

Abstract
The islet in type 2 diabetes is characterized by β-cell loss, increased β-cell apoptosis, and islet amyloid derived from islet amyloid polypeptide (IAPP). When protein misfolding protective mechanisms are overcome, human IAPP (h-IAPP) forms membrane permeant toxic oligomers that induce β-cell dysfunction and apoptosis. In humans with type 2 diabetes (T2D) and mice transgenic for h-IAPP, endoplasmic reticulum (ER) stress has been inferred from nuclear translocation of CCAAT/enhancer-binding protein homologous protein (CHOP), an established mediator of ER stress. To establish whether h-IAPP toxicity is mediated by ER stress, we evaluated diabetes onset and β-cell mass in h-IAPP transgenic (h-TG) mice with and without deletion of CHOP in comparison with wild-type controls. Diabetes was delayed in h-TG CHOP−/− mice, with relatively preserved β-cell mass and decreased β-cell apoptosis. Deletion of CHOP attenuates dysfunction of the autophagy/lysosomal pathway in β-cells of h-TG mice, uncovering a role for CHOP in mediating h-IAPP-induced dysfunction of autophagy. As deletion of CHOP delayed but did not prevent h-IAPP-induced β-cell loss and diabetes, we examined CHOP-independent stress pathways. JNK, a target of the IRE-1pTRAF2 complex, and the Bcl-2 family proapoptotic mediator BIM, a target of ATF4, were comparably activated by h-IAPP expression in the presence and absence of CHOP. Therefore, although these studies affirm that CHOP is a mediator of h-IAPP-induced ER stress, it is not the only one. Therefore, suppression of CHOP alone is unlikely to be a durable therapeutic strategy to protect against h-IAPP toxicity because multiple stress pathways are activated.
The islet in type 2 diabetes (T2D) is characterized by a deficit in β-cell mass, increased β-cell apoptosis, and islet amyloid (1). The amyloid is composed of islet amyloid polypeptide (IAPP), a protein coexpressed and cosecreted with insulin by pancreatic β-cells (2). This islet pathology is reproduced in other species vulnerable to T2D such as nonhuman primates (3).
A potential contribution of IAPP misfolding in the pathogenesis of β-cell dysfunction and loss in T2D is supported by studies employing transgenic rodents that express human IAPP (h-IAPP). In both h-IAPP transgenic (h-TG) mice and rats (4–7), diabetes develops as a consequence of a loss of β-cell mass with increased β-cell apoptosis. This pathology is in part due to membrane disruptive toxic oligomers of h-IAPP which form intracellularly in β-cells of both humans with T2D (8) and h-TG transgenic mice (9). The resulting β-cell dysfunction and apoptosis have been ascribed to both oxidative stress (10) and endoplasmic reticulum (ER) stress (7, 11). The importance of the propensity of h-IAPP to form intracellular toxic oligomers in the induction of ER stress, β-cell apoptosis and diabetes has been demonstrated by the absence of toxic oligomers and ER stress (revealed by nuclear translocation of CCAAT/enhancer-binding protein homologous protein [CHOP]) in mice with comparable transgenic overexpression of nonamyloidogenic rodent IAPP (7, 8). Sustained activation of the unfolded protein response (UPR)-induced transcription factor CHOP has been credited with playing a central role in the transition from cellular protection afforded by the UPR to induction of apoptosis by ER stress (12, 13). To this end, it has been proposed that demonstration of a delay in β-cell failure and diabetes onset by CHOP deletion is a requirement to establish ER stress as a mechanism subserving β-cell failure (12).
In the present studies our primary goal was to establish whether h-IAPP-induced β-cell toxicity meets that definition of ER stress. Specifically, we addressed this by testing the hypothesis that deletion of CHOP delays diabetes onset in h-TG mice. Having established that deletion of CHOP delays rather than prevents diabetes, we further investigated the pathways through which toxic h-IAPP induced β-cell loss in the absence of CHOP signaling.
Materials and Methods
Mouse models
Mice were bred and housed at the University of California, Los Angeles (UCLA) animal housing facility. The UCLA Institutional Animal Care and Use Committee approved all surgical and experimental procedures. Animals were maintained on a 12-hour light, 12-hour dark cycle with Harlan Teklad Rodent Diet 8604 and water ad libitum. Males were used for the experiments.
CHOP (B6.129S-Ddit3tm1Dron/J CHOP-10) knockout (CHOP−/−) mice were obtained from The Jackson Laboratory. CHOP−/− mice were backcrossed to the FVB background for 9 generations. Control wild-type FVB mice were originally purchased from Charles Rivers Laboratory and bred at UCLA. These CHOP−/− mice were then crossbred with the homozygous h-TG mouse model (h-TG: FVB-Tg(IAPP)6Jdm/Tg(IAPP)6Jdm) described previously (4). This breeding scheme generated 4 groups of mice: WT, CHOP−/−, h-TG, and h-TG CHOP−/− mice. h-TG and h-TG CHOP−/− mice were homozygous for h-IAPP; only males were used in the study. Mice were followed for 14 weeks obtaining fasting blood glucose and body weight, unless the blood glucose was greater than 300 mg/dL, in which case they were euthanized at that time point to avoid distress. For blood glucose measurement, mice were fasted overnight between 16–18 hours. Blood was taken from the tail of mice and measured using a FreeStyle Lite blood glucose meter (Abbot).
