

Abstract
AIM: To elucidate the mechanism(s) by which S-adenosyl-L-methionine (SAM) decreases hepatitis C virus (HCV) expression.
METHODS: We examined the effects of SAM on viral expression using an HCV subgenomic replicon cell culture system. Huh7 HCV-replicon cells were treated with 1 mmol/L SAM for different times (24-72 h), then total RNA and proteins were isolated. cDNA was synthesized and real time-PCR was achieved to quantify HCV-RNA, superoxide dismutase 1 and 2 (SOD-1, SOD-2) catalase, thioredoxin 1, methionine adenosyltransferase 1A and 2A (MAT1A, MAT2A) expression, and GAPDH and RPS18 as endogenous genes. Expression of cellular and viral protein was evaluated by western-blot analysis using antibodies vs HCV-NS5A, SOD-1, SOD-2, catalase, thioredoxin-1, MAT1A, MAT2A, GAPDH and actin. Total glutathione levels were measured at different times by Ellman’s recycling method (0-24 h). Reactive oxidative species (ROS) levels were quantified by the dichlorofluorescein assay (0-48 h); Pyrrolidin dithiocarbamate (PDTC) was tested as an antioxidant control and H2O2 as a positive oxidant agent.
RESULTS: SAM exposition decreased HCV-RNA levels 50%-70% compared to non-treated controls (24-72 h). SAM induced a synergic antiviral effect with standard IFN treatment but it was independent of IFN signaling. In addition, 1 mmol/L SAM exposition did not modify viral RNA stability, but it needs cellular translation machinery in order to decrease HCV expression. Total glutathione levels increased upon SAM treatment in HCV-replicon cells. Transcriptional antioxidant enzyme expression (SOD-1, SOD-2 and thioredoxin-1) was increased at different times but interestingly, there was no significant change in ROS levels upon SAM treatment, contrary to what was detected with PDTC treatment, where an average 40% reduction was observed in exposed cells. There was a turnover from MAT1A/MAT2A, since MAT1A expression was increased (2.5 fold-times at 48 h) and MAT2A was diminished (from 24 h) upon SAM treatment at both the transcriptional and translational level.
CONCLUSION: A likely mechanism(s) by which SAM diminish HCV expression could involve modulating antioxidant enzymes, restoring biosynthesis of glutathione and switching MAT1/MAT2 turnover in HCV expressing cells.
Keywords: Hepatitis C virus, S-adenosyl-L-methionine, Superoxide dismutase 1, Superoxide dismutase 2, Replication, Hepatitis C virus-RNA, NS5A, Oxidative stress, Antioxidants, Viral proteins, Reactive oxygen species, Pyrrolidine dithiocarbamate
Core tip: S-adenosyl-L-methionine (SAM) downregulates hepatitis C virus (HCV) expression by unknown mechanisms. We evaluated the effects of SAM on viral expression using an HCV subgenomic replicon cell culture system. We observed that SAM induces a synergic antiviral effect with standard interferon (IFN) treatment independently of IFN signaling pathways; it does not modify viral RNA stability, but it needs cellular translation machinery in order to decrease HCV expression. In addition, results demonstrated that a likely mechanism(s) by which SAM decreases HCV expression could involve modulating antioxidant enzyme systems, restoring biosynthesis of glutathione and switching methionine adenosyltransferase 1 (MAT1)/MAT2 turnover in HCV expressing cells.
INTRODUCTION
Chronic hepatitis C (CHC) is a global burden[1]; around 3% (170-200 million people) of the total population is infected with hepatitis C virus (HCV)[2] and CHC eventually may lead to cirrhosis and hepatocellular carcinoma (HCC)[3]. In over 70% of cases, the immune response is unable to clear the virus, resulting in viral persistence. Current standard of care is based on the mixed scheme of pegylated interferon 2α (PEG-IFN), ribavirin (RBV)[4] and a protease or a polymerase inhibitor[5].
