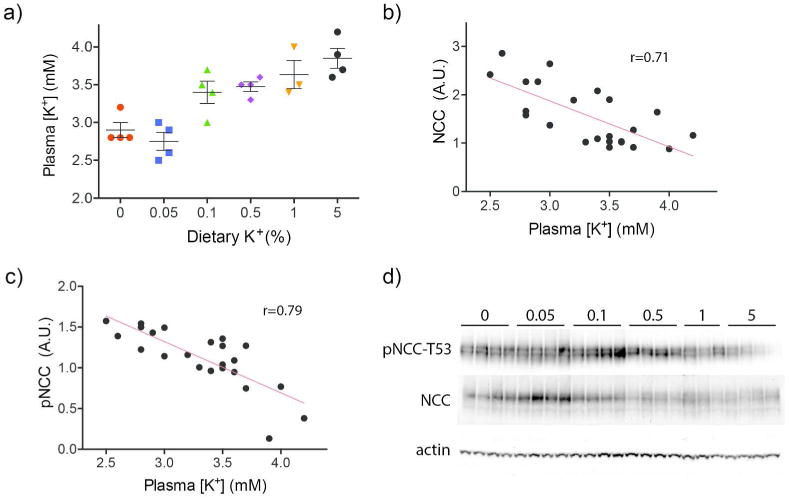
a) Gradual increases in dietary K+ content produced increases in plasma [K+] levels in mice. Colors are unrelated to colors used in Figure 1. b) NCC abundance and plasma [K+] were negatively correlated. p<0.001 for correlation. c) Phospho-NCC abundance and plasma [K+] were negatively correlated. p<0.0001 for correlation. d) Western blots used to quantify NCC and pNCC abundance for b and c. All values were normalized to actin. Animals were fed a K+-deficient diet (see Methods for details) supplemented with indicated amounts of KCl for 10 days. Animals used for Figure 2 are different from those used for Figure 1.
Chloride inhibits WNK4 kinase activity more potently than that of WNK1 or WNK3
These data suggest that plasma [K+] has a dominant effect on NCC activity across a range of values (we infer that pNCC abundance is a marker for NCC activity 9). We proposed previously that this response is mediated by changes in [Cl-]i, in response to altered membrane potential, and recapitulated it in HEK cells, grown in culture.4 Although these cells are not DCT cells, they can express NCC robustly, and phosphorylate it in a SPAK and WNK-dependent manner.4 In cultured HEK cells, where [Cl-]i is estimated to be greater than 40mM,4, 23 the response appears to be mediated by endogenous WNK1 and WNK3. In contrast, in mice on a low K+ diet, WNK4 abundance in the DCT is increased.4 Coupled with findings from WNK4 knockout studies,20, 21 this suggests that WNK4 may be especially important in vivo. Bazúa-Valenti et al.22 recently suggested that the effects of WNK4 are modulated by [Cl-]i, but this model requires that Cl- affects WNK4 at low physiological [Cl-]i, much lower than found in HEK cells grown in culture. They suggested that WNK4 has a different [Cl-] set point than WNK1 or WNK3, accounting for its unique behavior. Those experiments did not, however, examine the set point directly, as they used expression in Xenopus oocytes, and they tested the combined effects of modifying extracellular tonicity and [Cl-] together.
According to a model we previously developed, variations in extracellular [K+] such as those studied above would generate [Cl-]i concentrations in DCT cells of 10-20 mM. Given this, the data of Bazúa-Valenti et al.22, and the proposed importance of WNK4 for this effect, we determined the Cl- sensitivity of WNK4, and compared it to that of WNK1 and WNK3. In vitro kinase assays were performed using the various WNK kinase domains (WNKKD) and SPAK as the physiologically-relevant kinase target. Our approach was similar to that reported by Piala and colleagues, except that they employed the generic substrate, myelin basic protein (MBP) for most of their studies.8 We used E. coli to produce the three WNKs, a system that has been shown to yield phosphorylated and active WNKs.8 Although Cl- inhibited all 3 WNKs, it inhibited WNK4KD kinase activity at much lower concentrations than it did WNK1KD or WNK3KD (Fig 3a, b). WNK4KD was inhibited in the physiological range for DCT [Cl-]i 10, 24, 25 with the most potent effects between 0 and 40 mM (Fig 3e,f). WNK1KD and WNK3KD were inhibited at much higher levels, with WNK1KD being inhibited between 60 and 150 mM and WNK3KD inhibited between 100 and 150 mM (Fig 3c, d). Note, that a fraction of the GST fusion SPAK protein was truncated on its C-terminus during preparation (Supplemental Fig 1). Phosphorylation of both bands was quantified for these studies.
Figure 3. Effects of [Cl-] on in vitro WNK kinase activity using SPAK as a substrate.
