

Abstract
The Fritillaria imperialis is an ornamental flower that can be found in various parts of the world including Iraq, Afghanistan, Pakistan, and the Himalayas. The use of this plant as traditional remedy is widely known. This study aims to unveil the anti-cancer potentials of Isopimara-7,15-Dien-19-Oic Acid, extracted from the bulbs of F. imperialis in cervical cancer cell line, HeLa cells. Flow cytometry analysis of cell death, gene expression analysis via cDNA microarray and protein array were performed. Based on the results, Isopimara-7,15-Dien-19-Oic acid simultaneously induced cell death and promoted cell survival. The execution of apoptosis was apparent based on the flow cytometry results and regulation of both pro and anti-apoptotic genes. Additionally, the regulation of anti-oxidant genes were up-regulated especially thioredoxin, glutathione and superoxide dismutase- related genes. Moreover, the treatment also induced the activation of pro-survival heat shock proteins. Collectively, Isopimara-7,15-Dien-19-Oic Acid managed to induce cellular stress in HeLa cells and activate several anti- and pro survival pathways.
Keywords: HeLa, isopimara-7, 15-dien-19-oic acid, Fritillaria imperialis, antioxidant
Introduction
Cancer is a complex disease that is not fully understood yet. Cervical cancer is among the most diagnosed type of cancer in women today. Statistically, around 1 out of 154 will be diagnosed for cervical cancer (Siegel et al., 2014). The mechanism of cell death and cell survival often intertwines and involves a lot of variables. There is a delicate balance that plays a major role in cell sustenance and the tilt can lean either way, especially in reacting to external substances (Fulda et al., 2010). Unfortunately, a viable treatment for treating cervical cancer is yet to be found. Natural-derived molecules have become a promising target in finding the cure for major diseases including diabetes, cancer, and Alzheimer.
Fritillaria imperialis or commonly known as “crown imperial” is a species of flower from the Liliaceae family (Khare, 2007). This species can be found in various parts of the world specifically Iran, Turkey, Afghanistan, and some parts of the Himalaya (Khare, 2007; Badfar-Chaleshtori et al., 2012). This plant is considered an ornamental plant due to its large and attractive flowers. It is also known to have several medicinal properties such as becoming a diuretic, treating hypotensive, cardiotonic, and spasmolytic (Khare, 2007). There are several interesting molecules that can be extracted from this plant especially steroidal alkaloids (Atta-ur-Rahman et al., 2002; Akhtar et al., 2003; Khare, 2007). Additionally, another class of molecules that can also be extracted from the F. imperialis is sesquiterpenes (Atta-ur-Rahman et al., 2005). Sesquiterpenes are a class of natural products possessing various biological activities such as antimycobacterial (Abourashed et al., 2011), antifungal (Al-Ja'fari et al., 2013), anti-inflammatory, apoptosis-inducing, and immunosuppressant activities (Qi et al., 2015). Most of the sesquiterpene lactones impart a wide-range of pharmacological effects, including anti-cancer and immunomodulatory action (Lu et al., 2009; Choi et al., 2011; Ivanescu et al., 2015), antimicrobial, antioxidant, anti-inflammatory, and antinociceptive activities (Sulaiman et al., 2010; Dahham et al., 2015).
Diterpene Isopimara-7,15-dien-19-oic acid can be isolated from the bulbs of F. imperialis plant. The only known activity this molecule has is the prolyl endopeptidase inhibition (Atta-ur-Rahman et al., 2005). Other biological or chemical properties of this molecule are yet to be discovered. Thus, the aim of this study is to understand the molecular mechanism of HeLa cells, the most used cervical cancer cell line, upon induction with isopimara-7,15-dien-19-oic acid in vitro.
Materials and methods
Plant material and purification of compounds
Isopimara-7,15-dien-19-oic acid (Figure 1) was purified from a hexane fraction of Turkish plant F. imperialis. The hexane fraction was obtained as thick oil and subjected to column chromatography over silica gel by using acetone/petroleum ether as the solvent system. Isopimara-7,15-dien-19-oic acid was obtained as colorless prismatic crystals with melting point 159–160°C. The detailed extraction procedure and identification of isopimara-7,15-dien-19-oic acid were already published in our previous publication (Atta-ur-Rahman et al., 2005).
Figure 1.
The chemical structure of DIA isolated from the bulbs of Fritillaria imperialis.
Cell culture
HeLa cells were obtained from the ATCC Collection (ATCC, USA) and were maintained in RPMI-1640 (Sigma, USA) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Gibco, USA). The cells were incubated at 37°C equipped with 5% CO2.
MTT
The MTT analysis was performed as a preliminary cytotoxic study for DIA against HeLa cells. Cells were seeded in a 96 well plate at a density of 0.8 × 105 cells/ml overnight. Afterwards, the cells were treated with seven different doses of DIA ranging from 30 to 0.64 μg/mL and were left to incubate for 72 h. Upon reaching the allocated incubation time, 20 μl of 5 mg/mL of MTT (Sigma, USA) was added to each of the wells for 4 h at 37°C. Next, the media as well as the MTT were removed and 100 μl of DMSO was added to solubilize the resulting formazon crystals. Subsequently, the reading of the plate was obtained using a microplate reader (Bio-Tek Instruments, USA) at 570 nm.
Cell cycle flow cytometry analysis
HeLa cells were seeded in 6 well plates at a density of 2.4 × 105 cells/well overnight. The next day, 15 μg/mL of DIA was added into the designated wells and was left to incubate for 72 h in a humidified 37°C CO2 incubator. Afterwards, the cells were harvested, fixed in 500 μl of ice cold 80% ethanol and were stored in −20°C for 1 week. On the day of the analysis, the pellet was washed with 1 ml of PBS twice and was permeabilized and stained using 500 μl of RNAse-Propidium Iodide-PBS-Triton X100 for 15 min at room temperature. Next, the cells were analyzed using a FACS Calibur Flow Cytometry machine immediately (BD, USA).
Annexin V-FITC flow cytometry analysis
This assay was performed according to the protocol set by the Annexin V/FITC kit by BD, USA. Similar to the cell cycle analysis, HeLa cells were seeded in 6 well plates at a density of 2.4 × 105 cells/well overnight. The next day, 15 μg/mL of DIA was added into the designated wells and was left to incubate for 72 h in a humidified 37°C CO2 incubator. After the incubation time, the cells were harvested and washed twice with PBS. The cells were later stained with Annexin V-FITC and Propidium Iodide in 100 μl of 1X Binding Buffer. Then, the cells were analyzed using a FACS Calibur Flow Cytometry machine immediately (BD, USA).
JC-1 flow cytometry analysis
The JC-1 analysis was done using the BD Mitoscreen Kit (BD, USA). Similar to the cell cycle analysis, HeLa cells were seeded in 6 well plates at a density of 2.4 × 105 cells/well overnight. The next day, 15 μg/mL of DIA was added into the designated wells and was left to incubate for 72 h in a humidified 37°C CO2 incubator. After the incubation time, the cells were harvested and washed twice with PBS. Afterwards, the cells were stained and incubated with the JC-1 dye for 15 min at 37°C. Then the cells were washed twice with the provided washing buffer. The cells were later analyzed using a FACS Calibur Flow Cytometry machine immediately (BD, USA).
cDNA microarray
Three sets of biological replicates of untreated HeLa and DIA-treated HeLa were prepared. HeLa cells were seeded in 6 well plates at a density of 2.4 × 105 cells/well overnight. After 24 h, 15 μg/mL of DIA was added into the designated wells and was left to incubate for 48 h in a humidified 37°C CO2 incubator. After the incubation time, the cells were harvested and RNA was extracted using the QiagenRneasy Mini Kit (Qiagen, Germany). The quality of the RNA extracted was measured using the 2100 Bioanalyzer using a RNA Pico chip (Agilent, USA). In order to proceed to microarray, the RIN (RNA Integrity Number) should be more than eight. After all of the samples have passed the minimum requirement for microarray analysis the samples were then used for microarray. All of the samples were subjected to the SurePrint G3 Human Gene Expression 8x60K v2 microarray kit (Agilent Technologies, USA) according to manufacturer protocol, and scanned with Agilent DNA microarray scanner. The results from the microarray study has already been uploaded on the Gene Expression Omnibus with the accession number GSE72974.