Islet isolation
After overnight fast, animals were euthanized using isoflurane. The bile duct was clamped at the entrance to duodenum, cannulated, and pancreas was perfused with 2 mL of collagenase solution (HBSS [Invitrogen] supplemented with 25mM HEPES [Invitrogen], 0.23 mg/mL liberase [Roche], and 0.1 mg/mL DNase [Roche]). Pancreas was removed, transferred into a glass vial containing 2 mL of ice-cold collagenase solution (2 pancreata per vial), digested for 19 minutes at 37°C, then dispersed by shaking for 30 seconds. Islets were manually picked, washed with ice-cold PBS, and lysed in lysis buffer (50mM HEPES, 1% Nonidet P-40, 2mM Na3VO4, 100mM NaF, 10mM PyrPO4, 4mM EDTA, 1mM Phenylmethanesulfonyl fluoride, 1 μg/mL leupeptin, and 1 μg/mL aprotinin). After 20 minutes of incubation in lysis buffer on ice, islets were sonicated for 10 seconds and centrifuged at 10 000 rpm at 4°C for 10 minutes. Supernatant was stored at −80°C until use for subsequent protein determination by detergent compatible protein assay (Bio-Rad) and Western blotting. For each sample for Western blotting, islets from 2–4 mice were pooled.
Western blotting
Proteins (25–40 μg/lane) were separated on a 4%–12% Bis-Tris NuPAGE gel and blotted onto a Polyvinylidene fluoride membrane (FluoroTrans; Pall Life Sciences). Membranes were probed overnight at 4°C with primary antibodies against cleaved caspase-3, Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), JNK, p-JNK, Microtubule-associated proteins 1A/1B light chain 3B (LC3B), Bcl-2 interacting mediator of cell death (BIM), Phosphorylated BIM (p-BIM, Cell Signaling Technology), growth arrest and DNA damage-inducible protein (GADD)34 (Santa Cruz Biotechnology, Inc), and p62 (GP62-C, Progen Biotechnik). Horseradish peroxidase-conjugated secondary antibodies were from Zymed Laboratories or Invitrogen. Proteins were visualized by enhanced chemiluminescence (Millipore), and protein expression levels were quantified using the Labworks software (UVP).
RNA isolation, RT-PCR, and real-time quantitative PCR
Total RNA was extracted using the RNeasy Mini kit (QIAGEN) performing on-column DNase digestion with RNase-Free DNase Set (QIAGEN), according to the manufacturer's instructions. A total of 0.25 μg of RNA was used for preparation of single stranded cDNA using Superscript III Reverse Transcriptase (Invitrogen) by the oligo-dT priming method. Real-time quantitative PCR was performed with 700 ng of cDNA using the LightCycler FastStart DNA MasterPLUS SYBR Green I kit (Roche) and the LightCycler PCR equipment (Roche). The oligonucleotide primers were: 5′-GCTGCCCTATACCCACATCT-3′ and 5′-CGCCTTCATCCGAGAAAC-3′ for CHOP; 5′-CTGCCCTATACCCACATCT-3′ and 5′-CGCCTTCATCCGAGAAAC-3′ for p62/SQSTM1; 5′-ATGATGGCTTGGCCAGTG-3′ and 5′-CCATTTTCTCCAACATCCAATC-3′ for activating transcription factor 4 (ATF4); and 5′-GTCTCCTTCGAGCTGTTTGC-3′ and 5′-AGCCAAATCCTTTCTCTCCA-3′ for Cyclophilin. All measurements were normalized to the housekeeping gene Cyclophilin.
Tissue collection
After an overnight fast, animals were euthanized using isoflurane. Pancreas was immediately resected, blotted dry, weighed, and then fixed in 4% paraformaldehyde for 24 hours at 4°C followed by paraffin embedding. Subsequently, 4-μm tissue sections were obtained through the maximal plane of pancreas possible and mounted on glass slides.
Immunofluorescent and immunohistochemistry staining
To confirm the deletion of CHOP in CHOP−/− mice, sections were immunostained for CHOP and insulin as follows. Slides were rehydrated in 2 toluene washes followed by 3 ethanol washes and water. Then, slides were blocked for 1 hour in 3% BSA/TBS/0.2% Triton X-100 and incubated for 72 hours at 4°C with rabbit anti-CHOP (1:200, catalog number sc-575; Santa Cruz Biotechnology, Inc) diluted in 3% BSA/TBS/0.2% Tween 20, followed by 1-hour treatment at room temperature (RT) with secondary antibody conjugated to Cy3 (1:200; Jackson ImmunoResearch). Slides were then stained for insulin using guinea pig anti-insulin antibody (1:200, ON, +4°C, catalog number ab7842; Abcam) and donkey-derived secondary antibody conjugated to FITC (1:200, RT, 1 hour; Jackson ImmunoResearch). Slides were mounted with Vectashield with DAPI (Vector Laboratories), viewed and imaged using a Leica DM6000 microscope (Leica Microsystems) at ×200 magnification (×20 objective) and OpenLab 5.0 software (Improvision). The frequency of CHOP-positive β-cells was assessed as described below for Ki67 and TUNEL-positive cells.