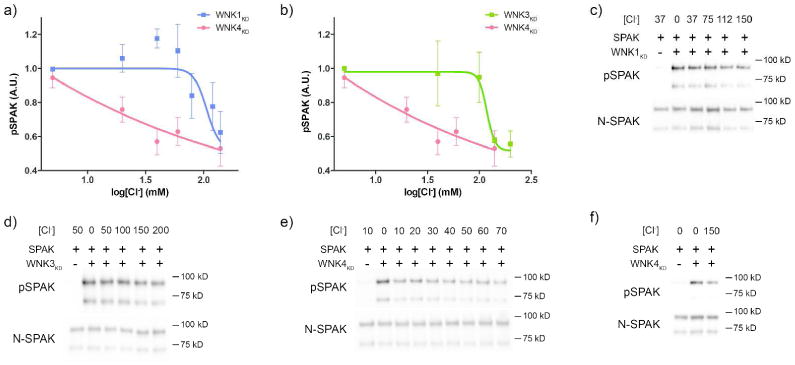
a) Effects of increasing [Cl-] on kinase activity on the WNK1 kinase domain (WNK1KD) and the WNK4 kinase domain (WNK4KD) in vitro. SPAK was used as the substrate. WNK4KD activity was inhibited at much lower [Cl-] than WNK1KD. For WNK4KD, N=4 for all data points except log[Cl-]=2.2mM where N=3. N=7 for WNK1KD. b) Effects of increasing [Cl-] on kinase activity of the WNK3 kinase domain (WNK3KD) and the WNK4 kinase domain (WNK4KD) in vitro as in a. WNK4KD activity was inhibited at much lower [Cl-] than WNK3KD. N=5 for WNK3KD. WNK4KD data is the same as shown in a. c) Representative Western blots for pSPAK and total SPAK abundance from a WNK1KD kinase assay with increasing [Cl-] (mM). d) Representative Western blots for pSPAK and total SPAK abundance from a WNK3KD kinase assay with increasing [Cl-] (mM). e) Representative Western blots for pSPAK and total SPAK abundance from a WNK4KD kinase assay with increasing [Cl-] between 0 and 70 mM. f) Same as e, but for 0mM and 150mM [Cl-].
Mutating the WNK Cl-binding motif has different effects on WNK1, WNK3, and WNK4 in mammalian cells
In cultured cells and Xenopus oocytes, WNK4 has been observed commonly to inhibit SPAK (or oxidative stress response kinase, OxSR1; sometimes called OSR1) and NCC activity. This is in contrast to WNK1 and WNK3, which typically stimulate SPAK and NCC activity in the same model conditions. WNK4 inhibition of NCC activity in oocytes has been shown to require its ability to interact with other WNKs via its HQ domain.18 Thus, Bazua-Valenti and colleagues suggested that, in oocytes, WNK4 is inactivated by ambient [Cl-] and thereby exerts a dominant-negative effect.22 To test this hypothesis, we mutated the WNK4 Cl--sensing domain that is homologous to the WNK1 domain.8 Although wild-type WNK4 decreased pSPAK in HEK cells, consistent with prior reports, WNK4 L322F L324F increased it (Fig 4a, Supplemental Fig 2). The effects of WNK4 L322F L324F were kinase-dependent because mutation of the putative catalytic lysine (WNK4 L322F L324F K186M) returned pSPAK abundance to levels similar to that observed with WT WNK4 (Fig 4a). Mutating the Cl--sensing motif in the three different WNKs was also shown to have different effects on NCC phosphorylation. Consistent with the Cl-sensitivity reported by Piala et al.8, expression of the WNK1 mutant (WNK1 L369F L371F) increased pNCC by 140%, compared to expression of WT WNK1 (Fig 4b, e). In contrast, expression of the WNK3 Cl--sensing mutant (WNK3 L295F L297F) had little effect on pNCC abundance compared to expression of WT WNK3 (Fig 4c, e), supporting our observation (Fig 3) that WNK3 is the least sensitive to [Cl-]. Expression of WNK4 L322F L324F, on the other hand, increased pNCC abundance by 339% of that resulting from WT WNK4 expression (Fig 4d, e). Thus, the effects of WNK4 chloride-sensing mutations were substantially larger than those of WNK1 and WNK3 (Fig 4e).
Figure 4. Effects of mutating the WNK Cl--sensing motif on SPAK and NCC in HEK cells.
