

Abstract
Cellular DNA is constantly exposed to oxidative stress from both exogenous and endogenous sources, creating lesions that lead to aging related diseases, including cancer. 8-oxo-guanine (8OG) is one of the most common forms of oxidative DNA damage and failure to repair this lesion results in G:C to T:A transversion. Another common lesion, 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapydG), shares the same precursor as 8OG. In E. coli, both lesions are recognized and excised by the DNA glycosylase Fpg. X-ray crystallographic studies have shown that FapydG and 8OG adopt different conformations in the active site of Fpg. Our simulations suggest that the different binding modes observed for 8OG and FapydG arise directly from response to the non-conserved E77 present in the thermophilic Fpg sequences used for the crystallography experiments. In simulations with consensus S77, these lesions adopt very similar binding modes.
Cellular DNA is constantly exposed to oxidative stress from both exogenous and endogenous sources, creating lesions that lead to aging related diseases, including cancer1, 2. 8-oxo-guanine (8OG)3 is one of the most common forms of oxidative DNA damage2 and failure to repair this lesion results in G:C to TA transversion4. Another common lesion, 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapydG), shares the same precursor as 8OG (Figure 1)5. 8OG differs from guanine at the N7 and O8 positions; N7 is protonated and the C8 hydrogen atom is replaced by oxygen. FapydG contains an open imidazole ring (Figure 1). In E. coli, both lesions are recognized and excised by the DNA glycosylase Fpg6 (also known as MutM).
Figure 1.
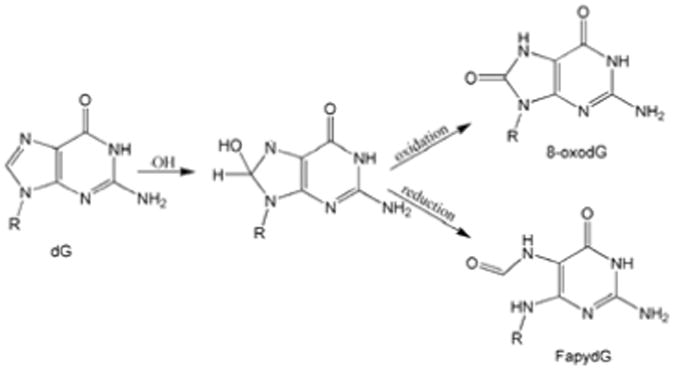
The formation of 8-oxodG and Fapy-dG by hydroxyl radicals. Note that the imidazole ring is open in FapydG.
While the catalytic mechanism by which 8OG is excised from DNA has been extensively investigated by biochemical and structural methods7-13, relatively little is known regarding the mechanism by which Fpg recognizes its cognate lesion and whether discrimination between the oxidized base and guanine occurs during one or several stages of binding. X-ray crystallographic studies have revealed the conformations of both lesions in the active site of Fpg, with specific interactions with protein residues that could contribute, at least partially, to recognition. Such studies have shown that FapydG and 8OG adopt different conformations in the active site of Fpg. In the structure of B. st. (Bacillus stearothermophilus) Fpg bound to duplex DNA containing 8OG (pdb id: 1R2Y), the extrahelical 8OG adopts the syn conformation with no direct contacts to O810. In the structure of L. lactis (Lactococcus lactis) Fpg bound to DNA containing cFapydG (a stable structural analog of FapydG, Figure S1), cFapydG assumes its anti conformation, with a highly non-planar open imidazole ring while O8 interacts with the sidechain of Tyr238 through a bridging water (pdb id: 1XC8)14. These differences raise the question of whether recognition in the active site truly differs for these lesions, or whether the observed difference relates to the use of carba-Fapy as an analog substrate, mutations used to inactivate the enzyme, or differences in the DNA sequences used for the 2 structures (Tables S1 and S2).