Differential expression analysis for microarray
The results from the microarray analysis were analyzed using the Genespring GX Software Version 13.1 (Agilent, USA). The differential expression comparison was made between the untreated HeLa cells and the DIA-treated HeLa cells. Final results were analyzed based on gene ontology with expression level having p < 0.05.
Quantitative real-time PCR
To validate the results obtained from the microarray study, real-time PCR was performed on the same samples using different sets of primers. Around 1 μg of RNA from each of the samples were converted to cDNA using the Quantitect Reverse Transcription Kit according to the manufacturer's protocol (Qiagen, Germany). Then, real-time PCR was conducted using the SYBR Select Master Mix (Invitrogen, USA) on the iCycler IQ5 (Bio-rad, USA). Table 1 illustrates the name of the gene, accession number, and sequence of the primers used in this assay (http://pga.mgh.harvard.edu/primerbank/).
Table 1.
Accession number and the sequence of the primers used to validate the microarray results.
| Gene name | Accession number | Sequence |
|---|---|---|
| HMOX1 | NM_002133.2 | F: 5-AAGACTGCGTTCCTGCTCAAC-3 |
| R: 5-AAAGCCCTACAGCAACTGTCG-3 | ||
| DDIT3 | NM_001195057.1 | F: 5-GAACGGCTCAAGCAGGAAATC-3 |
| R: 5-TTCACCATTCGGTCAATCAGAG-3 | ||
| GPX3 | NM_002084.3 | F: 5-AGAGCCGGGGACAAGAGAA-3 |
| R: 5-ATTTGCCAGCATACTGCTTGA-3 | ||
| GADD45A | NM_001924.3 | F: 5-GAGAGCAGAAGACCGAAAGGA-3 |
| R: 5-CAGTGATCGTGCGCTGACT-3 | ||
| ACTB | NM_001101.3 | F: 5-AGAGCTACGAGCTGCCTGAC-3 |
| R: 5-AGCACTGTGTTGGCGTACAG-3 | ||
| GAPDH | NM_002046.4 | F: 5- GGATTTGGTCGTATTGGGC-3 |
| R: 5- TGGAAGATGGTGATGGGATT-3 | ||
| 18S RRNA | HQ387008.1 | F: 5- GTAACCCGTTGAACCCCATT-3 |
| R: 5- CCATCCAATCGGTAGTAGCG -3 |
Proteome profiler array TM
The proteome profiler antibody array was employed to determine the effects of DIA on the activation of several cell stress-related proteins. This assay was done according to the manufacturer's protocol. The cell lysates were incubated with the designated membranes overnight at 4°C. The following day, the membranes were washed three times and were then incubated with the freshly prepared antibody cocktail for 2 h. Afterwards, the membranes were washed for three times again, before being incubated with the streptavidin-horseradish-peroxidase for 30 min at room temperature. Then, the membranes were developed under chemiluminescence conditions using the ChemiDOC XRS (Bio-rad, USA).
Superoxide dismutase (SOD) and glutathione (GSH) quantification
Total proteins were extracted from the untreated HeLa and DIA-treated HeLa and were measured using the Bradford assay (Sigma, USA). For SOD, 100 μL of extracted protein was mixed with 200 μL of working solution (0.1 mol/L phosphate buffer, 0.15 mg/mL sodium cyanide in 0.1 mol/L ethylenediaminetetraacetic acid, 1.5 mmol/L nitrobluetetrazolium and 0.12 mmol/L riboflavin). On the other hand, GSH was quantified using Glutathione assay kit (Sigma, USA), where 10 μL of protein was added with 150 μL of working solution (1.5 mg/mL DTNB, 6 U/mL glutathione reductase, and 1 × assay buffer). After 5 min of incubation, 50 μL of NADPH solution (0.16 mg/mL) was added to the mixture. The absorbance for SOD and GSH were measured using ELISA plate reader (Bio-Tek Insturments, USA) at respective wavelengths of 560 and 420 nm.
Statistical analysis
All experiments were done in three biological replicates and expressed as mean ± standard deviation. Results with statistical significant (p < 0.05) was assayed by student t-test comparing to the untreated control.
Results
DIA inhibited the proliferation of HeLa cells and induced apoptosis in vitro
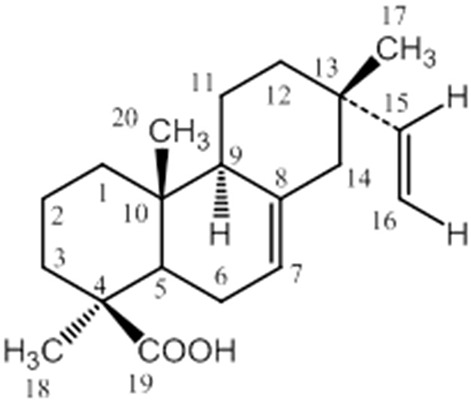
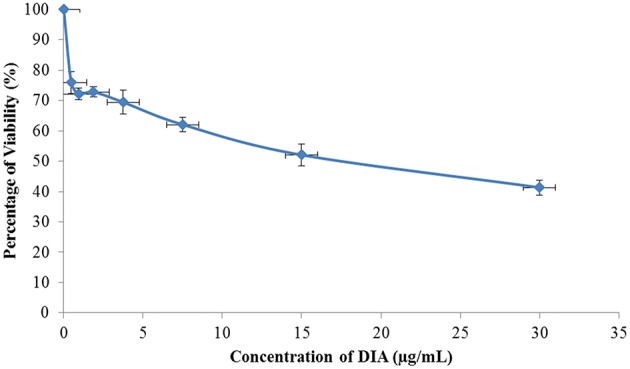
Based on the MTT results, DIA only showed HeLa cells viability (Figure 1) in a dose-dependent manner but not on breast cancer (MCF-7 and MDA-MB231), colon cancer (HT-29), and hepatoblastoma (HepG2) cell lines (results not shown) at concentration up to 30 μg/mL. As in Figure 2, the half-maximal inhibitory concentration (IC50) of DIA against HeLa cells after 48 h was 15 μg/mL. Moreover, as in Figure 3A, the effects of DIA on HeLa cells was apparent as it induces an increase in the Sub G0/G1 phase. The percentage of cell population in the Sub G0/G1 phase for the untreated HeLa cells was 8.2%, while for the DIA-treated cells was 29.77%. Additionally, as evidenced by the Annexin V assay, DIA-treated HeLa cells had an increase in the early apoptosis and late apoptosis populations, coupled with a decrease in the viable cell population, comparing to the untreated cells. As shown in Figure 3B, the percentage of viable cells for the untreated cells was 97.97%, this was followed by a decrease to 33.19% after 48 h of treatment with DIA. For the early apoptosis population, the percentage in the untreated cells was 0.05%, while in the DIA-treated cells, the percentage of the population increased to 34.11%. A similar pattern can also be observed in the late apoptosis population, from 0.02% in the untreated cells to 26.17% in the DIA-treated cells. Furthermore, based on the JC-1 assay, the percentage of monomers in the DIA-treated cells (47.52%) was higher than the untreated cells (5.25%) as displayed in Figure 3C.