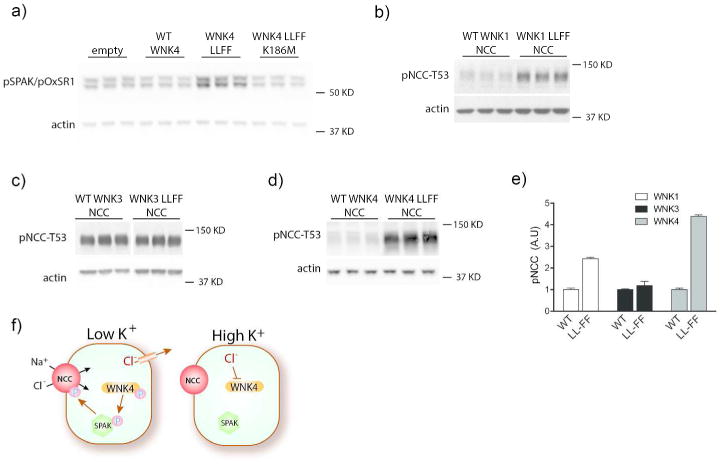
a) Western blot for pSPAK/pOxSR1 on HEK cells transfected with empty vector, WT WNK4, WNK4 L322F L324F (WNK4 LLFF), or kinase-dead WNK4 LLFF K186M. WT WNK4 reduced pSPAK/pOxSR1 abundance and WNK4 LLFF increased it. WNK4 LLFF K186M reduced pSPAK/pOxSR1 abundance to levels similar to WT WNK4. All effects were significant (p<0.05) by one-way ANOVA with Dunnett's multiple comparisons test. b-d) Western blots for pNCC-T53 in HEK cells expressing NCC and b) WT WNK1 or WNK1 L369F L371F, c) WT WNK3 or WNK3 L295F L297F, or d) WT WNK4 or WNK4 L322F L324F. LLFF mutations in WNK1 and WNK4 increased pNCC abundance. p<0.05 by unpaired t-test for both. WNK3 LLFF did not affect pNCC differently than WT WNK3. e) Bar chart comparing relative effects of the different WNK LLFF mutants versus their WT forms. p<0.05 by two-way ANOVA. Representative images are shown. f) Cartoon suggesting how K+ intake affects NCC. When plasma [K+] is low, chloride effluxes from cells, activating WNK4 kinase, which phosphorylates SPAK and therefore NCC. When plasma [K+] is high, higher intracellular chloride concentration inhibits WNK4 kinase activity so it cannot activate SPAK or NCC. Note that, for clarity, the cartoon shows phosphorylation events as ‘all or none’. These effects are likely highly graded in vivo.
Discussion
In humans, plasma [K+] is normally maintained within the narrow physiological range of 3.5 mM to 5.0 mM. Although substantial changes in dietary K+ intake are known to modulate NCC abundance and activity in animals,4, 11, 13-15, 18 it has not been clear whether K+ intake is physiologically important, or if non-physiological dietary K+ manipulations are required to affect the cotransporter. Here, we show that even modest changes in dietary K+ intake modify plasma [K+], and that these modest changes in plasma [K+] alter the abundance of phosphorylated NCC to a physiologically important degree.26, 27
Our pooled data indicate that the relationship between plasma [K+] and NCC is continuous across the physiological range of [K+]. By our calculations, a decrease in plasma [K+] from 4.5mM to 3mM is accompanied by an increase in pNCC abundance of 3-4 fold. Such a change in plasma [K+] would be predicted to reduce [Cl-]i from 18 to 13 mM.4 While [K+] changes of this magnitude are unlikely to cause arrhythmias or muscle weakness, such increases in pNCC abundance do affect electrolyte and blood pressure homeostasis.26, 27 Our data also suggest that dietary fluctuations in K+ content are able to influence plasma [K+] within this range. Several studies demonstrating diurnal variation in plasma [K+]28-30 indicate that plasma [K+] may indeed fluctuate around a given individual set point. Additionally, while population plasma [K+] values between 3.5mM and 5.0mM are normal, there may be individual plasma [K+] set points, as has been shown for plasma [Na+].31
The WNKs are key mediators of K+-sensing by DCT cells, owing in part to their Cl- responsiveness. Here, we identified a key difference between the WNKs expressed in the DCT. WNK4 kinase activity was highly Cl--sensitive, being inhibited at [Cl-] often present in DCT cells (10-20 mM).10, 24 WNKs 1 and 3 were inhibited only when [Cl-] was much. As WNK4 is highly expressed along the DCT,32 it appears to be essential for [K+] sensing there. The present results also explain how WNK1 can respond to extracellular K+ in our HEK cell model, which does not express WNK4. The [Cl-]i reported for HEK cells4, 23 is above 40 mM, much higher than that of DCT cells. It is near the range that we observed for WNK1 inhibition by Cl-. Therefore, in HEK cells, low [K+] can reduce [Cl-]i to activate WNK1, while in the DCT, with [Cl-]i in the range of 10-20 mM,4 WNK1 will not be inhibited; instead, WNK4 will act as the primary sensor.