To address this issue, we performed simulations in solution for the two systems, both starting from the 1R2Y crystallographic coordinates of the B. st. Fpg/DNA complex 10,14 and differing only in that one system replaced 8OG with FapydG. The E3Q inactivating mutation in the crystal structure was reverted to E3 for the simulation; likewise, the cFapydG was simulated as the natural FapydG. In both cases no significant conformational changes were observed in the several nanoseconds simulations and the difference in binding modes for the two lesions were maintained15, 16. Furthermore, the simulations reproduced the bridging water seen in the cFapydG crystal structure even though the initial structure did not retain crystallographic water positions.
Since MD simulations of finite length can be kinetically trapped near the initial conformation, we supplemented these with umbrella sampling calculations to obtain the potential of mean force (PMF) for changing key aspects of lesion conformation. In the case of 8OG, we obtained the energy profile for rotation of the glycosidic bond through the full 360° (Figure 2), obtaining two well-defined energy minima. One, located at about 55°, represents the syn conformation of 8OG. The other, at ∼ -67°, corresponds to a high anti 8OG and is ∼2.7 kcal/mol higher in energy than syn, consistent with observation of the syn conformation in the 1R2Y structure with this lesion. For FapydG we calculated the free energy for rotation about the C4-C5-N7-C8 dihedral that results in the non-planar conformation and water bridge (Figure 3). Multiple minima were present, with a global minimum at -95°, in good agreement with the value of -103° in the 1XC8 structure.
Figure 2.
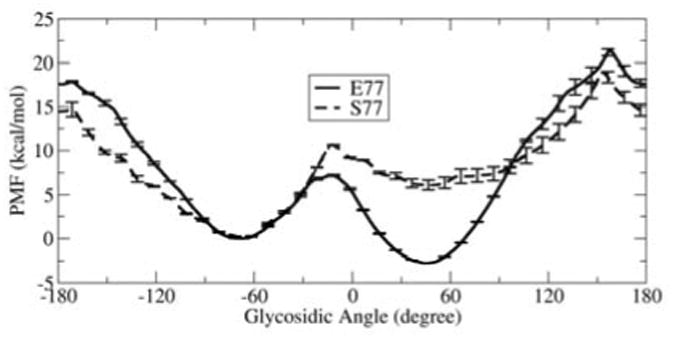
Free energy profiles for rotation around the 8OG glycosidic bond in the Fpg active site. Data is shown for Fpg with E77 (solid line) and S77 (dashed line). The free energy of the anti minimum was assigned a value of zero for both curves.
Figure 3.
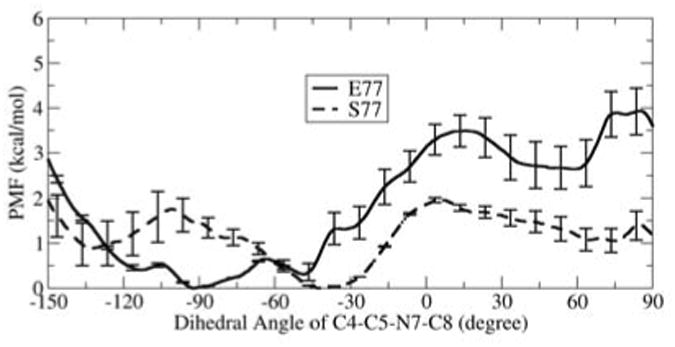
Free energy profiles for rotation around C4-C5-N7-C8. Data is shown for Fpg with E77 (solid line) and S77 (dashed line).
In both cases, we found that the free energy minimum indeed corresponded closely to the value observed in the respective crystal structures, with 8OG preferring the syn conformation and FapydG a highly non-planar conformation with a water-bridged interaction between O8 and Tyr238. Since we used identical protein and DNA sequences and coordinates (other than the lesion) for the 2 systems, our data suggests that those are not responsible for the experimentally observed difference in lesion conformation. Likewise, the close agreement between simulations and experiments suggests that the modifications used to facilitate the experiments (E3Q inactivating mutation, cFapydG vs. FapydG) do not affect the preferred lesion binding modes.