The mechanisms by which HCV causes cell damage are poorly elucidated and different metabolic pathways have been proposed in its pathogenesis, including the active participation of oxidative stress in chronic HCV infection. A major increases in several markers of oxidative stress are reported in HCV infection, such as inflammation, iron overload and mitochondrial injury, induced by viral proteins[6]. Oxidative stress has been described to be present in CHC patients. To date, reactive oxygen species (ROS) induction has been a subject of study for understanding HCV pathogenesis[7]. Direct measures in liver tissue show that patients with CHC have an increase in ROS[8]. Cells get protection against oxidative damage by natural antioxidant molecules, notably glutathione, and several antioxidant enzymes, such as superoxide dismutases (SOD1 and SOD2), catalase (CAT), glutathione peroxidase (GPx) and thioredoxin 1 (TRX1)[9]. Studies in subjects with chronic HCV infection have found decreased levels of glutathione[10,11] and other antioxidants and enzymatic activity[12-14]. Levent et al[15], published a clinical study in which different oxidative stress markers were measured in CHC patients, showing that plasmatic SOD1 levels had a significant reduction compared with healthy subjects. A reduction in SOD1 and GPx reflects a decrease in liver synthesis and antioxidant power in patients with HCV, and this might be considered an early marker of oxidative stress. At the same time, when markers of oxidative stress were compared in patients infected with HCV, before and after treatment, the levels showed a recovery[15]. However, more data are needed in order to understand the role of HCV expression in the disturbance of oxidative stress levels in infected cells.
Recently, it has been reported that S-adenosyl-L-methionine (SAM), decreases HCV RNA levels; however, the mechanism(s) involved are unknown. SAM is the main precursor of glutathione synthesis. Methionine adenosyltransferase 1A (MAT1A) enzyme is responsible of its biosynthesis in normal liver, but it is replaced by MAT2A in liver regeneration and hepatic carcinoma[16-18]. Based on this, we evaluated the effects of SAM exposition on HCV viral replication and protein expression and analyzed possible mechanisms involved using a hepatoma cell line expressing HCV proteins. Furthermore, the regulation of MAT1A and MAT2A enzymes involved in endogenous SAM synthesis was analyzed. Our findings suggest that SAM is able to diminish HCV expression at least in part through modulation of antioxidant enzymes, biosynthesis of glutathione and switching MAT1A/MAT2A turnover in HCV expressing cells. These findings suggest the possibility that SAM modulation could help to counteract HCV-induced damage.
MATERIALS AND METHODS
Cell culture and treatments
We used a genotype 1b HCV subgenomic Huh7-replicon cell culture system (harboring the subgenomic HCV replicon I389/NS3-3), described previously[19,20]. Huh7-replicon cells were cultured in advanced Dulbecco’s modified Eagle’s medium (GIBCO-BRL; Grand Island, NY, United States) supplemented with 2% fetal bovine serum, 1% nonessential amino acids, 200 IU of penicillin G and 200 mg of streptomycin/mL at 37 °C. For each experimental condition, cells were plated and 24 h later, were treated either with SAM (1-5 mmol/L), a combined treatment with PEG-IFNα + RBV (double therapy), or 5 μmol/L PDTC (time zero) and then incubated until 72 h. Cell viability was calculated by comparing the different conditions with untreated cells (100% viability). Upon each experimental condition, total RNA and proteins were extracted from the cell cultures and subjected to real-time reverse transcriptase polymerase chain reaction (RT-qPCR) and western blot analysis, respectively. S-(5′-Adenosyl)-L-methionine p-toluenesulfonate SAM) salt was purchased from Sigma (St. Louis MO, United States).
RNA extraction
Upon each experimental condition, total RNA was isolated from cells using Trizol reagent (Life Technologies, Carlsbad, CA, United States) as described by the manufacturer’s specification[21]. Isolated RNA was washed with 75% ethanol and diluted in 30 μL of RNase-free water.