These findings do not, however, negate a role for WNKs 1 and 3 in regulating NCC or other transport proteins, in vivo, but they do suggest that the nature of the effects may differ. While WNK4 has the characteristics of a physiological Cl- sensor, WNK1 and WNK3 do not, at least within cells of typical tetrapods. Thus, WNK1 and WNK3 may act as if they are constitutively active in mammalian cells, regulating transport based primarily on their abundance or localization. Even in neuronal cells, where WNKs have been postulated to modulate [Cl-]i, [Cl-]i does not lie within a range that would turn off WNK1 or WNK3.33 Conversely, as WNKs can heterodimerize and trans-autophosphorylate,34 interactions between WNK4 and WNK1 or WNK3 may participate in Cl--sensing, perhaps explaining the redistribution of WNKs 1 and 3 in the DCT in response to low K+ diet that we reported.4
Our findings generally accord with reports from other groups, but there are some key differences. In studies by Piala and colleagues,8 WNK1 kinase activity was inhibited by Cl-, with an IC50 of 40 mM, but the authors concluded that this property does not mediate Cl- sensitivity in vivo, because it was shared with other kinases that do not mediate Cl- signaling. Interestingly, the primary results were derived using a generic substrate, MBP, yet this group also tested the kinase activity of WNK1 against a physiological substrate, OxSR1 (also called OSR1). In this case, they observed an IC50 of 530 mM, a value outside the range of any mammalian cell. Because they found striking inhibition of WNK1 autophosphorylation by Cl-, in a concentration range compatible with physiological activity, they concluded that effects on autophosphorylation mediate effects of Cl- on kinase activity.
Our results were focused primarily on activity as the physiologically relevant parameter, so they do not provide direct information about autophosphorylation or trans-autophosphorylation. Several groups have documented that WNK kinases, as produced in E. coli, are phosphorylated and active.8, 35, 36 Instead, our results point to clear differences in kinase Cl- sensitivity between WNK4, WNK1, and WNK3. Our experimental approach differed somewhat from that taken by Piala and colleagues.8 First, an increase in [Cl-] in their experiments was also accompanied by a rise in tonicity, as they did not separate the two variables. We used an equimolar design in which either gluconate or Cl- was added to maintain tonicity. In this manner, our results support the conclusion of Uchida and colleagues that tonicity alone is not sensed by WNK kinases.37 Second, while Piala and colleagues demonstrated that Cl- modulated WNK1 activity toward MBP, we used the substrate, SPAK, the primary mediator of signaling to NCC.38, 39 Thus, our HEK cell culture results support the recent report by Bazúa-Valenti et al. in which they showed that WNK4 activity in Xenopus oocytes is modulated by [Cl-]i.22 Consistent with their hypothesis, our data indicate that disruption of the WNK4 Cl--sensing domain activates it to phosphorylate SPAK and NCC in a mammalian cell model.
The present results may provide insight into mechanisms of type 4 Renal Tubular Acidosis, as occurs in chronic kidney disease,40 sickle cell nephropathy,41 or urinary tract obstruction.42 In these settings, patients typically present with hyperkalemia and mild hyperchloremic acidosis, suggesting aldosterone deficiency. Yet the arterial pressure is often elevated and extracellular fluid volume is typically normal to high. Some patients exhibit low concentrations of both renin and aldosterone, but in others, aldosterone concentrations are normal. The current results suggest that a failure of the DCT cells to sense a rise in plasma [K+] may participate in this scenario. In this case, persistent electroneutral Na+-Cl-reabsorption by NCC would contribute to extracellular volume expansion, while impaired distal Na+ delivery would limit K+ secretion, even though aldosterone may be present.
Taken in sum, the data help to resolve the confusion surrounding WNK4. In cell and oocyte models, where [Cl-]i is high, WNK4 kinase activity is inhibited, while WNKs 1 and 3 are active. In this case, exogenously expressed WNK4 can bind to WNK1 or WNK3 and inhibit their activities.36, 43 The WNK4 carboxyl terminal domain, expressed by itself, can inhibit NCC, either by associating with it6, 44 or with protein phosphatases;45 thus, in the absence of kinase activity, WNK4 can be inhibitory suggesting additional negative regulatory mechanisms in vitro. In vivo, however, DCT [Cl-]i is low and WNK4 would tend to be more active; therefore, genetic ablation of WNK4 results in reduced NCC activity. As the effects of Cl- on WNK4 occur within the concentration range in the DCT, WNK4 is poised to sense plasma [K+] (Fig 4f). As suggested by Figure 2, however, the effects of plasma [K+] on pNCC will be graded, forming a molecular rheostat.26 The results indicate that quite modest changes in dietary K+ intake affect plasma [K+] and NCC activity. These effects are mediated largely by WNK4, as this kinase exhibits unique Cl--sensitive properties.