Both B. st. and L. lactis have a non-conserved Glu in the Fpg active site (residue 76 and 77 in L. lactis and B. st., respectively; we employ E77 since we simulated the B. st. sequence). In E. coli and many other Fpgs (Table S3), Ser occupies position 77. We previously reported16 that the acidic Glu at this position can affect the conformation of bound 8OG. To further investigate the role of this non-conserved residue, we repeated the umbrella sampling calculations after replacing E77 with the more frequently observed S77. In the case of 8OG, we observed that replacement of E77 with S77 results in a change of ∼9 kcal/mol in the relative energies of anti and syn; while syn was preferred by 2.7 kcal/mol for E77, anti becomes more stable by ∼6.1 kcal/mol with the consensus S77.
We also repeated our umbrella sampling calculations for the E77S variant when bound to FapydG (Figure 3). In the structure containing the non-conserved E77, the open imidazole ring is non-planar in simulations and the 1XC8 crystal structure (Figure 4A). With S77 Fpg, the preferred C4-C5-N7-C8 dihedral angle is near -40°, significantly more planar than the -95° global minimum with E77 Fpg15. Furthermore, the water bridge between Tyr238 and FapydG O8 observed in the 1XC8 crystal structure and simulations with E77 is no longer present with S77.
Figure 4.
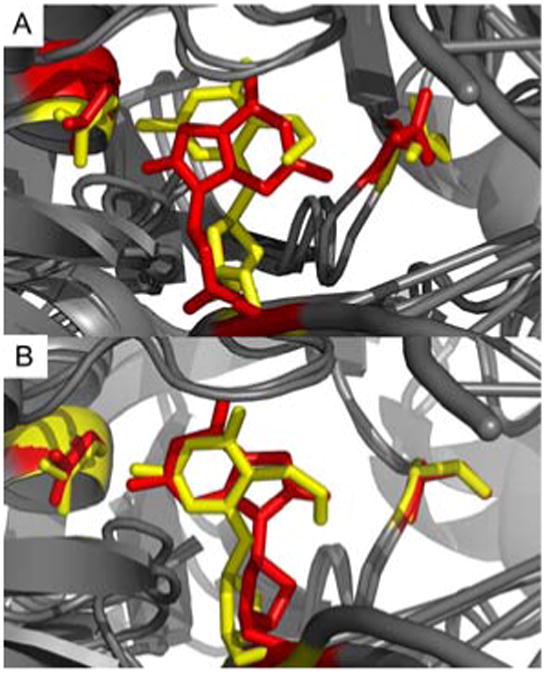
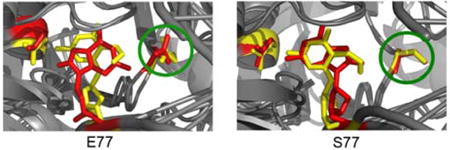
Overlap of preferred structures for Fpg bound to DNA containing 8OG and FapydG. A: E77 Fpg, B: S77 Fpg. FapydG, 8OG, E5 and E77/S77 are shown in stick representation using red for 8OG complexes and yellow for FapydG.
We can rationalize both changes in lesion conformation from S77 to E77 as a response to unfavorable electrostatics in the active site16. The non-planar FapydG conformation increases the distance between O8 and the E77 side chain (from ∼ 2.9 Å in the planar conformation to ∼ 4.8 Å in the non-planar conformation), reducing the electrostatic repulsion between these groups. Since the closed ring prevents a non-planar 8OG, it responds to the unfavorable electrostatics by adopting the syn conformation16. Future simulations will investigate whether FapydG also has a local minimum for the syn conformation, and whether these syn structures play a role in the lesion eversion pathway, and whether the E3Q inactivating mutation used for crystallography also affects the relative free energies of these minima.
Figure 4A shows the two Fpg/lesion complexes from simulations with E77 Fpg. Both are consistent with their corresponding crystal structures, with syn 8OG and non-planar FapydG. Figure 3B shows the two complexes simulated with the consensus S77. In this case, both lesions adopt the anti conformation and the FapydG is more nearly planar, lying in the plane occupied by 8OG. These results suggest that the different binding modes observed for 8OG and FapydG arise directly from response to the non-conserved E77 present in both of the thermophilic Fpg sequences used for the crystallography experiments. In simulations with consensus S77, the lesions adopt very similar binding modes. The role of this mutation remains unclear, however it has been reported that thermophiles rely more heavily on charged amino acids than mesophilic homologs17. Since Ser is more commonly observed at position 77, it is likely that the unusual conformations observed in these crystal structures may not have direct relevance for lesion recognition18.