Quantitative real-time RT-qPCR for HCV-RNA and mRNA of antioxidant enzymes
Total RNA was then subjected to RT using a high- capacity complementary DNA (cDNA) archive kit (Applied Biosystems, Foster City, CA, United States) as described by the manufacturer’s specifications. Two hundred ng of cDNA were used to perform real-time PCR for HCV-RNA, antioxidant genes, MAT1A, MAT2A, RPS18 and GAPDH mRNA quantification. HCV amplifications were done in triplicate with the following oligonucleotides: HCV-Forward (+75-93 nt), 5’-GCGTCTAGCCATGGCGTTA-3’; HCV-Reverse (+138-157 nt), 5’-GGTTCCGCAGACCACTATGG-3’; and the TaqMan probe (+94-110 nt), 5-FAM-CTGCACGACACTCATAC-NFQ-3’. For each PCR reaction, 10 μL of TaqMan PCR Master Mix, 1 μL of 20 × Assay Mix, and 9 μL of cDNA diluted in RNase free-water were used. For cellular genes analyzed, Sybr green chemistry was used for detection, and the following oligonucleotides were used: for SOD-1 gene forward 5’-AGGGCATCATCAATTTCGAG-3’, reverse 5’-TGCCTCTCTTCATCCTTTGG-3’; SOD-2, forward 5’-GGAACGGGGACACTTACAAA-3’, reverse 5’-CACCCTCGTGCGAATGATGG-3’; Thioredoxin-1 forward 5’-CTGCTTTTCAGGAAGCCTTG-3’, reverse 5’-TGTTGGCATGCATTTGACTT-3’; MAT1A forward 5’-ACATCGGAGTCTGTGGGAGA-3’, reverse 5’-CGGTCTTGCACACTGTCTCA-3’; MAT2A forward 5’-GGTAGCCTTGGAGCAACAGT-3’, reverse 5’-CTGGTCTCCAGCACCAATGT-3’; CAT forward 5’-TAAGACTGACCAGGGCATC-3’, reverse 5’-CAAACCTTGGTGAGATCGAA-3’; Ribosomal-18S (RPS18) forward 5’-TGTGGTGTTGAGGAAAGCAG-3’, reverse 5’-AAGTGACGCAGCCCTCTATG-3’. For each PCR reaction, 10 μL of SYBR-Green PCR Master Mix, 1 μL of each oligonucleotide 10 μmol/L, and 8 μL of cDNA diluted in RNase-free water were added. Conditions for temperature cycling were: setup at 50 °C for 2 min, then 95 °C for 10 min, 40 cycles of 95 °C for 15 s and 60 °C for 60 s. Data were recorded and plots were generated. GAPDH and RPS18 expression was monitored and used to normalize assays. For GAPDH-RNA expression, we used a GAPDH (20 ×) assay (Applied Biosystems, Foster City, CA, United States) as described by the manufacturer’s specifications.
Protein extraction and immunoblot assay
After each treatment, cells were lysed and proteins were extracted and quantified as we described before[22]. Fifty micrograms of protein were resolved by 12% SDS-PAGE and transferred onto PVDF membranes (Amersham Biosciences, Freiburg, Germany). Membranes were incubated with one of the following antibodies: anti-NS5A antibody (Abcam, Cambridge, MA, United States), anti-MAT1, anti-MAT2, anti-PKR, anti-STAT1 (Abcam, Cambridge, MA, United States) or anti-actin MAb (MP Biomedicals, Aurora, OH, United States). Upon washing with TBS/Tween, the membranes were exposed to horseradish-peroxidase conjugated anti-mouse IgG, anti-rabbit IgG or anti-goat IgG (Promega, Madison, WI, United States). Detection was made using Western-Blotting Chemiluminescence Luminol Reagent (Santa Cruz Biotech). We performed each experiment in triplicate, and densitometric analysis was done to quantify relative expression of each protein detected by immunoblotting. The ratios of NS5A and actin were quantified with ImageJ 1.46r 2011 software. ANOVA was used to determine statistical differences. The differences were considered significant if P < 0.05.
Total GSH and GSSG
To determine oxidative stress levels in Huh7-replicon cells upon SAM treatment, two major indicators were evaluated at different time points and concentrations: glutathione levels and ROS production. The detection of GSH and GSSG was performed using a specific kit (GSH Assay Kit; Ann Arbor, MI, United States). Huh7 HCV-replicon and parental cells were exposed with 1 mmol/L SAM for 1, 2, 6, 12 and 24 h. Cells were disrupted with freeze and unfreeze cycles. Supernatant was collected for the analysis and stored at -80 °C until the assay was done. The supernatants were low in protein (< 1 mg/mL) and were devoid of particulates so they were assayed directly without deproteinization, according to the manufacturer indications. GSSG was quantified by derivatizing GSH with 2-vinyl pyridine. The xMark™ Microplate Absorbance Spectrophotometer (Bio-Rad, Hercules, CA, United States) was used for the absorbance measure using a 415 nm filter.