To assess β-cell proliferation and apoptosis, we performed immunofluorescent staining of adjacent sections of pancreas with 1) mouse anti-Ki67 antibody (1:50, ON, +4°C, catalog number 550609; BD Pharmingen) followed by guinea pig antiinsulin antibody; or 2) stained by TUNEL (In Situ Cell Death Detection AP kit, catalog number 11684809910; Roche) and guinea pig anti-insulin antibody.
For β-cell mass measurements, pancreatic sections were immunostained for insulin and counterstained with hematoxylin. Briefly, slides were rehydrated as above. They were then blocked in 3% H2O2/10% methanol for 30 minutes at RT. Slides were washed in TBS/0.1% Tween followed by a second blocking step in Tris B buffer (0.1M Trizma base, 2% BSA, 0.85% NaCl, and 0.1% Triton X-100) for 1 hour at RT. Slides were incubated with guinea pig anti-insulin antibody diluted in Tris B (1:100). The next day slides were incubated in Biotin donkey anti-guinea pig diluted in Tris B (1:100) and ABC peroxide solution was added for 1 hour at RT. Following this was the addition of 3,3′-diaminobenzidine (DAB) solution for approximately 1–2 minutes (when brown deposits were observed). Slides were then incubated in hematoxylin for 7 seconds, followed by washes in ethanol and toluene before they were cover slipped.
β-Cell mass
The entire pancreatic section stained for insulin by IHC was imaged at ×40 magnification (×4 objective) using an Olympus IX70 inverted microscope. The pancreatic cross-sectional area positive for insulin and the total pancreas cross-sectional area were digitally quantified using Image Pro Plus software (Image Pro Plus version 4.5.1; Media Cybernetics) to determine the ratio of β-cell area to pancreas area. The resulting ratio was multiplied by the pancreas weight to obtain the β-cell mass in each mouse.
Replication and apoptosis
We quantified the impact of CHOP deletion on β-cell turnover in 5 mice per group (WT, CHOP−/−, h-TG, and h-TG CHOP−/− mice) at age 9 weeks; this age was chosen to avoid the confounding effects of varying blood glucose concentrations. All islets in the section were imaged using a Leica DM6000 microscope using ×20 objective (×200 magnification) and OpenLab 5.0 software. The frequency of β-cell replication and apoptosis were determined by manually counting 1) all β-cells contained in each pancreatic cross-section, 2) Ki67-positive nuclei present within β-cells, and 3) TUNEL-positive nuclei within β-cells. Strict criteria were used for determining Ki67 or TUNEL positivity in β-cells: more than 2/3 of the perimeter of the nucleus must be surrounded by insulin, the nuclear contours of the Ki67 or TUNEL stain must match those of DAPI in addition to exactly overlapping, and the nucleus must be present in the same plane as the β-cell in question. The mean number of β-cells counted per pancreatic cross section was 3304 ± 564 for WT, 2850 ± 503 for CHOP−/− 2191 ± 304 for h-TG and 2389 ± 640 for h-TG CHOP−/− mice. The frequency of Ki67 and TUNEL-positive nuclei were expressed as a percentage of total number of β-cells in the pancreatic cross-section. The same approach was used for assessment of frequency of β-cells with nuclear CHOP.
Statistical analysis
Results are expressed as the mean ± SEM for n independent experiments, as indicated in figure legends. Statistical analyses were carried out by Student's t test or ANOVA followed by Fisher's post hoc test for multiple comparisons using Statistica (StatSoft). P < .05 was taken as evidence of statistical significance; *, P < .05; **, P < .01; ***, P < .001.
Results
Deletion of CHOP delays the onset of diabetes in h-TG mice
Deletion of CHOP was confirmed by real time RT-PCR analysis for CHOP mRNA in isolated islets (Figure 1A) and for CHOP protein expression by immunohistochemistry in pancreas sections 9-week-old WT, CHOP−/−, h-TG, and h-TG CHOP−/− mice (Figure 1, B and C). Deletion of CHOP delayed diabetes onset in h-TG mice but had no effect on body weight over the period of the study (Figure 2, A and B, respectively). By 12 weeks of age, 89% of h-TG mice developed diabetes (had fasting blood glucose 126 mg/dL or higher) in contrast to 40% of h-TG CHOP−/− mice (Figure 2C). By the terminal point of the experiment at 14 weeks of age, 100% of h-TG mice and 67% of mice in h-TG CHOP−/− group were diabetic. Note that because mice with blood glucose greater than 300 mg/dL were euthanized to avoid distress, the differences in blood glucose values between the groups in Figure 2A are therefore conservative because more mice in the h-TG group were euthanized before 14 weeks of age than in the h-TG CHOP−/−.
Figure 1.

CHOP knockdown in mice expressing h-IAPP. A, Quantitative RT-PCR for CHOP was performed in isolated islets from 9-week-old mice (WT n = 4, CHOP−/− n = 3, h-TG n = 4, and h-TG CHOP−/− n = 3 with fasting blood glucose levels 68.8 ± 2.8, 71.2 ± 5.1, 79.3 ± 8.0, and 60.3 ± 6.2 mg/dL, respectively). B, Frequency of cells double-positive for CHOP and insulin in 9-week-old mice (n = 4 per group with fasting blood glucose levels 76.5 ± 3.0, 52.6 ± 3.0, 68.8 ± 5.0, and 58.6 ± 4.5 mg/dL for WT, CHOP−/−, h-TG, and h-TG CHOP−/−, respectively). C, Representative immunofluorescence images: CHOP in red, insulin in green, and nuclei in blue (DAPI). Inset is a higher magnification of a nucleus stained for CHOP. Data are expressed as mean ± SEM; **, P < .01; ***, P < .001.