Figure 2.
MTT analysis of HeLa cells after being treated with DIA for 48 h at 30 μg/mL followed by seven 2-serial dilutions.
Figure 3.
Flow cytometry analysis of (A) Cell cycle analysis by staining the DNA using propidium iodide, (B) Annexin V analysis for the detection of externalization of phosphatidylserine (LL, viable; LR, Early apoptosis; UR, Late apoptosis), and (C) JC-1 analysis for the detecting the change of mitochondrial membrane potential in HeLa cells (Red: Aggregates; Green: Monomers) after 48 h of treatment with 15 μg/mL of DIA.
DIA regulated cellular stress-related proteins in HeLa cells
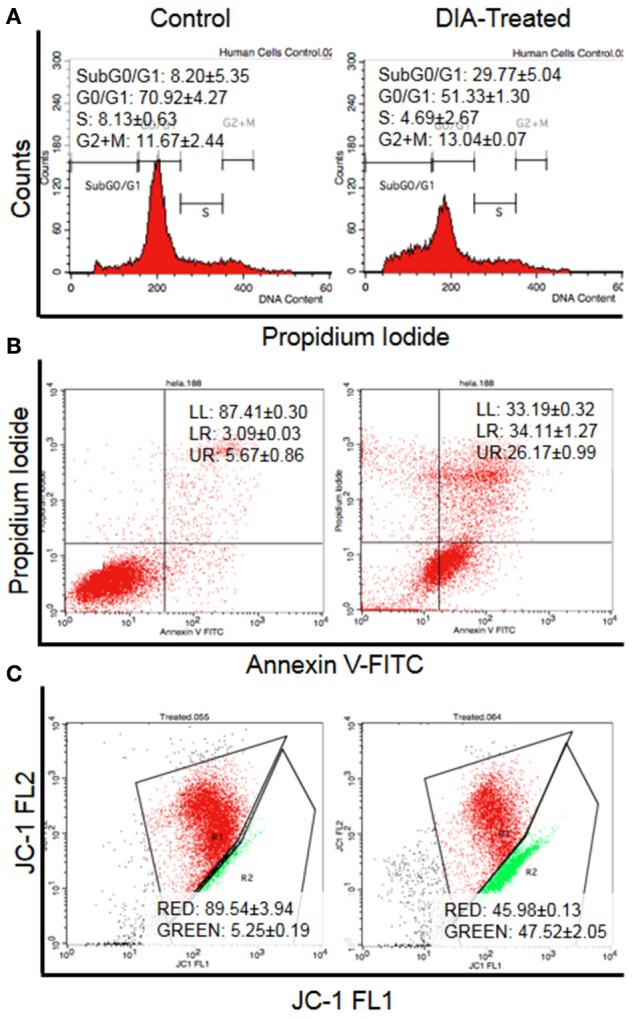
Figure 4 illustrates the proteome profiler analysis for cell-stress related proteins as well as the quantification values. DIA-treated cells managed to increase the regulation of several heat shock proteins including hsp27 and hsp70. Moreover, the expression of cytochrome C, SOD2, thioredoxin, carbonic anhydrase IX, p38, and HIF-1a were also increased upon induction with DIA in comparison with the control. The validation of both the microarray and proteome results is presented in Figure 5. Similar pattern of expression could be seen for all of the proteins in the proteome to the same genes in the microarray differential analysis.
Figure 4.
Representative image of the proteome profiler analysis for cell stress-related images as well as the quantification values for HeLa cells; both control (untreated) and DIA-treated cells (15 ug/mL) for 48 h.
Figure 5.

Validation of the proteome profiler results by comparing the expression of proteins to the respective genes in microarray (p-value for the microarray results are p < 0.05).
DIA regulated the expression of apoptosis, oxidative stress, and chaperone-related genes
cDNA microarray study was done to determine the effects of DIA on the mRNA expression of HeLa cells. Based on Table 2, DIA managed to regulate a large number of genes related to apoptosis, oxidative stress, and heat shock proteins. DIA affected the expression of 96 genes that are involved in either cell death or cell survival.
Table 2.
Differentially regulated genes related to oxidative stress and MAPK pathway in HeLa cells after 48 h of treatment with 15 μg/mL DIA with p < 0.05.
| Accession number | Gene symbol | Gene name | Log fold change | Regulation |
|---|---|---|---|---|
| NM_002501 | NFIX | Nuclear factor I/X (CCAAT-binding transcription factor) | 2.64 | Survival |
| NM_000499 | CYP1A1 | Cytochrome P450, family 1, subfamily A, polypeptide 1 | 2.38 | Survival |
| NM_145791 | MGST1 | Microsomal glutathione S-transferase 1 | 7.61 | Survival |
| NM_003998 | NFKB1 | Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 | 4.23 | Survival |
| NM_201397 | GPX1 | Glutathione peroxidase 1 | 3.51 | Survival |
| NM_000854 | GSTT2 | Glutathione S-transferase theta 2 | 5.67 | Survival |
| NM_001752 | CAT | Catalase | 3.01 | Survival |
| NM_002133 | HMOX1 | Hemeoxygenase (decycling) 1 | 2.44 | Survival |
| NM_001261445 | TXNRD1 | Thioredoxinreductase 1 | 4.88 | Survival |
| NM_001270458 | MAOA | Monoamine oxidase A | 1.56 | Survival |
| NM_000101 | CYBA | Cytochrome b-245, alpha polypeptide | 6.63 | Survival |
| NM_002084 | GPX3 | Glutathione peroxidase 3 (plasma) | –2.59* | Survival |
| NM_006440 | TXNRD2 | Thioredoxin reductase 2 | 2.04 | Survival |
| NM_138980 | MAPK10 | Mitogen-activated protein kinase 10 | –3.62* | Survival |
| NM_000379 | XDH | Xanthine dehydrogenase | –1.48* | Survival |
| NM_016931 | NOX4 | NADPH oxidase 4 | 1.60 | Survival |
| NM_000454 | SOD1 | Superoxide dismutase 1, soluble | 9.92 | Survival |
| NM_201397 | GPX1 | Glutathione peroxidase 1 | 10.58 | Survival |
| NM_012473 | TXN2 | Thioredoxin 2 | 2.37 | Survival |
| NM_002229 | JUNB | Jun B proto-oncogene | 1.05 | Survival |
| NM_000637 | GSR | Glutathionereductase | 1.99 | Survival |
| NM_005952 | MT1X | Metallothionein 1X | 5.07 | Survival |
| NM_001024465 | SOD2 | Superoxide dismutase 2, mitochondrial | 2.63 | Survival |
| NM_006164 | NFE2L2 | Nuclear factor, erythroid 2-like 2 | –3.82* | Survival |
| NM_001072 | UGT1A6 | UDP glucuronosyltransferase 1 family, polypeptide A6 | 2.39 | Survival |
| NM_001025366 | VEGFA | Vascular endothelial growth factor A | –5.57* | Survival |
| NM_001216 | CA9 | Carbonic anhydrase IX | 1.67 | Survival |
| NM_004380 | CREBBP | CREB binding protein | 3.67 | Survival |
| NM_181054 | HIF1A | Hypoxia inducible factor 1, alpha subunit | 2.67 | Survival |
| NM_005345 | HSPA1A | Heat shock 70 kDa protein 1A | 7.92 | Survival |
| NM_001540 | HSPB1 | Heat shock 27 kDa protein 1 | 10.71 | Survival |
| NM_000043 | FAS | Fas cell surface death receptor | 1.7873325 | Death |
| NM_001226 | CASP6 | Caspase 6, apoptosis-related cysteine peptidase | 2.8654807 | Death |
| NM_003810 | TNFSF10 | Tumor necrosis factor (ligand) superfamily, member 10 | –1.0368137* | |
| NM_021975 | RELA | v-rel avian reticuloendotheliosis viral oncogene homolog A | 1.0848213 | |
| NM_213566 | DFFA | DNA fragmentation factor, 45kDa, alpha polypeptide | 1.9827862 | Death |