ROS level quantification.
Huh7 HCV replicon cells (2 × 104 cells) were incubated with 1 mmol/L SAM at different time points (0.5, 1, 3, 12, 24 and 48 h). ROS levels were assessed by DCFH-DA assay. Fluorescence was detected at 503 nm and 530 nm, excitation and emission wavelengths respectively, by GloMax®-Multi Microplate Multimode Reader (Promega, Fitchburg, WI, United States). Hydrogen peroxide (H2O2, 1 μmol/L) was used as a positive damage control and pyrrolidine dithiocarbamate (PDTC, 5 μmol/L) as antioxidant control.
Statistical analysis
All variables were evaluated in triplicate and experimental conditions were performed at least three times. All values were scored as means ± SD. One-way analysis of variance was done to evaluate for differences in means and the t-test was performed for comparisons. In all experiments, if P < 0.05, the differences were considered significant.
RESULTS
SAM treatment downregulates HCV expression
First, cell viability experiments demonstrated that there were no cytotoxic effects of SAM at the concentrations of 2.5 mmol/L or less on HCV-replicon cells as demonstrated by MTT assay (Figure 1A). Also, as we previously reported, there were no cytotoxic effects of PDTC at the concentrations used. Based on this, we evaluated the effect of SAM on HCV-expression in HCV-replicon cells. We incubated cells with 1 mmol/L SAM at three different time points (24, 48 and 72 h), then cells were lysed and total proteins were extracted and subjected to western blot analysis. We observed that SAM dramatically inhibited HCV-NS5A protein levels compared with untreated cells (around 90% inhibition). In addition, this effect was time dependent because we observed a higher viral protein decrease in SAM-treated cells at 72 h post-treatment (Figure 1B). To determine if the effect of SAM on viral replication was due to the cytotoxic effect on treated cells, we evaluated cell viability and total cell count on SAM-treated cells. Figure 1A demonstrates that no significant difference in cell number and viability was present among unexposed (100% viability) and exposed cells with 1 to 5 mmol/L SAM concentration (98% and 85%, respectively) and this effect was time-dependent. Based on this data, we used 1 mmol/L SAM for subsequent experiments.
Figure 1.
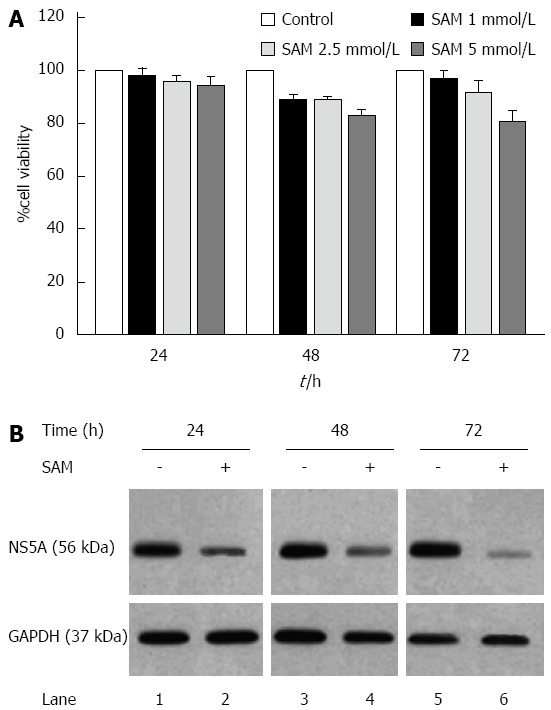
Effect of S-adenosyl-L-methionine in hepatitis C virus expression and cell viability. A: Cell viability in HCV-replicon cells exposed to SAM. Huh7 HCV replicon cells were treated with SAM at indicated concentrations for 24, 48 and 72 h. Cell viability was then determined by MTT assay and the % viability was calculated by comparing the different conditions with untreated cells (100% viability); B: HCV NS5A protein levels in Huh7 HCV replicon cells treated with SAM. Huh7 HCV replicon cells (2 × 105 cells) were incubated with SAM 1 mmol/L. Cell lysates were prepared, and equal amounts of protein extracts (30 μg) were subjected to immunoblot analysis to detect NS5A. GAPDH was used as a control. SAM: S-adenosyl-L-methionine; HCV: Hepatitis C virus.