Methods
Animals
Studies were approved by Oregon Health and Science University's Animal Care and Usage Committee (Protocol IS3286). Mice were 12–24 weeks BALB/c or a C57Bl/6.
Pooled data presented in Figure 1 are from five mouse experiments. For figure 1, plasma [K+] and pNCC data were obtained from the following groups of animals: 1) 7-day aldosterone-infused (240 μg/kg/d, Alzet osmotic minipump model 1002) and sham-treated C57Bl/6 wild-type animals. 2) Same as 1), but BALB/c wild-type animals. 3) 7-day aldosterone-infused and sham-treated C57Bl/6 wild-type animals. Sham-treated mice were fed a normal K+ diet (1%). Aldosterone-infused animals were fed either normal or high K+ (5% K+) diet. Normal and high K+ diets were prepared by adding KCl to a K+-deficient diet (Harlan Laboratories K+-deficient diet; TD.88239) 4) 5 day normal (0.49% NaCl) and low NaCl diet- (Harlan Laboratories Na+-deficient (0.01-0.02% Na+) diet; TD.90228) fed C57Bl/6 wild-type animals. NaCl was added to the Na+-deficient to 0.49% for normal NaCl diet. 5) 3 day normal and high K+ diet-fed C57Bl/6 wild-type animals.
Data for figure 2 was obtained from a single mouse experiment using C57Bl/6 wild-type animals. Six groups of animals were each fed different amounts of K+ for 10 days. KCl was added to the K+-deficient diet to achieve indicated dietary K+ content for each group. Animals for Figure 2 were different from those for Figure 1.
Antibodies
Custom antibodies against NCC46 and pNCC-T5347 have been reported before. Commercial antibodies used were directed against β-actin (Abcam), myc (Sigma), pSPAK-S373/pOxSR1-S325 (Millipore), GST (Santa Cruz), and N-terminal SPAK (Santa Cruz). The C-terminal SPAK antibody was a generous gift of E. Delpire (Vanderbilt). The peptide used to block the N-terminal SPAK antibody was purchased from Santa Cruz.
Blood Analysis
Whole blood was collected via cardiac puncture at the completion of experiments. Plasma K+ values were obtained by iSTAT just after collection by loading whole blood into a chem 8 cartridge (Abbot Point of Care, Inc).
Immunoblotting
At the completion of experiments, kidneys were harvested and snap-frozen in liquid nitrogen followed by transfer to a -80°C freezer. Kidneys were then homogenized on ice in chilled buffer containing protease and phosphatase inhibitors. Protein (50 μg) was separated on 4-12% Bis-Tris gel (Invitrogen). Densitometry was performed using ImageJ (http://rsbweb.nih.gov/ij/).
Cell Culture
HEK 293 cells were cultured in DMEM with high glucose, sodium pyruvate, and L-Glutamine (HyClone) supplemented with 10% fetal bovine serum and penicillin/streptomycin. Cells were transfected with indicated constructs using Lipofectatime (Invitrogen), followed by harvesting and lysis 48 hours later. Total protein quantification and Western blots were then performed.
All WT WNK and NCC mammalian expression vectors have been previously reported.18 Human WNKs 1, 3, and 4 cDNAs were subcloned into pMO-myc, which uses the SV40 promoter to drive expression. Mouse NCC was subcloned into pcDNA3.1. All point mutations were made using a QuikChange Lightning kit (Agilent Technologies).
In vitro Kinase Assays
GST fusion proteins were made by subcloning rat WNK1 (4-1473)26, human WNK3 (4-1260),26 mouse WNK4 (4-1329),48 and full-length mouse SPAK cDNAs into pGex6P-1. Using a QuikChange Lightning kit (Agilent Technologies), the SPAK catalytic lysine (K104) was then mutated to an arginine, which has been shown to completely inhibit its kinase activity.49 All kinase assays used this kinase dead GST-SPAK K104R construct. Protein expression was induced in BL21 cells (Novagen).
Kinase assays were then performed by combining 0.25 ug of SPAK K104R with either 0.5μg of WNK1, 0.5μg of WNK3, or 5μg of WNK4 in kinase assay buffer (10mM HEPES, pH 8.0, 1mM benzamadine, 1mM DTT, and MgCl2/Mg gluconate) with 0.15mM cold ATP. MgCl2 and Mg gluconate were added to vary the chloride concentration for a given experiment while maintaining all reactions at the same osmolality. Chloride was added in the amount indicated in figures. Reactions were incubated at 37°C for 45 minutes then stopped by adding SDS PAGE sample buffer and heating for 5 mins at 95°C. Samples were then loaded onto a 3-8% tris-acetate gel (Invitrogen) or a 4-12% Bis-tris gel (Bio-Rad) and Western blots were performed to quantify phospho-SPAK Ser383 and total SPAK (N-terminal).