Figure 2.

CHOP deficiency results in the delay in diabetes onset in mice expressing h-IAPP. Fasting blood glucose (A) and fasting body weight (B) over the course of study. Each bar represents n = 3–18 for h-TG (n = 1 at 14 wk) and n = 9–26 for h-TG CHOP−/− mice. Data are expressed as mean ± SEM; *, P < .05 (2-tailed unpaired Student's t test). C, Percent of diabetic mice over the course of study. Diabetes development was monitored in 18 h-TG and 27 h-TG CHOP−/− mice by weekly measurements of blood glucose after overnight fasting. A mouse was considered diabetic if the blood glucose level was 126 mg/dL or higher.
Deletion of CHOP preserves β-cell mass in h-TG mice by providing protection against β-cell apoptosis
In order to address the hypothesis that CHOP signaling contributes to h-IAPP-induced ER stress-induced β-cell apoptosis, we quantified β-cell mass in each group of mice at 10 weeks of age. As expected, there was a deficit in β-cell mass in h-TG mice compared with WT mice (0.45 ± 0.13 vs 2.16 ± 0.33 mg, P < .001) (Figure 3A). However, β-cell mass was relatively preserved in h-TG CHOP−/− vs h-TG mice (1.09 ± 0.14 vs 0.45 ± 0.13 mg, P < .05).
Figure 3.

CHOP deficiency delays β-cell loss in h-TG mice. A, β-cell mass in 10-week-old mice (n = 6 per group). B, The frequency of TUNEL-positive β-cells in 9-week-old mice in WT, CHOP−/−, h-TG, and h-TG CHOP−/− mice (n = 5 per group). C, Cleaved caspase-3 levels in islet lysates from 9-week-old mice detected by Western blotting (n = 4 per group). Actin or GAPDH were used as loading control. Data are expressed as mean ± SEM; *, P < .05; ***, P < .001.
To establish whether the preservation of β-cell mass was due to attenuation of β-cell apoptosis, we quantified the frequency of TUNEL-positive β-cells in pancreatic sections from WT, CHOP−/−, h-TG, and h-TG CHOP−/− mice (n = 5 per group). To avoid possible confounding effects of elevated glucose levels on β-cell apoptosis, for analysis we selected pancreata from 9-week-old mice with similar fasting blood glucose levels within the normal range (76.5 ± 3.0, 52.6 ± 3.0, 68.8 ± 5.0, and 58.6 ± 4.5 mg/dL for WT, CHOP−/−, h-TG, and h-TG CHOP−/−, respectively). In accordance with the previously reported data on the increase in β-cell apoptosis in h-TG rodents (5–7), the frequency of TUNEL-positive β-cells in h-TG mice was 0.36 ± 0.09% (Figure 3B) while hardly detectable in WT mice. Although TUNEL-positive β-cells in h-TG CHOP−/− mice were detectable, β-cell apoptosis was decreased by 50% compared with h-TG mice (0.17 ± 0.08%, P < .05). The attenuation of apoptosis in h-TG CHOP−/− mice was confirmed by the levels of cleavage of caspase-3 in isolated islets from each group measured by Western blotting. The level of cleaved caspase-3 was increased in h-TG mice compared with WT mice (3.5 ± 1.7-fold, P < .05). In support of the hypothesis that h-IAPP-induced β-cell apoptosis is mediated in part through induction of CHOP, cleavage of caspase-3 was decreased in the absence of CHOP (P < .05) (Figure 3C).
To ensure that the relative preservation of β-cell mass with deletion of CHOP was not due to an unexpected increase in β-cell replication, we also quantified β-cell replication in the same 9-week-old mice we used for TUNEL analysis (n = 5 per group). β-Cell replication, measured by Ki67+ nuclear immunostaining, was comparable in h-TG and h-TG CHOP−/− mice (1.22 ± 0.18% vs 1.33 ± 0.25%, P = .71). β-Cell replication was also comparable in CHOP−/− and WT mice (1.05 ± 0.15% vs 0.70 ± 0.23%, P = .24). In summary, these data support the hypothesis that h-IAPP-induced β-cell apoptosis, and consequent loss of β-cell mass, is mediated at least in part through induction of CHOP and thus through the ER stress pathway.