SAM induces a synergic antiviral effect with standard IFN treatment but it is independent of IFN signaling
We further investigated whether the effect of SAM on HCV expression is similar to IFN-standard treatment (PEG-IFN + RBV). We incubated Huh7-replicon cells either with 1 mmol/L SAM, PEG-IFN + RBV or combined treatment, and then cells were harvested at three different time points. Real-time RT-PCR assay showed that viral RNA was downregulated in a time dependent fashion reaching the highest inhibition value at 72 h post-SAM treatment in HCV replicon cells (60% of inhibition at 48 h and 75% of inhibition at 72 h, dP < 0.01) (Figure 2A). As we expected PEG-IFN + RBV treatment declined HCV-RNA levels at 24, 48 and 72 h upon treatment (around 80%). Interestingly, combined treatment of SAM + PEG-IFN + RBV showed a maximal inhibition of HCV-RNA at the three times of exposition (around 92% of inhibition at 24, 48 and 72 h, dP < 0.01) (Figure 2A). Taken together, these results demonstrated that SAM possesses a synergic antiviral effect in this replicon cell culture system.
Figure 2.
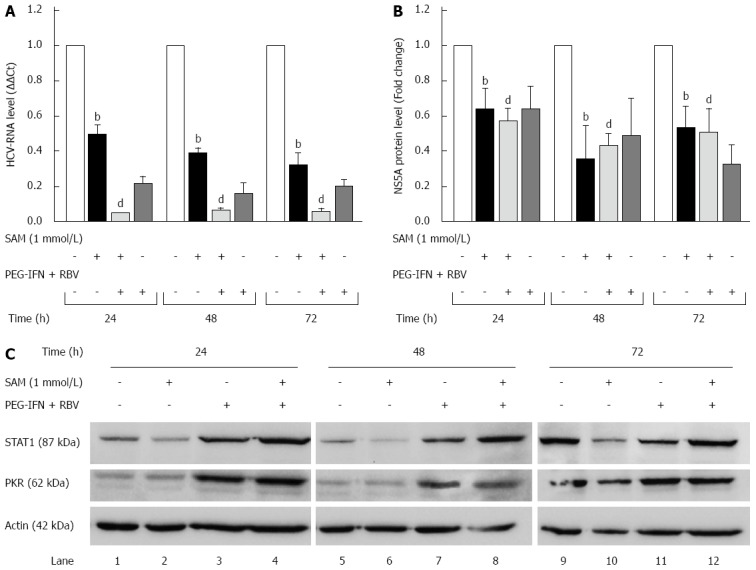
Synergic antiviral effect of S-adenosyl-L-methionine plus standard interferon treatment. A: Effect of SAM and PEG-IFN + RBV on HCV-RNA levels. Huh7 HCV replicon cells (2 × 105 cells) were incubated with 1 mmol/L SAM alone or SAM plus PEG-IFN (1000 UI/mL) and RBV (50 μmol/L). Cells were harvested at 24, 48 and 72 h and HCV-RNA levels were quantified by real-time RT-PCR and normalized with GAPDH and RPS18 using delta delta Ct method. Mean results from three independent experiments are shown. bP ≤ 0.01 comparison of SAM vs untreated cells; dP ≤ 0.01 comparison of SAM + PEG-IFN + RBV vs untreated cells; B: HCV NS5A protein levels in Huh7 HCV replicon cells treated with SAM PEG-IFN + RBV. Huh7 HCV replicon cells (2 × 105) cells were incubated with 1 mmol/L SAM alone or SAM plus PEG-IFN (1000 UI/mL) and RBV (50 μmol/L). Cell lysates were prepared, and equal amounts of protein extracts (40 μg) were subjected to immunoblot analysis to detect NS5A and actin levels. The ratios of NS5A and actin were quantified with ImageJ 1.46r 2011 software, means of three blots are represented in the graphic. bP ≤ 0.01 comparison of SAM vs untreated cells; dP ≤ 0.01 comparison of SAM + PEG-IFN + RBV vs untreated cells; C: Huh7 HCV replicon cells (1.5 × 104 cells) were incubated with 1 mmol/L SAM alone, or SAM plus PEG-IFN (1000 UI/mL) and RBV (50 μmol/L). Cells were harvested at 24, 48 and 72 h and total proteins were obtained. Cell lysates were prepared, and equal amounts of protein extracts (40 μg) were subjected to immunoblot analysis to detect STAT1, PKR and actin levels. SAM: S-adenosyl-L-methionine; HCV: Hepatitis C virus; IFN: Interferon; RBV: Ribavirin.