Statistics
A linear regression model (Prism) was used to fit data in figures 1 and 2. For WNK4 kinase assay data, a one phase exponential decay model was used. A four parameter log(inhibitor) vs response with variable slope model was used for WNK1 and WNK3 kinase assay data. For WNK1 and WNK3, the model was constrained to values less than 1.0. One-way ANOVA with Dunnett's multiple comparison was used to determine significance of HEK experiments using only WNK4. Unpaired student's t-test was used to determine significance of effects of the different WNK LLFF mutations on pNCC compared with their wild-type counterparts. Two-way ANOVA was used to determine if there was a difference in the effect of LLFF mutations for the different WNKs.
Supplementary Material
Supplemental Figure 1: A fraction of GST-SPAK K104R produced in E. coli is truncated on its C-terminus. Western blots performed on recombinant GST-tagged SPAK K104R produced in E. coli. Blots were performed using antibodies directed at a) SPAK C-terminus, b) SPAK N-terminus, c) GST, and d) SPAK N-terminus with and without blocking peptide.
Supplemental Figure 2: Effects of WT and WNK4 LLFF on SPAK/OxSR1 phosphorylation in HEK cells. Western blot for pSPAK/pOxSR1 on HEK cells transfected with empty vector, WT WNK4, or WNK4 L322F L324F (WNK4 LLFF). WT WNK4 reduced pSPAK/pOxSR1 abundance and WNK4 LLFF increased it. Both effects were significant (p<0.05) by one-way ANOVA with Dunnett's multiple co mparisons test.
Acknowledgments
This work was funded by grants from the NIH (2R01DK051496-15A1 and 5T32DK067864-10 to DHE), and the Department of Veterans Affairs (1I0BX002228-01A1 to DHE). AST was the recipient of an American Heart Association pre-doctoral fellowship award (3PRE14090030). This work was performed by AST in partial fulfillment for a Ph.D. in Cell & Developmental Biology from Oregon Health and Science University.
Footnotes
Disclosure: The authors have no financial interests to disclose.
References
- 1.Addison WLT. The use of sodium cholride, potassium chloride, sodium bromide, and potassium bormide in cases of arterial hypertension which are amenable to potassium chloride. Can Med Assoc J. 1928;18:281–285. [PMC free article] [PubMed] [Google Scholar]
- 2.Adrogue HJ, Madias NE. Sodium and potassium in the pathogenesis of hypertension. The New England journal of medicine. 2007;356:1966–1978. doi: 10.1056/NEJMra064486. [DOI] [PubMed] [Google Scholar]
- 3.Young DB. Analysis of long-term potassium regulation. Endocrine reviews. 1985;6:24–44. doi: 10.1210/edrv-6-1-24. [DOI] [PubMed] [Google Scholar]
- 4.Terker AS, Zhang C, McCormick JA, et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 2015;21:39–50. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson FH, Disse-Nicodeme S, Choate KA, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 6.Wilson FH, Kahle KT, Sabath E, et al. Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc Natl Acad Sci U S A. 2003;100:680–684. doi: 10.1073/pnas.242735399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang CL, Angell J, Mitchell R, et al. WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J Clin Invest. 2003;111:1039–1045. doi: 10.1172/JCI17443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piala AT, Moon TM, Akella R, et al. Chloride Sensing by WNK1 Involves Inhibition of Autophosphorylation. Science signaling. 2014;7:ra41. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pacheco-Alvarez D, Cristobal PS, Meade P, et al. The Na+:Cl- cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem. 2006;281:28755–28763. doi: 10.1074/jbc.M603773200. [DOI] [PubMed] [Google Scholar]
- 10.Weinstein AM. A mathematical model of rat distal convoluted tubule. I. Cotransporter function in early DCT. American journal of physiology Renal physiology. 2005;289:F699–720. doi: 10.1152/ajprenal.00043.2005. [DOI] [PubMed] [Google Scholar]
- 11.Vitzthum H, Seniuk A, Schulte LH, et al. Functional coupling of renal K+ and Na+ handling causes high blood pressure in Na+ replete mice. The Journal of physiology. 2014;592:1139–1157. doi: 10.1113/jphysiol.2013.266924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castaneda-Bueno M, Cervantes-Perez LG, Rojas-Vega L, et al. Modulation of NCC activity by low and high K+ intake: Insights into the signaling pathways involved. American journal of physiology Renal physiology. 2014 doi: 10.1152/ajprenal.00255.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Lubbe N, Moes AD, Rosenbaek LL, et al. K+-induced natriuresis is preserved during Na+ depletion and accompanied by inhibition of the Na+-Cl- cotransporter. American journal of physiology Renal physiology. 2013;305:F1177–1188. doi: 10.1152/ajprenal.00201.2013. [DOI] [PubMed] [Google Scholar]