Deletion of CHOP attenuates the action of high expression h-IAPP to induce a defect in the autophagy/lysosomal pathway
The autophagy/lysosomal pathway is critical for clearance of misfolded protein aggregates, and has been shown to be critical in defense against h-IAPP β-cell toxicity (14–17). In keeping with other misfolded protein diseases caused by intracellular accumulation of amyloidogenic proteins, cellular toxicity induced by h-IAPP leads to defective clearance of these aggregated proteins as best demonstrated by accumulation of the scaffold protein p62 that is usually delivered with its cargo of misfolded proteins through the autophagy/lysosomal pathway (14). Previous studies have proposed that sustained activation of CHOP contributes to suppression of the autophagy/lysosomal pathway (18). To investigate whether activation of CHOP contributes to the defect in the autophagy/lysosomal pathway induced by h-IAPP, we evaluated this pathway in h-TG CHOP−/− vs h-TG mice (aged 9 wk). In islets of h-TG mice, accumulation of the autophagosome marker LC3II (Figure 4, A and B) and the sequestosome-1/p62 protein (Figure 4, A and C), supports previous reports of defective flux and protein clearance through the autophagy/lysosomal pathway under conditions of h-IAPP toxicity (14). The accumulation of LC3II and p62 proteins was partially relieved by deletion of CHOP in h-TG mice (Figure 4, A–C). The comparable p62 mRNA levels in h-TG and h-TG CHOP−/− mice (Figure 4D) affirms that the decrease of p62 protein levels in h-TG CHOP−/− islets reflects increased clearance rather than decreased synthesis. We conclude that CHOP activation, under conditions of h-IAPP-induced ER stress, might play a previously unrecognized role in constraining flux through the autophagy/lysosomal pathway.
Figure 4.

CHOP deficiency ameliorates autophagy/lysosomal defect induced by h-IAPP. A, Western blotting for autophagy markers: LC3II and p62 in islets from 9-week-old mice. GAPDH is used as a loading control. B, Quantification of Western blotting for LC3II (n = 4–6 per group). C, Quantification of Western blottings for p62 (n = 4–6 per group). D, p62 mRNA levels were assessed by quantitative PCR (n = 4 per group). Data are expressed as mean ± SEM; **, P < .01; ***, P < .001; n.s., not significant.
Mechanisms underlying sustained β-cell apoptosis despite CHOP deletion in h-TG mice
An increased burden of ER misfolded proteins induces activation of the protein kinase-like ER kinase (PERK) limb of the UPR that transiently decreases the ER protein synthetic burden by PERK induced inactivation (by phosphorylation) of eukaryotic initiation factor 2-α subunit (eIF2α) (19). This transient protection is then reversed by the paradoxical increased translation of ATF4 mRNA that induces GADD34, an eIF2α phosphatase (20, 21). Sustained activation of CHOP and ATF4 may thus induce ER stress induced β-cell apoptosis by permitting overloading of the ER with misfolded proteins, a particular problem in the case of a professional secretory cell expressing an amyloidogenic protein such as h-IAPP.
To address this, we measured ATF4 and GADD34 expression in WT, CHOP−/−, h-TG, and h-TG CHOP−/− mice. The ATF4 mRNA was increased in h-TG mice compared with WT (Figure 5A) and this increase was sustained in h-TG CHOP−/− mice. The protein levels of GADD34 measured by Western blotting followed the same pattern (Figure 5B). The increased GADD34 despite deletion of CHOP is consistent with the recent findings that ATF4 independently activates GADD34 (21).
Figure 5.
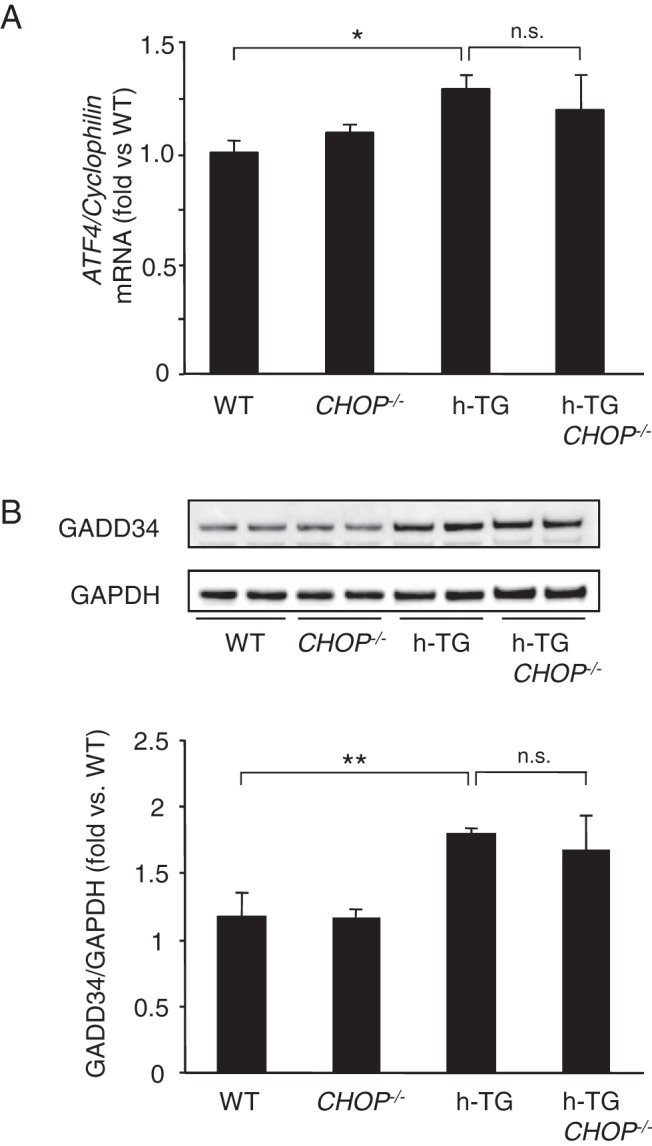
UPR sustained in CHOP-deficient h-TG mice. A, Quantification of ATF4 mRNA in islets from 9-week-old mice (n = 4 per group). Data are expressed as mean ± SEM; *, P < .05; **, P < .01; n.s., not significant. B, Protein level of GADD34 in islet lysates from 9-week-old mice (n = 4 per group).