| NM_138578 | BCL2L1 | BCL2-like 1 | 1.5335723 | Death |
| NM_033306 | CASP4 | Caspase 4, apoptosis-related cysteine peptidase | 1.2512972 | Death |
| NM_001012271 | BIRC5 | Baculoviral IAP repeat containing 5 | 7.95958 | |
| NM_019887 | DIABLO | Diablo, IAP-binding mitochondrial protein | 5.7222176 | |
| NM_004346 | CASP3 | Caspase 3, apoptosis-related cysteine peptidase | 5.705864 | Death |
| NM_002467 | MYC | v-myc avian myelocytomatosis viral oncogene homolog | 3.997664 | |
| NM_138764 | BAX | BCL2-associated X protein | 1.3814521 | Death |
| NM_138621 | BCL2L11 | BCL2-like 11 (apoptosis facilitator) | 1.6899183 | |
| NM_020529 | NFKBIA | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha | 5.042662 | |
| NM_058197 | CDKN2A | Cyclin-dependent kinase inhibitor 2A | 7.5630064 | |
| NM_032977 | CASP10 | Caspase 10, apoptosis-related cysteine peptidase | 3.1102889 | Death |
| NM_000639 | FASLG | Fas ligand (TNF superfamily, member 6) | –2.4349697* | Death |
| NM_033355 | CASP8 | Caspase 8, apoptosis-related cysteine peptidase | 2.0166228 | Death |
| NM_001127184 | CFLAR | CASP8 and FADD-like apoptosis regulator | 4.176869 | Death |
| NM_002392 | MDM2 | MDM2 proto-oncogene, E3 ubiquitin protein ligase | 2.3782425 | Survival |
| NM_000546 | TP53 | Tumor protein p53 | 1.6474063 | Death |
| NM_021138 | TRAF2 | TNF receptor-associated factor 2 | 7.908759 | Death |
| NM_018947 | CYCS | Cytochrome c, somatic | 6.6717825 | Death |
| AK094730 | HRK | Harakiri, BCL2 interacting protein | 1.1366951 | |
| NM_207002 | BCL2L11 | BCL2-like 11 (apoptosis facilitator) | 1.0382731 | Death |
| NM_004346 | CASP3 | Caspase 3, apoptosis-related cysteine peptidase | 1.0135665 | Death |
| NM_003639 | IKBKG | Inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase gamma | 5.08775 | |
| NM_003824 | FADD | Fas (TNFRSF6)-associated via death domain | 7.09289 | Death |
| NM_181523 | PIK3R1 | Phosphoinositide-3-kinase, regulatory subunit 1 (alpha) | 1.7552358 | |
| NM_000043 | FAS | Fas cell surface death receptor | 1.2629685 | Death |
| NM_004322 | BAD | BCL2-associated agonist of cell death | 6.317196 | Death |
| NM_021138 | TRAF2 | TNF receptor-associated factor 2 | 2.5242937 | Death |
| NM_014452 | TNFRSF21 | Tumor necrosis factor receptor superfamily, member 21 | 1.2962595 | |
| NM_003804 | RIPK1 | Receptor (TNFRSF)-interacting serine-threonine kinase 1 | 1.8435402 | |
| NM_000612 | IGF2 | Insulin-like growth factor 2 | –1.0878063* | Survival |
| NM_001188 | BAK1 | BCL2-antagonist/killer 1 | 3.254304 | |
| NM_000875 | IGF1R | insulin-like growth factor 1 receptor | 2.7272189 | |
| NM_145725 | TRAF3 | TNF receptor-associated factor 3 | –5.650675* | Death |
| NM_000657 | BCL2 | B-cell CLL/lymphoma 2 | –2.1980934* | Death |
| AK309150 | BAD | BCL2-associated agonist of cell death | 2.1382089 | Death |
| NM_197966 | BID | BH3 interacting domain death agonist | 3.6607141 | Death |
| NM_002228 | JUN | Jun proto-oncogene | 1.9206382 | Survival |
| NM_001289072 | HELLS | Helicase, lymphoid-specific | 2.2962887 | |
| NM_033292 | CASP1 | Caspase 1, apoptosis-related cysteine peptidase | 3.3557472 | Death |
| NM_003842 | TNFRSF10B | Tumor necrosis factor receptor superfamily, member 10b | 4.5481834 | Death |
| NM_207002 | BCL2L11 | BCL2-like 11 (apoptosis facilitator) | –1.1714572* | Death |
| NM_003789 | TRADD | TNFRSF1A-associated via death domain | 5.638668 | |
| NM_004131 | GZMB | Granzyme B (granzyme 2, cytotoxic T-lymphocyte-associated serine esterase 1) | –4.478533* | |
| NM_001229 | CASP9 | Caspase 9, apoptosis-related cysteine peptidase | 4.309328 | Death |
| NM_003805 | CRADD | CASP2 and RIPK1 domain containing adaptor with death domain | 4.569709 | Death |
| NM_002392 | MDM2 | MDM2 proto-oncogene, E3 ubiquitin protein ligase | –1.910822* | Death |
| NM_004031 | IRF7 | Interferon regulatory factor 7 | 4.7208776 | |
| NM_033338 | CASP7 | Caspase 7, apoptosis-related cysteine peptidase | 4.725479 | Death |
| NM_001556 | IKBKB | Inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase beta | 3.2848263 | |
| NM_005658 | TRAF1 | TNF receptor-associated factor 1 | –1.2607836* | Death |
| NM_001278 | CHUK | Conserved helix-loop-helix ubiquitous kinase | 4.082881 | |
| NM_003810 | TNFSF10 | Tumor necrosis factor (ligand) superfamily, member 10 | –1.1297648* | |
| NM_181861 | APAF1 | Apoptotic peptidase activating factor 1 | 1.063245 | Death |
| NM_001282669 | DFFB | DNA fragmentation factor, 40kDa, beta polypeptide (caspase-activated DNase) | 1.0859745 | Death |
| NM_002198 | IRF1 | Interferon regulatory factor 1 | 2.7887836 | |
| NM_002503 | NFKBIB | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, beta | 2.073728 | |
| NM_197966 | BID | BH3 interacting domain death agonist | 6.265839 | |
| NM_004324 | BAX | BCL2-associated X protein | 1.0556245 | |
| NM_032515 | BOK | BCL2-related ovarian killer | 4.000063 | |
| NM_001202519 | CFLAR | CASP8 and FADD-like apoptosis regulator | 2.3586068 | Death |
Negative values represent down-regulated genes upon treatment.