- 14.Vallon V, Schroth J, Lang F, et al. Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. American journal of physiology Renal physiology. 2009;297:F704–712. doi: 10.1152/ajprenal.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wade JB, Liu J, Coleman RA, et al. SPAK Mediated NCC Regulation in Response to Low K+ Diet. American journal of physiology Renal physiology. 2015 doi: 10.1152/ajprenal.00388.2014. ajprenal 00388 02014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rinehart J, Kahle KT, de Los Heros P, et al. WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl- cotransporters required for normal blood pressure homeostasis. Proc Natl Acad Sci U S A. 2005;102:16777–16782. doi: 10.1073/pnas.0508303102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bazua-Valenti S, Gamba G. Revisiting the NaCl cotransporter regulation by With No-lysine Kinases. American journal of physiology Cell physiology. 2015 doi: 10.1152/ajpcell.00065.2015. ajpcell 00065 02015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chávez-Canales M, Zhang C, Soukaseum C, et al. The WNK-SPAK-NCC cascade revisited: WNK1 stimulates the activity of the NaCl cotransporter via SPAK, an effect antagonized by WNK4. Hypertension. 2014 doi: 10.1161/HYPERTENSIONAHA.114.04036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ko B, Mistry AC, Hanson L, et al. A new model of the distal convoluted tubule. American journal of physiology Renal physiology. 2012;303:F700–710. doi: 10.1152/ajprenal.00139.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N, et al. Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:7929–7934. doi: 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi D, Mori T, Nomura N, et al. WNK4 is the major WNK kinase positively regulating NCC in the mouse kidney. Bioscience reports. 2014;9:e00107. doi: 10.1042/BSR20140047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bazua-Valenti S, Chavez-Canales M, Rojas-Vega L, et al. The Effect of WNK4 on the Na+-Cl- Cotransporter Is Modulated by Intracellular Chloride. Journal of the American Society of Nephrology : JASN. 2014 doi: 10.1681/ASN.2014050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamprecht G, Schaefer J, Dietz K, et al. Chloride and bicarbonate have similar affinities to the intestinal anion exchanger DRA (down regulated in adenoma) Pflugers Archiv : European journal of physiology. 2006;452:307–315. doi: 10.1007/s00424-006-0049-6. [DOI] [PubMed] [Google Scholar]
- 24.Boettger T, Hubner CA, Maier H, et al. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature. 2002;416:874–878. doi: 10.1038/416874a. [DOI] [PubMed] [Google Scholar]
- 25.Beck FX, Dorge A, Rick R, et al. The distribution of potassium, sodium and chloride across the apical membrane of renal tubular cells: effect of acute metabolic alkalosis. Pflugers Archiv : European journal of physiology. 1988;411:259–267. doi: 10.1007/BF00585112. [DOI] [PubMed] [Google Scholar]
- 26.Yang CL, Zhu X, Ellison DH. The thiazide-sensitive Na-Cl cotransporter is regulated by a WNK kinase signaling complex. J Clin Invest. 2007;117:3403–3411. doi: 10.1172/JCI32033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vidal-Petiot E, Elvira-Matelot E, Mutig K, et al. WNK1-related Familial Hyperkalemic Hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:14366–14371. doi: 10.1073/pnas.1304230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Statland BE, Winkel P, Bokelund H. Factors contributing to intra-individual variation of serum constituents. 1. Within-day variation of serum constituents in healthy subjects. Clinical chemistry. 1973;19:1374–1379. [PubMed] [Google Scholar]
- 29.Pocock SJ, Ashby D, Shaper AG, et al. Diurnal variations in serum biochemical and haematological measurements. J Clin Pathol. 1989;42:172–179. doi: 10.1136/jcp.42.2.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fijorek K, Puskulluoglu M, Tomaszewska D, et al. Serum potassium, sodium and calcium levels in healthy individuals - literature review and data analysis. Folia Med Cracov. 2014;54:53–70. [PubMed] [Google Scholar]
- 31.Zhang Z, Duckart J, Slatore CG, et al. Individuality of the plasma sodium concentration. American journal of physiology Renal physiology. 2014;306:F1534–1543. doi: 10.1152/ajprenal.00585.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohno M, Uchida K, Ohashi T, et al. Immunolocalization of WNK4 in mouse kidney. Histochemistry and cell biology. 2011;136:25–35. doi: 10.1007/s00418-011-0827-x. [DOI] [PubMed] [Google Scholar]