To further investigate mechanisms underlying ongoing β-cell apoptosis in h-TG CHOP−/− mice, we examined other ER stress related signals known to activate the apoptosis cascade. The phosphorylation of JNK (p-JNK), a target of the IRE-1pTRAF2 complex (22), was similarly increased in β-cells of h-TG and h-TG CHOP−/− mice compared with WT mice (Figure 6A). In addition, the B Bcl-2 homology domain 3-only protein BIM that competes with the anti-apoptotic Bcl-2 family members to potentially liberate mitochondrial membrane permeant proteins that initiate apoptotic pathways, was comparably increased and phosphorylated in h-TG and h-TG CHOP−/− islets (Figure 6, B and C).
Figure 6.

CHOP deletion does not block activation of JNK- and BIM-dependent proapoptotic pathways in h-TG mice. Quantitative analysis of Western blottings of protein lysates obtained from islets of 9-week-old mice. A, Ratio of p-JNK to total JNK (n = 4 per group). B, Quantification of Western blotting for total BIM (n = 4 per group). C, Quantification of Western blotting for p-BIM (n = 4 per group). Data are expressed as mean ± SEM; *, P < .05; ***, P < .001.
In conclusion, deletion of CHOP delayed β-cell loss and diabetes onset induced by high expression of h-IAPP, affirming that h-IAPP acts in part through ER stress. However, alternative pathways of ER stress, and likely other mechanisms of h-IAPP-induced stress, bypass deletion of CHOP and ultimately lead to loss of β-cell mass and diabetes.
Discussion
The present studies were designed to test the hypothesis that genetic deletion of CHOP delays diabetes onset in mice expressing high levels of h-IAPP, and thus establish that h-IAPP toxicity is mediated at least in part through ER stress. That hypothesis was affirmed, but also brought focus on CHOP-independent mechanisms of h-IAPP-induced β-cell toxicity, because deletion of CHOP delays but does not prevent h-IAPP-induced diabetes.
The ER is responsible for synthesis, proper folding and transport of secretory proteins (12, 19, 20). Pancreatic β-cells are long lived secretory cells that are required (in humans) to express, fold, and process approximately 20 million proinsulin molecules a day per cell (23). IAPP, the second major client protein of the ER in β-cells, is one of the most aggregate-prone amyloidogenic proteins known (24). Of interest, the vulnerability of species for developing T2D is defined by the propensity of IAPP to form toxic oligomers, which is high in humans, nonhuman primates and cats, but low in rodents (25, 26). Moreover, a genetic mutation in IAPP that increases its propensity to form oligomers is associated with an increased risk of T2D (27). Under conditions of insulin resistance, the burden of insulin and IAPP protein folding and processing per cell is increased further, particularly in adult humans with minimal capacity to expand β-cell mass (28, 29). Perhaps not surprisingly therefore, obesity induced insulin resistance is an important risk factor for development of T2D in humans, and has been ascribed to both the increased burden of protein folding as well as lipotoxicity, either of which may induce ER stress (23, 30). Both h-IAPP toxicity and lipotoxicity have also been reported to compromise the function of the autophagy/lysosomal pathway that has been shown to be critical in defending β-cells from h-IAPP toxicity (14–17, 31). The interplay between the autophagy and apoptotic pathways is complex, and depends on both the context and duration of the imposed stress (18).
We reconfirmed here that mice with high expression of h-IAPP develop diabetes due to loss of β-cells through β-cell apoptosis, characterized by activation and nuclear translocation of CHOP, reproducing the islet pathology in humans with T2D (7, 11). Moreover, we report that in the absence of CHOP, h-IAPP-induced β-cell loss through apoptosis is partially attenuated, resulting in a delay, but not prevention, of diabetes onset. These data are consistent with a partial contributory role of ER stress in the mediation of h-IAPP toxicity (19). We further establish that the critical autophagy/lysosomal pathway for β-cell protection against h-IAPP-induced β-cell toxicity is partially preserved by deletion of CHOP. This finding uncovers a previously unappreciated role of CHOP in mediating the h-IAPP-induced defect in the autophagy/lysosomal pathway. In the context of amino acid deprivation induced ER stress, CHOP has been shown to have a dual role in the cross talk between autophagy and apoptosis, initially activating autophagy by transcriptionally up-regulating key autophagy genes but, with prolonged amino acid deprivation, CHOP suppresses autophagy and thus contributes to apoptosis (18). Although it is well established that the autophagy/lysosomal pathway is important in the clearance of misfolded amyloidogenic proteins, and that cellular toxicity induced by these proteins induces a defect in this pathway (14–17, 32), the mechanisms by which this defect occurs are yet to be fully elucidated.