Validation of microarray results with quantitative real-time PCR
To validate the results of differentially regulated genes from the microarray analysis, a set of genes were selected and analyzed using qPCR. All of the validated genes in qPCR had a similar pattern of expression in the microarray results, as presented in Figure 6.
Figure 6.
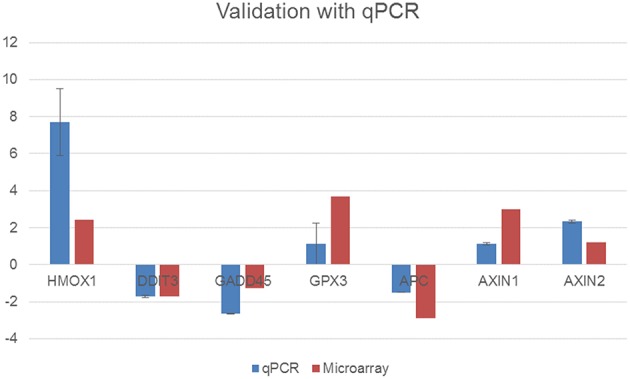
Validation of the microarray results using quantitative real-time PCR with selected genes in total RNA samples extracted from the control cells and 15 μg/mL DIA-treated cells (p-value for the microarray results are p < 0.05).
DIA-treated cells have a higher amount of SOD and GSH
Based on Figure 7, the production of both SOD and GSH were elevated in DIA-treated cells comparing to the untreated cells (control). DIA-treated cells have a 1 fold and 1.57 fold change difference respectively from the control cells.
Figure 7.
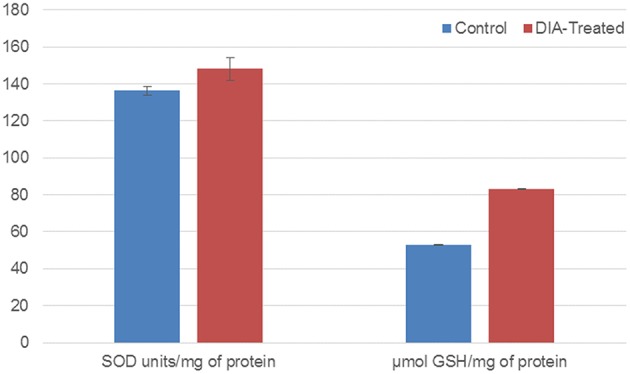
Bar chart analysis of the SOD units/mg of protein and μmol GSH/mg of protein in control cells and DIA-treated HeLa cells after 48 h of treatment with 15 μg/mL of DIA.
Discussion
Cellular stress plays an important role in response to chemotherapeutic agents, and this has been one of the major concerns in finding the perfect treatment for cancer, even for cervical cancer (Portt et al., 2011; Kim et al., 2014). F. Imperialis has been long known to possess medicinal properties. The extracts of this plant have not been extensively studied on yet especially on the diterpene group. There are several notable diterpenes that possess promising anti-cancer activities such as carnosol, crispene e, and taxol (Stahlhut et al., 1999; Chun et al., 2014; Mantaj et al., 2015). To the best of our knowledge, the anti-cancer effects of DIA extracted from the bulbs of F. Imperialis on HeLa cells has not been reported yet.
There are various phytochemicals that possess anti-cancer properties by modulating the cellular stress pathway (Kim et al., 2014). Additionally, there are also several agents used for cancer therapy that cause cellular ROS stress including cisplatin, ascorbic acid, and emodin (Pelicano et al., 2004). The cellular stress mechanism is diverged into multiple responses and one of the responses that could be initiated is cell death, including apoptosis (Martindale and Holbrook, 2002; Fulda et al., 2010; Portt et al., 2011). Cell death can be measured through various parameters such as DNA damage through cell cycle analysis, cellular membrane changes, and mitochondrial potential changes (Schmitt et al., 2007; Branzei and Foiani, 2008). At the molecular level, based on Table 2, the activation of pro-apoptotic genes is prevalent. The expression of the FADD and FAS gene in DIA-treated cells increased comparing to the control cells. The activation of FADD and FAS triggered a downstream of execution of apoptosis-related proteins such as caspase 8, caspase 3, BID, and JNK (Gupta et al., 2004; Clarke and Tyler, 2009), which subsequently regulate the BCL2-family proteins. The BCL-2 family is crucial in a cellular response mechanism as it can contribute to the switch of cell death vs. cell survival (Gross et al., 1999; Cory et al., 2003; Schmitt et al., 2007). Pro-apoptotic BCL-2 family genes such as BAX, BAD, and BAK were increased in DIA-treated cells. These proteins can cause mitochondrial dysfunction, which in turns affect the permeability transition pore, increased in radical oxygen species (ROS) and the release of cytochrome C (Gross et al., 1999; Cory et al., 2003). Cytochrome C is a pro-apoptotic protein that recruits the activation of apaf-1 and caspase 9 (Gross et al., 1999; Cory et al., 2003). As evidenced by the increase in gene regulation of cytochrome c, apaf-1, and caspase 9, as well as the changes in the mitochondrial membrane potential in the JC-1 assay, DIA treatment induced apoptosis through the mitochondrial pathway. Based on the cell cycle analysis, DIA increased the percentage of population in SubG0/GI which suggests that the treatment may induce DNA fragmentation in the execution of apoptosis. The regulation of the AIF gene and caspase 3 may be involved in the DNA fragmentation process. All these results suggest that DIA induced apoptosis in HeLa cells through DNA fragmentation and mitochondrial membrane potential changes.
Besides cell death, cellular stress response may also trigger the cell survival motion within the cancer cells (Martindale and Holbrook, 2002; Fulda et al., 2010). Both the protein and gene expression of HSP27 and HSP70 were elevated in HeLa cells upon treatment with DIA, which suggests that the cells were attempting at recovering from the damage induced by regulating these chaperones. The heat shock protein response is a classic retaliation to stress (Feder and Hofmann, 1999; Fulda et al., 2010; Seigneuric et al., 2011; Calderwood et al., 2012). Heat shock proteins are a highly conserved set of protein chaperones that promote cell survival in stressful conditions (Calderwood et al., 2006; Seigneuric et al., 2011). These proteins are known to be overexpressed in cancer especially HSP27, HSP70, and HSP90 (Calderwood et al., 2006, 2012; Seigneuric et al., 2011). Hence, there has been development of cancer biomarkers and vaccine for these proteins for the treatment of cancer (Calderwood et al., 2006, 2012; Seigneuric et al., 2011). HSP27 and HSP70 were thought to interact in the anti-cell death process as it can inhibit cytochrome c, caspase 9 and eventually the whole apoptotic cascade (Calderwood et al., 2006; Fulda et al., 2010).