Supplementary Material
Acknowledgments
Support by NIH GM6167803 (CS), CA047995 (CDS) and CA17395 (APG) and by the National Computational Science Alliance grant MCA02N028 (CS).
Footnotes
Supporting information available: Detailed methods, sequence conservation data, DNA sequences. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. 3rd. Oxford University Press; Oxford: 1999. pp. xxxi–936. [Google Scholar]
- 2.Bjelland S, Seeberg E. Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis. 2003;531:37–80. doi: 10.1016/j.mrfmmm.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Kasai H, Nishimura S. Nucleic Acids Research. 1984(12):2137–2145. doi: 10.1093/nar/12.4.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grollman AP, Moriya M. Trends in Genetics. 1993;9:246–249. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- 5.Pouget JP, Douki T, Richard MJ, Cadet J. Chemical Research in Toxicology. 2000;13:541–549. doi: 10.1021/tx000020e. [DOI] [PubMed] [Google Scholar]
- 6.Frieberg EC, Walker GC, Siede W, Wood RD, Shultz R, Ellenberger T. DNA Repair Mutagenesis. ASM Press; Washington, D.C.: 2006. [Google Scholar]
- 7.Zharkov DO, Shoham G, Grollman AP. DNA Repair. 2003;2:839–862. doi: 10.1016/s1568-7864(03)00084-3. [DOI] [PubMed] [Google Scholar]
- 8.Sugahara M, Mikawa T, Kumasaka T, Yamamoto M, Kato R, Fukuyama K, Inoue Y, Kuramitsu S. Embo Journal. 2000;19:3857–3869. doi: 10.1093/emboj/19.15.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fromme JC, Verdine GL. Nature Structural Biology. 2002;9:544–552. doi: 10.1038/nsb809. [DOI] [PubMed] [Google Scholar]
- 10.Fromme JC, Verdine GL. Journal of Biological Chemistry. 2003;278:51543–51548. doi: 10.1074/jbc.M307768200. [DOI] [PubMed] [Google Scholar]
- 11.Gilboa R, Zharkov DO, Golan G, Fernandes AS, Gerchman SE, Matz E, Kycia JH, Grollman AP, Shoham G. Journal of Biological Chemistry. 2002;277:19811–19816. doi: 10.1074/jbc.M202058200. [DOI] [PubMed] [Google Scholar]
- 12.Serre L, de Jesus KP, Boiteux S, Zelwer C, Castaing B. Embo Journal. 2002;21:2854–2865. doi: 10.1093/emboj/cdf304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Francis AW, Helquist SA, Kool ET, David SS. Journal of the American Chemical Society. 2003;125:16235–16242. doi: 10.1021/ja0374426. [DOI] [PubMed] [Google Scholar]
- 14.Coste F, Ober M, Carell T, Boiteux S, Zelwer C, Castaing B. Journal of Biological Chemistry. 2004;279:44074–44083. doi: 10.1074/jbc.M405928200. [DOI] [PubMed] [Google Scholar]
- 15.Song K, Hornak V, Santos CD, Grollman AP, Simmerling C. Journal of Computational Chemistry. doi: 10.1002/jcc.20625. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song K, Hornak V, Santos CD, Grollman AP, Simmerling C. Biochemistry. 2006;45:10886–10894. doi: 10.1021/bi060380m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas AS, Elcock AH. Journal of the American Chemical Society. 2004;126:2208–2214. doi: 10.1021/ja039159c. [DOI] [PubMed] [Google Scholar]
- 18.Perlow-Poehnelt RA, Zharkov DO, Grollman AP, Broyde S. Biochemistry. 2004;43:16092–105. doi: 10.1021/bi048747f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.