Although h-IAPP-induced β-cell loss and diabetes onset were delayed by deletion of CHOP, consistent with other inducers of β-cell ER stress (13), h-IAPP toxicity ultimately prevailed leading to diabetes. CHOP did not protect against the activation of JNK, a target of the IRE-1pTRAF2 complex, previously reported to be an effector of h-IAPP-induced cytotoxicity (33). Because the PERK-phosphorylated eIF2α-ATF4 pathway increases expression of the P-eIF2α phosphatase GADD34, the temporary relief of the ER protein folding burden following activation of PERK is overcome. Also, the proapoptotic Bcl-2 family protein BIM is a target of PERK-ATF4 mediated repression of miR-106b-25 cluster (34), offering an explanation that despite CHOP deletion, BIM was comparably increased by transgenic h-IAPP expression. Also, BIM degradation is dependent on the ubiquitin proteasome system that is dysfunctional under conditions of h-IAPP toxicity due, at least in part, to h-IAPP-induced deficiency of Ubiquitin carboxy-terminal hydrolase L1 (35, 36). Another ER stress-induced mediator of cell stress predicted to elude CHOP deletion is ATF4-induced Nucleotide-binding NACHT domain and Leucine-Rich Repeat containing proteins (NLRP) inflammasome activation (37), a contributor to h-IAPP-induced β-cell stress (38, 39). Furthermore, oxidative stress, a known mediator of h-IAPP-induced apoptosis (10, 17), eludes deletion of CHOP (40).
Taken together, the present studies affirm that h-IAPP toxicity in a mouse model of T2D is mediated at least in part through ER stress induced by CHOP activation. In keeping with other aggregate prone misfolded protein induced cellular dysfunction, the downstream pathways that lead to cellular dysfunction and stress are numerous and complex. Strategies to accomplish sustained amelioration of cytotoxicity mediated by misfolded protein aggregates will likely require multiple simultaneous interventions, such as those targeted to suppress ER and oxidative stress, and those to enhance mitochondrial repair and oligomer elimination by increasing flux through the autophagy (mitophagy)/lysosomal pathway. Efforts to block initiation of these stress pathways by preventing toxic oligomer formation might offer a more promising therapeutic strategy.
Acknowledgments
This work was supported by the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases (NIH/NIDDK) Grant DK059579 and the Larry L. Hillblom Foundation Grant 2014-D-001-NET (to P.C.B.) and by the NIH/NIDDK Grant DK090995 (to J.F.R.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ATF4
- activating transcription factor 4
- Bcl-2
- B cell lymphoma-2
- BIM
- Bcl-2 interacting mediator of cell death
- CHOP
- CCAAT/enhancer-binding protein homologous protein
- eIF2α
- eukaryotic initiation factor 2-α subunit
- ER
- endoplasmic reticulum
- GADD
- growth arrest and DNA damage-inducible protein
- GAPDH
- Glyceraldehyde 3-phosphate dehydrogenase
- h-IAPP
- human IAPP
- h-TG
- h-IAPP transgenic
- IAPP
- islet amyloid polypeptide
- IRE-1pTRAF2
- Inositol-requiring enzyme 1 Phosphorylated TNF receptor-associated factor 2
- JNK
- c-Jun N-terminal kinase
- p-BIM
- Phosphorylated BIM
- p-JNK
- Phosphorylated JNK
- PERK
- protein kinase-like ER kinase
- RT
- room temperature
- T2D
- type 2 diabetes
- TUNEL
- TdT-mediated dUTP nick-end labeling
- UCLA
- University of California, Los Angeles
- UPR
- unfolded protein response
- WT
- wild type.
References
- 1. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. [DOI] [PubMed] [Google Scholar]
- 2. Butler PC, Chou J, Carter WB, et al. Effects of meal ingestion on plasma amylin concentration in NIDDM and nondiabetic humans. Diabetes. 1990;39:752–756. [DOI] [PubMed] [Google Scholar]
- 3. Wagner JD, Carlson CS, O'Brien TD, Anthony MS, Bullock BC, Cefalu WT. Diabetes mellitus and islet amyloidosis in cynomolgus monkeys. Lab Anim Sci. 1996;46:36–41. [PubMed] [Google Scholar]
- 4. Janson J, Soeller WC, Roche PC, et al. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci USA. 1996;93:7283–7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Butler AE, Janson J, Soeller WC, Butler PC. Increased β-cell apoptosis prevents adaptive increase in β-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes. 2003;52:2304–2314. [DOI] [PubMed] [Google Scholar]
- 6. Butler AE, Jang J, Gurlo T, Carty MD, Soeller WC, Butler PC. Diabetes due to a progressive defect in β-cell mass in rats transgenic for human islet amyloid polypeptide (HIP Rat): a new model for type 2 diabetes. Diabetes. 2004;53:1509–1516. [DOI] [PubMed] [Google Scholar]
- 7. Huang CJ, Haataja L, Gurlo T, et al. Induction of endoplasmic reticulum stress-induced β-cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide. Am J Physiol Endocrinol Metab. 2007;293:E1656–E1662. [DOI] [PubMed] [Google Scholar]
- 8. Gurlo T, Ryazantsev S, Huang CJ, et al. Evidence for proteotoxicity in β cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am J Pathol. 2010;176:861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin CY, Gurlo T, Kayed R, et al. Toxic human islet amyloid polypeptide (h-IAPP) oligomers are intracellular, and vaccination to induce anti-toxic oligomer antibodies does not prevent h-IAPP-induced β-cell apoptosis in h-IAPP transgenic mice. Diabetes. 2007;56:1324–13 [DOI] [PubMed] [Google Scholar]