Another reaction to cell stress is the response to oxidative stress (Martindale and Holbrook, 2002; Fulda et al., 2010; Reuter et al., 2010; Portt et al., 2011). Based on the microarray and proteome results, DIA managed to induce oxidative stress in HeLa cells upon treatment. The introduction of anti-cancer agents can trigger the production of ROS substantially (Reuter et al., 2010; Fiaschi and Chiarugi, 2012). The production of ROS is a normal metabolic process in a given cellular system, nevertheless, the balance between ROS and anti-oxidants play a pivotal role in the progression of cancer (Reuter et al., 2010; Fiaschi and Chiarugi, 2012; Ma, 2013). Moreover, the hypoxic conditions of the microenvironment could also contribute to the sustain release of ROS. One of the hallmark of cancer is that cancer cells thrive under hypoxic conditions (Bartrons and Caro, 2007). This will usually lead to the activation of HIF1 protein which in turns will affect the accumulation of ROS (Bartrons and Caro, 2007; Reuter et al., 2010). Additionally, the activation of HIF1 could also affect the activation of carbonic anhydrase IX and VEGFA, which both proteins are inclined to participate in the progression of tumorigenesis (Hui et al., 2002; Jubb et al., 2004; Dungwa et al., 2011). The presence of ROS could affect both the cell death and cell survival mode. One of the pathways that is activated in an oxidative stress state is the KEAP1/NRF-2 stress pathway (Reuter et al., 2010; Gorrini et al., 2013; Ma, 2013). Moreover, the sustained production of ROS will also activate several other signaling pathways such as JNK, MAPK, and ERK pathways. The MAPK pathway could also contribute to the activation of the NRF2 pathway adding to the ROS-mediated initiation of NRF2.
The expression of NRF2-related genes in DIA-induced HeLa cells is significant and this may give way to the underlying mechanism of DIA. The NRF2 is the key player to several antioxidant pathways including the iron sequestration pathway, quinone detoxification, GSH production, and thioredoxin production (Nguyen et al., 2009; Gorrini et al., 2013; Ma, 2013). Once the NRF2 is stimulated it will further activate phase II detoxification enzymes (Kwak and Kensler, 2010). The thioredoxin (trx) and glutathione pathway are among the antioxidants pathway that can cross-talk with multiple other pathways and with each other (Brigelius-Flohé et al., 2012; Isaac Harris et al., 2014; Lu and Holmgren, 2014; Vriend and Reiter, 2015). Both systems are dependent on NADPH and are involved in the antioxidant defensive mechanism, redox regulation and cell growth (Arnér and Holmgren, 2006; Peng et al., 2012, 2014). There are various ways as to how the thioredoxin system contributes toward the progression of cancer (Arnér and Holmgren, 2006). There are two trx systems in the mammalian cells; the cytosolic trx and mitochondrial trx (Lu and Holmgren, 2014). Both the cytosolic and mitochondrial trx systems are dependent on peroxidases and eventually involved in the redox regulation (Lu and Holmgren, 2014). Thioredoxin reductase 1 is known to be overexpressed in most malignant cancer cells (Miyazaki et al., 1998; Yoo et al., 2006; Karlenius and Tonissen, 2010). In fact, besides being involved in the defensive mechanism of cells, thioredoxin peroxidase (TRx) has been reported to inhibit apoptosis by interfering with p53 and p21 (Zhang et al., 1997; Ueno et al., 1999; Brigelius-Flohé et al., 2012). Targeting players of the thioredoxin pathway such as thioredoxin reductase, peroxidase or thioredoxin has been an interest for cancer therapy (Arnér and Holmgren, 2006; Lu et al., 2007; Karlenius and Tonissen, 2010; Penney and Roy, 2013). Moreover, in most cancer cells, the level of GSH is often upregulated and can contribute to the drug-resistance mechanism (Balendiran et al., 2004; Traverso et al., 2013). GSH is a non-protein molecule that has several important physiological properties (Balendiran et al., 2004). Moreover, it is known that GSH can contribute toward the initiation of cancer. In phase II detoxification process, GST plays an important role as it assists in the conjugation of GSH with different cancer-promoting electrophiles (Balendiran et al., 2004). The high levels of GSH and GST has become one of the important properties in various types of cancer (Balendiran et al., 2004). Besides GSH, the level of SOD was also increased in DIA-treated cells. SOD is known to be higher in cancer cells than normal cells (Oberley and Buettner, 1979). The role of SOD as an antioxidant is by converting the radical O2 to the less radical H2O2 (Matés, 2000; Kowald and Klipp, 2004; Valko et al., 2006). Catalase, another antioxidant enzyme will later on convert the produced H2O2 to water and oxygen (Matés, 2000; Kowald and Klipp, 2004; Valko et al., 2006;). Additionally, the GPx enzyme will also convert H2O2 to water and GSSG (Matés, 2000; Valko et al., 2006). Another important phase II detoxification enzyme is the pro-survival HMOX-1, which is heavily involved in the inactivation of the pro-oxidant heme to ferrous iron, carbon monoxide and bilirubin (Lau et al., 2008; Yim et al., 2011). Activation of heat shock protein and antioxidant mechanism by this diterpene may protect the cancer cell and thus reduce the killing efficacy of DIA. Overall, Figure 8 summarizes the proposed schematic of mechanism of action of DIA in HeLa cells.
Figure 8.

Proposed schematic of the mechanism of action of DIA in HeLa cells by regulating both the anti- and pro-survival pathways.
Conclusion
Overall, DIA managed to induce cellular stress in HeLa cells as evidenced by the results above. DIA induced cellular death via the apoptosis pathway by regulating the FAS and BCL-2 family genes. On the other hand, DIA also activated several pro-survival pathways including the heat shock protein response and anti-oxidant pathways. The dual regulation of DIA in HeLa cells could further benefit the understanding to the molecular mechanism of DIA. Future work using this compound can be applied in an in vivo setting to promote a deeper understanding of the function of DIA as an anti-cancer agent.
Author contributions
NA, SY, NBA: Designed the experiments. NA, AM, MN, NZ, SY: Performed the experiments. MN, SZ, JI: Preparation and identification of the compound. SY, NA, NR: Analyzed the results. NA, SY, MN: Prepared the manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge the efforts of Prof. Dr. Bilge Sener for providing and identifying the plant. This project was supported by the University Malaysia Pahang (internal grant No. RDU 120389 and 150349) and Universiti Putra Malaysia (internal grant; vote number: 9399200).