- 33.Glykys J, Dzhala V, Egawa K, et al. Local impermeant anions establish the neuronal chloride concentration. Science. 2014;343:670–675. doi: 10.1126/science.1245423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thastrup JO, Rafiqi FH, Vitari AC, et al. SPAK/OSR1 regulate NKCC1 and WNK activity: analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. The Biochemical journal. 2012;441:325–337. doi: 10.1042/BJ20111879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu BE, Min X, Stippec S, et al. Regulation of WNK1 by an autoinhibitory domain and autophosphorylation. J Biol Chem. 2002;277:48456–48462. doi: 10.1074/jbc.M207917200. [DOI] [PubMed] [Google Scholar]
- 36.Vitari AC, Thastrup J, Rafiqi FH, et al. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem J. 2006;397:223–231. doi: 10.1042/BJ20060220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naito S, Ohta A, Sohara E, et al. Regulation of WNK1 kinase by extracellular potassium. Clin Exp Nephrol. 2011;15:195–202. doi: 10.1007/s10157-010-0378-9. [DOI] [PubMed] [Google Scholar]
- 38.McCormick JA, Mutig K, Nelson JH, et al. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab. 2011;14:352–364. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grimm PR, Taneja TK, Liu J, et al. SPAK isoforms and OSR1 regulate sodium-chloride co-transporters in a nephron-specific manner. The Journal of biological chemistry. 2012;287:37673–37690. doi: 10.1074/jbc.M112.402800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeFronzo RA. Hyperkalemia and hyporeninemic hypoaldosteronism. Kidney international. 1980;17:118–134. doi: 10.1038/ki.1980.14. [DOI] [PubMed] [Google Scholar]
- 41.Batlle D, Grupp M, Gaviria M, et al. Distal renal tubular acidosis with intact capacity to lower urinary pH. AmJMed. 1982;72:751–758. doi: 10.1016/0002-9343(82)90540-x. [DOI] [PubMed] [Google Scholar]
- 42.Batlle DC, Arruda JAL, Kurtzman NA. Hyperkalemic distal renal tubular acidosis associated with obstructive uropathy. NEnglJMed. 1981;304:373–380. doi: 10.1056/NEJM198102123040701. [DOI] [PubMed] [Google Scholar]
- 43.Yang CL, Zhu X, Ellison DH. The thiazide-sensitive Na-Cl cotransporter is regulated by a WNK kinase signaling complex. J Clin Invest. 2007;117:3403–3411. doi: 10.1172/JCI32033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang CL, Zhu X, Wang Z, et al. Mechanisms of WNK1 and WNK4 interaction in the regulation of thiazide-sensitive NaCl cotransport. J Clin Invest. 2005;115:1379–1387. doi: 10.1172/JCI22452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin DH, Yue P, Rinehart J, et al. Protein phosphatase 1 modulates the inhibitory effect of With-no-Lysine kinase 4 on ROMK channels. American journal of physiology Renal physiology. 2012;303:F110–119. doi: 10.1152/ajprenal.00676.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bostanjoglo M, Reeves WB, Reilly RF, et al. 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. Journal of the American Society of Nephrology : JASN. 1998;9:1347–1358. doi: 10.1681/ASN.V981347. [DOI] [PubMed] [Google Scholar]
- 47.McCormick JA, Nelson JH, Yang CL, et al. Overexpression of the Sodium Chloride Cotransporter Is Not Sufficient to Cause Familial Hyperkalemic Hypertension. Hypertension. 2011;58:888–894. doi: 10.1161/HYPERTENSIONAHA.110.167809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Z, Yang CL, Ellison DH. Comparison of WNK4 and WNK1 kinase and inhibiting activities. Biochem Biophys Res Commun. 2004;317:939–944. doi: 10.1016/j.bbrc.2004.03.132. [DOI] [PubMed] [Google Scholar]
- 49.Ushiro H, Tsutsumi T, Suzuki K, et al. Molecular cloning and characterization of a novel Ste20-related protein kinase enriched in neurons and transporting epithelia. Arch Biochem Biophys. 1998;355:233–240. doi: 10.1006/abbi.1998.0736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: A fraction of GST-SPAK K104R produced in E. coli is truncated on its C-terminus. Western blots performed on recombinant GST-tagged SPAK K104R produced in E. coli. Blots were performed using antibodies directed at a) SPAK C-terminus, b) SPAK N-terminus, c) GST, and d) SPAK N-terminus with and without blocking peptide.
Supplemental Figure 2: Effects of WT and WNK4 LLFF on SPAK/OxSR1 phosphorylation in HEK cells. Western blot for pSPAK/pOxSR1 on HEK cells transfected with empty vector, WT WNK4, or WNK4 L322F L324F (WNK4 LLFF). WT WNK4 reduced pSPAK/pOxSR1 abundance and WNK4 LLFF increased it. Both effects were significant (p<0.05) by one-way ANOVA with Dunnett's multiple co mparisons test.