- 10. Zraika S, Hull RL, Udayasankar J, et al. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced β cell apoptosis. Diabetologia. 2009;52:626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang CJ, Lin CY, Haataja L, et al. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated β-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes. 2007;56:2016–2027. [DOI] [PubMed] [Google Scholar]
- 12. Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rivera JF, Gurlo T, Daval M, et al. Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic β-cells: protective role of p62-positive cytoplasmic inclusions. Cell Death Differ. 2011;18:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shigihara N, Fukunaka A, Hara A, et al. Human IAPP-induced pancreatic β cell toxicity and its regulation by autophagy. J Clin Invest. 2014;124:3634–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim J, Cheon H, Jeong YT, et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient β cells induces diabetes. J Clin Invest. 2014;124:3311–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rivera JF, Costes S, Gurlo T, Glabe CG, Butler PC. Autophagy defends pancreatic β cells from human islet amyloid polypeptide-induced toxicity. J Clin Invest. 2014;124:3489–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. B'chir W, Chaveroux C, Carraro V, et al. Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell Signal. 2014;26:1385–1391. [DOI] [PubMed] [Google Scholar]
- 19. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. [DOI] [PubMed] [Google Scholar]
- 20. Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J Cell Biol. 2001;153:1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ma Y, Hendershot LM. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem. 2003;278:34864–34873. [DOI] [PubMed] [Google Scholar]
- 22. Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. [DOI] [PubMed] [Google Scholar]
- 23. Costes S, Langen R, Gurlo T, Matveyenko AV, Butler PC. β-Cell failure in type 2 diabetes: a case of asking too much of too few? Diabetes. 2013;62:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cao P, Abedini A, Raleigh DP. Aggregation of islet amyloid polypeptide: from physical chemistry to cell biology. Curr Opin Struct Biol. 2013;23:82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Westermark P, Engström U, Johnson KH, Westermark GT, Betsholtz C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci USA. 1990;87:5036–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ohagi S, Nishi M, Bell GI, Ensinck JW, Steiner DF. Sequences of islet amyloid polypeptide precursors of an Old World monkey, the pig-tailed macaque (Macaca nemestrina), and the dog (Canis familiaris). Diabetologia. 1991;34:555–558. [DOI] [PubMed] [Google Scholar]
- 27. Sakagashira S, Sanke T, Hanabusa T, et al. Missense mutation of amylin gene (S20G) in Japanese NIDDM patients. Diabetes. 1996;45:1279–1281. [DOI] [PubMed] [Google Scholar]
- 28. Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic β-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab. 2008;10(suppl 4):32–42. [DOI] [PubMed] [Google Scholar]
- 29. Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-Cell mass and turnover in humans: effects of obesity and aging. Diabetes Care. 2013;36:111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang H, Kouri G, Wollheim CB. ER stress and SREBP-1 activation are implicated in β-cell glucolipotoxicity. J Cell Sci. 2005;118:3905–3915. [DOI] [PubMed] [Google Scholar]
- 31. Chu KY, O'Reilly L, Ramm G, Biden TJ. High-fat diet increases autophagic flux in pancreatic β cells in vivo and ex vivo in mice. Diabetologia. 2015;58:2074–2078. [DOI] [PubMed] [Google Scholar]
- 32. Maiese K. Targeting molecules to medicine with mTOR, autophagy, and neurodegenerative disorders. Br J Clin Pharmacol. 2015. October 15 doi: 10.1111/bcp.12804. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 33. Subramanian SL, Hull RL, Zraika S, Aston-Mourney K, Udayasankar J, Kahn SE. cJUN N-terminal kinase (JNK) activation mediates islet amyloid-induced β cell apoptosis in cultured human islet amyloid polypeptide transgenic mouse islets. Diabetologia. 2012;55:166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gupta S, Read DE, Deepti A, et al. Perk-dependent repression of miR-106b-25 cluster is required for ER stress-induced apoptosis. Cell Death Dis. 2012;3:e333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Costes S, Huang CJ, Gurlo T, et al. β-Cell dysfunctional ERAD/ubiquitin/proteasome system in type 2 diabetes mediated by islet amyloid polypeptide-induced UCH-L1 deficiency. Diabetes. 2011;60:227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Costes S, Gurlo T, Rivera JF, Butler PC. UCHL1 deficiency exacerbates human islet amyloid polypeptide toxicity in β-cells: evidence of interplay between the ubiquitin/proteasome system and autophagy. Autophagy. 2014;10:1004–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. D'Osualdo A, Anania VG, Yu K, et al. Transcription factor ATF4 induces NLRP1 inflammasome expression during endoplasmic reticulum stress. PLoS One. 2015;10:e0130635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010;11:897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Westwell-Roper C, Dunne A, Kim ML, Verchere CB, Masters SL. Activating the NLRP3 inflammasome using the amyloidogenic peptide IAPP. Methods Mol Biol. 2013;1040:9–18. [DOI] [PubMed] [Google Scholar]
- 40. Han J, Song B, Kim J, et al. Antioxidants complement the requirement for protein chaperone function to maintain β-cell function and glucose homeostasis. Diabetes. 2015;64:2892–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]