References
- Abourashed E. A., Galal A. M., Shibl A. M. (2011). Antimycobacterial activity of ferutinin alone and in combination with antitubercular drugs against a rapidly growing surrogate of Mycobacterium tuberculosis. Nat. Prod. Res. 25, 1142–1149. 10.1080/14786419.2010.481623 [DOI] [PubMed] [Google Scholar]
- Akhtar M. N., Atta-ur-Rahman Choudhary, M. I., Sener B., Erdogan I., Tsuda Y. (2003). New class of steroidal alkaloids from Fritillaria imperialis. Phytochemistry 63, 115–122. 10.1016/S.0031-9422(02)00569-1 [DOI] [PubMed] [Google Scholar]
- Al-Ja'fari A.-H., Vila R., Freixa B., Costa J., Cañigueral S. (2013). Antifungal compounds from the rhizome and roots of Ferula hermonis. Phytother. Res. 27, 911–915. 10.1002/ptr.4806 [DOI] [PubMed] [Google Scholar]
- Arnér E. S. J., Holmgren A. (2006). The thioredoxin system in cancer. Semin. Cancer Biol. 16, 420–426. 10.1016/j.semcancer.2006.10.009 [DOI] [PubMed] [Google Scholar]
- Atta-ur-Rahman Akhtar, M. N., Choudhary M. I., Tsuda Y., Sener B., Khalid A., et al. (2002). New steroidal alkaloids from Fritillaria imperialis and their cholinesterase inhibiting activities. Chem. Pharm. Bull. 50, 1013–1016. 10.1248/cpb.50.1013 [DOI] [PubMed] [Google Scholar]
- Atta-ur-Rahman Nadeem Akhtar, M., Iqbal Choudhary M., Tsuda Y., Yasin A., Sener B., et al. (2005). New diterpene isopimara-7,15-dien-19-oic acid and its prolyl endopeptidase inhibitory activity. Nat. Prod. Res. 19, 13–22. 10.1080/14786410310001643885 [DOI] [PubMed] [Google Scholar]
- Badfar-Chaleshtori S., Shiran B., Kohgard M., Mommeni H., Hafizi A., Khodambashi M., et al. (2012). Assessment of genetic diversity and structure of Imperial Crown (Fritillaria imperialis L.) populations in the Zagros region of Iran using AFLP, ISSR and RAPD markers and implications for its conservation. Biochem. Syst. Ecol. 42, 35–48. 10.1016/j.bse.2011.12.027 [DOI] [Google Scholar]
- Balendiran G. K., Dabur R., Fraser D. (2004). The role of glutathione in cancer. Cell Biochem. Funct. 22, 343–352. 10.1002/cbf.1149 [DOI] [PubMed] [Google Scholar]
- Bartrons R., Caro J. (2007). Hypoxia, glucose metabolism and the Warburg's effect. J. Bioenerg. Biomembr. 39, 223–229. 10.1007/s10863-007-9080-3 [DOI] [PubMed] [Google Scholar]
- Branzei D., Foiani M. (2008). Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 9, 297–308. 10.1038/nrm2351 [DOI] [PubMed] [Google Scholar]
- Brigelius-Flohé R., Müller M., Lippmann D., Kipp A. P. (2012). The yin and yang of Nrf2-regulated selenoproteins in carcinogenesis. Int. J. Cell Biol. 2012, 8. 10.1155/2012/486147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood S. K., Khaleque M. A., Sawyer D. B., Ciocca D. R. (2006). Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem. Sci. 31, 164–172. 10.1016/j.tibs.2006.01.006 [DOI] [PubMed] [Google Scholar]
- Calderwood S. K., Stevenson M. A., Murshid A. (2012). Heat shock proteins, autoimmunity, and cancer treatment. Autoimmune Dis. 2012, 10. 10.1155/2012/486069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi E.-J., Lee S., Chae J.-R., Lee H.-S., Jun C.-D., Kim H. (2011). Eupatilin inhibits lipopolysaccharide-induced expression of inflammatory mediators in macrophages. Life Sci. 88, 1121–1126. 10.1016/j.lfs.2011.04.011 [DOI] [PubMed] [Google Scholar]
- Chun K.-S., Kundu J., Chae I. G., Kundu J. K. (2014). Carnosol: a phenolic diterpene with cancer chemopreventive potential. J. Cancer Prev. 19, 103–110. 10.15430/JCP.2014.19.2.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke P., Tyler K. L. (2009). Apoptosis in animal models of virus-induced disease. Nat. Rev. Micro. 7, 144–155. 10.1038/nrmicro2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory S., Huang D. C., Adams J. M. (2003). The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 22, 8590–8607. 10.1038/sj.onc.1207102 [DOI] [PubMed] [Google Scholar]
- Dahham S., Tabana Y., Iqbal M., Ahamed M., Ezzat M., Majid A., et al. (2015). The anticancer, antioxidant and antimicrobial properties of the sesquiterpene β-Caryophyllene from the essential oil of Aquilaria crassna. Molecules 20, 11808. 10.3390/molecules200711808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungwa J., Hunt L., Ramani P. (2011). Overexpression of carbonic anhydrase and HIF-1α in Wilms tumours. BMC Cancer 11:390. 10.1186/1471-2407-11-390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder M. E., Hofmann G. E. (1999). Heat-shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu. Rev. Physiol. 61, 243–282. 10.1146/annurev.physiol.61.1.243 [DOI] [PubMed] [Google Scholar]
- Fiaschi T., Chiarugi P. (2012). Oxidative stress, tumor microenvironment, and metabolic reprogramming: a diabolic liaison. Int. J. Cell Biol. 2012, 8. 10.1155/2012/762825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S., Gorman A. M., Hori O., Samali A. (2010). Cellular stress responses: cell survival and cell death. Int. J. Cell Biol. 2010, 23. 10.1155/2010/214074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C., Harris I. S., Mak T. W. (2013). Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947. 10.1038/nrd4002 [DOI] [PubMed] [Google Scholar]
- Gross A., McDonnell J. M., Korsmeyer S. J. (1999). BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13, 1899–1911. 10.1101/gad.13.15.1899 [DOI] [PubMed] [Google Scholar]
- Gupta S., Kim C., Yel L., Gollapudi S. (2004). A Role of Fas-Associated Death Domain (FADD) in increased apoptosis in aged humans. J. Clin. Immunol. 24, 24–29. 10.1023/B:JOCI.0000018059.56924.99 [DOI] [PubMed] [Google Scholar]
- Hui E. P., Chan A. T. C., Pezzella F., Turley H., To K.-F., Poon T. C. W., et al. (2002). Coexpression of hypoxia-inducible factors 1α and 2α, carbonic anhydrase IX, and vascular endothelial growth factor in nasopharyngeal carcinoma and relationship to survival. Clin. Cancer Res. 8, 2595–2604. [PubMed] [Google Scholar]
- Isaac Harris S., Aislinn Treloar E., Inoue S., Sasaki M., Gorrini C., Kim Lee C., et al. (2014). Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222. 10.1016/j.ccell.2014.11.019 [DOI] [PubMed] [Google Scholar]
- Ivanescu B., Miron A., Corciova A. (2015). Sesquiterpene lactones from Artemisia genus: biological activities and methods of analysis. J. Anal. Methods Chem. 2015, 21. 10.1155/2015/247685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubb A. M., Pham T. Q., Hanby A. M., Frantz G. D., Peale F. V., Wu T. D., et al. (2004). Expression of vascular endothelial growth factor, hypoxia inducible factor 1α, and carbonic anhydrase IX in human tumours. J. Clin. Pathol. 57, 504–512. 10.1136/jcp.2003.012963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlenius T. C., Tonissen K. F. (2010). Thioredoxin and cancer: a role for thioredoxin in all states of tumor oxygenation. Cancers 2, 209–232. 10.3390/cancers2020209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare C. P. (2007). Indian Medicinal Plants: An Illustrated Dictionary. New York, NY: Springer Science and Business. [Google Scholar]
- Kim M.-K., Suh D., Kim B., Song Y.-S. (2014). Cellular stress responses and cancer: new mechanistic insights on anticancer effect by phytochemicals. Phytochem. Rev. 13, 207–221. 10.1007/s11101-013-9307-3 [DOI] [Google Scholar]
- Kowald A., Klipp E. (2004). Alternative pathways might mediate toxicity of high concentrations of superoxide dismutase. Ann. N.Y. Acad. Sci. 1019, 370–374. 10.1196/annals.1297.065 [DOI] [PubMed] [Google Scholar]
- Kwak M.-K., Kensler T. W. (2010). Targeting NRF2 signaling for cancer chemoprevention. Toxicol. Appl. Pharmacol. 244, 66–76. 10.1016/j.taap.2009.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A., Villeneuve N. F., Sun Z., Wong P. K., Zhang D. D. (2008). Dual roles of Nrf2 in cancer. Pharmacol. Res. 58, 262–270. 10.1016/j.phrs.2008.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., Holmgren A. (2014). The thioredoxin antioxidant system. Free Radic. Biol. Med. 66, 75–87. 10.1016/j.freeradbiomed.2013.07.036 [DOI] [PubMed] [Google Scholar]
- Lu J., Chew E.-H., Holmgren A. (2007). Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. 104, 12288–12293. 10.1073/pnas.0701549104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.-Y., Chen T.-S., Qu J.-L., Pan W.-L., Sun L., Wei B. (2009). Dihydroartemisinin (DHA) induces caspase-3-dependent apoptosis in human lung adenocarcinoma ASTC-a-1 cells. J. Biomed.Sci. 16:16. 10.1186/1423-0127-16-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q. (2013). Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 53, 401–426. 10.1146/annurev-pharmtox-011112-140320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantaj J., Rahman S. M. A., Bokshi B., Hasan C. M., Jackson P. J. M., Parsons R. B., et al. (2015). Crispene E, a cis-clerodane diterpene inhibits STAT3 dimerization in breast cancer cells. Org. Biomol. Chem. 13, 3882–3886. 10.1039/C5OB00052A [DOI] [PubMed] [Google Scholar]
- Martindale J. L., Holbrook N. J. (2002). Cellular response to oxidative stress: signaling for suicide and survival. J. Cell. Physiol. 192, 1–15. 10.1002/jcp.10119 [DOI] [PubMed] [Google Scholar]
- Matés J. M. (2000). Effects of antioxidant enzymes in the molecular control of reactive oxygen species toxicology. Toxicology 153, 83–104. 10.1016/S0300-483X(00)00306-1 [DOI] [PubMed] [Google Scholar]
- Miyazaki K., Noda N., Okada S., Hagiwara Y., Miyata M., Sakurabayashi I., et al. (1998). Elevated serum level of thioredoxin in patients with hepatocellular carcinoma. Biotherapy 11, 277–288. 10.1023/A:1008032703468 [DOI] [PubMed] [Google Scholar]
- Nguyen T., Nioi P., Pickett C. B. (2009). The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 284, 13291–13295. 10.1074/jbc.R900010200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberley L. W., Buettner G. R. (1979). Role of superoxide dismutase in cancer: a review. Cancer Res. 39, 1141–1149. [PubMed] [Google Scholar]
- Pelicano H., Carney D., Huang P. (2004). ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat. 7, 97–110. 10.1016/j.drup.2004.01.004 [DOI] [PubMed] [Google Scholar]
- Peng X., Mandal P. K., Kaminskyy V. O., Lindqvist A., Conrad M., Arnér E. S. J. (2014). Sec-containing TrxR1 is essential for self-sufficiency of cells by control of glucose-derived H2O2. Cell Death Dis. 5, e1235. 10.1038/cddis.2014.209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X., Xu J., Elias Arnér S. J. (2012). Thiophosphate and selenite conversely modulate cell death induced by glutathione depletion or cisplatin: effects related to activity and Sec contents of thioredoxin reductase. Biochem. J. 447, 167–174. 10.1042/BJ20120683 [DOI] [PubMed] [Google Scholar]
- Penney R. B., Roy D. (2013). Thioredoxin-mediated redox regulation of resistance to endocrine therapy in breast cancer. Biochim. Biophys. Acta 1836, 60–79. 10.1016/j.bbcan.2013.02.005 [DOI] [PubMed] [Google Scholar]
- Portt L., Norman G., Clapp C., Greenwood M., Greenwood M. T. (2011). Anti-apoptosis and cell survival: a review. Biochim. Biophys. Acta 1813, 238–259. 10.1016/j.bbamcr.2010.10.010 [DOI] [PubMed] [Google Scholar]
- Qi Q.-Y., Ren J.-W., Sun L.-W., He L.-W., Bao L., Yue W., et al. (2015). Stucturally diverse sesquiterpenes produced by a chinese tibet fungus Stereum hirsutum and their cytotoxic and immunosuppressant activities. Org. Lett. 17, 3098–3101. 10.1021/acs.orglett.5b01356 [DOI] [PubMed] [Google Scholar]
- Reuter S., Gupta S. C., Chaturvedi M. M., Aggarwal B. B. (2010). Oxidative stress, inflammation, and cancer: how are they linked? Free Radic. Biol. Med. 49, 1603–1616. 10.1016/j.freeradbiomed.2010.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt E., Paquet C., Beauchemin M., Bertrand R. (2007). DNA-damage response network at the crossroads of cell-cycle checkpoints, cellular senescence and apoptosis. J. Zhejiang Univ. Sci. B 8, 377–397. 10.1631/jzus.2007.B0377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneuric R., Mjahed H., Gobbo J., Joly A.-L., Berthenet K., Shirley S., et al. (2011). Heat shock proteins as danger signals for cancer detection. Front. Oncol. 1:37. 10.3389/fonc.2011.00037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R., Ma J., Zou Z., Jemal A. (2014). Cancer statistics, 2014. CA Cancer J. Clin. 64, 9–29. 10.3322/caac.21208 [DOI] [PubMed] [Google Scholar]
- Stahlhut R., Park G., Petersen R., Ma W., Hylands P. (1999). The occurrence of the anti-cancer diterpene taxol in Podocarpus gracilior Pilger (Podocarpaceae). Biochem. Syst. Ecol. 27, 613–622. 10.1016/S0305-1978(98)00118-5 [DOI] [Google Scholar]
- Sulaiman M. R., Mohd Padzil A., Shaari K., Khalid S., Shaik Mossadeq W. M., Mohamad A. S., et al. (2010). Antinociceptive activity of Melicope ptelefolia ethanolic extract in experimental animals. J. Biomed. Biotechnol. 2010, 6. 10.1155/2010/937642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverso N., Ricciarelli R., Nitti M., Marengo B., Furfaro A. L., Pronzato M. A., et al. (2013). Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 10 10.1155/2013/972913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M., Masutani H., Arai R. J., Yamauchi A., Hirota K., Sakai T., et al. (1999). Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J. Biol. Chem. 274, 35809–35815. 10.1074/jbc.274.50.35809 [DOI] [PubMed] [Google Scholar]
- Valko M., Rhodes C. J., Moncol J., Izakovic M., Mazur M. (2006). Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 160, 1–40. 10.1016/j.cbi.2005.12.009 [DOI] [PubMed] [Google Scholar]
- Vriend J., Reiter R. J. (2015). The Keap1-Nrf2-antioxidant response element pathway: a review of its regulation by melatonin and the proteasome. Mol. Cell. Endocrinol. 401, 213–220. 10.1016/j.mce.2014.12.013 [DOI] [PubMed] [Google Scholar]
- Yim M.-S., Ha Y.-S., Kim I. Y., Yun S.-J., Choi Y. H., Kim J. (2011). HMOX1 is an important prognostic indicator of nonmuscle invasive bladder cancer recurrence and progression. J. Urol. 185, 701–705. 10.1016/j.juro.2010.09.081 [DOI] [PubMed] [Google Scholar]
- Yoo M. H., Xu X. M., Carlson B. A., Gladyshev V. N., Hatfield D. L. (2006). Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J. Biol. Chem. 281, 13005–13008. 10.1074/jbc.C600012200 [DOI] [PubMed] [Google Scholar]
- Zhang P., Liu B., Kang S. W., Seo M. S., Rhee S. G., Obeid L. M. (1997). Thioredoxin peroxidase is a novel inhibitor of apoptosis with a mechanism distinct from that of Bcl-2. J. Biol. Chem. 272, 30615–30618. 10.1074/jbc.272.49.30615 [DOI] [PubMed] [Google Scholar]