Next, we evaluated the effect of these treatments on HCV protein expression. Cell lysates were prepared and subjected to western blot analysis to detect NS5A protein. Results from three immunoblots were analyzed by densitometry and graphics showed similar results (Figure 2B). Basically, we found that NS5A protein levels were also decreased in a time depended manner. We also observed a synergistic effect of SAM + PEG-IFN + RBV treatment to decrease viral proteins at 24, 48 and 72 h (40%, 60% and 55% respectively) (dP < 0.01).
To investigate if this synergic antiviral effect is mediated by a higher stimulation of IFN signaling by SAM, we examined the STAT1 and PKR protein levels by western blot analysis. We found that 1 mmol/L SAM-treatment does not induce STAT1 and PKR protein expression (Figure 2C, lanes 2, 6 and 10) in Huh7-replicon cells at 24, 48 and 72 h. Contrary to this, it seems that SAM decreases STAT1 protein expression at the three times of exposure (Figure 2C, lanes 2, 6 and 10). On the other hand, immunoblot analysis of protein extracts from IFN + RBV or SAM-combined treated cells clearly showed an increase in PKR and STAT1 proteins (Figure 2C lanes 3, 4, 7, 8, 11 and 12). Together these data suggest that interferon signaling seems not stimulated by SAM exposition and therefore, inhibition of viral replication and protein synthesis by SAM may be independent of IFN-induced status.
SAM exposition does not modify viral RNA stability, but it needs cellular translation machinery in order to decrease HCV expression
The aforementioned results suggest the possibility that SAM-treatment may either diminish the transcription rate of viral RNA or decrease viral RNA or protein stability in addition of the negative effect on HCV-RNA levels observed before (Figure 2). To investigate this point we next explored whether SAM modifies HCV-RNA levels upon actinomycin D (Act D) and cycloheximide (CHX) treatment.
Huh7 HCV-replicon cells were treated either with Act D (to block cellular mRNA transcription by RNA polymerase II) or CHX (to block the cellular translation rate of proteins) and 2 h later 1 mmol/L SAM was added. Cells were incubated for sixteen hours and then harvested in order to perform HCV-RNA quantification. We found that in Act D-treated cells, the viral RNA level was stabilized and accumulated (it was not degraded), which explains that after 16 h of treatment there was an increase in the basal levels of HCV-RNA (steady-state levels). When cells were treated simultaneously with SAM and Act D, SAM treatment was not able to decrease increased basal levels of viral RNA stabilized by Act D. On the basis of this finding, we can conclude at this point that the reduction on viral RNA mediated by SAM is an independent mechanism that does not involve alterations in its half-life and reduction of HCV-RNA stability (Figure 3A).
Figure 3.
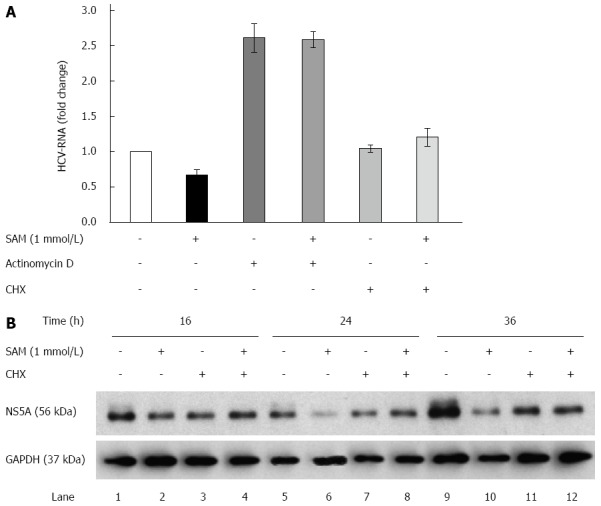
Role of actinomycin D and cycloheximide on the antiviral effect of S-adenosyl-L-methionine. A: SAM decreases replicon RNA levels without affecting its stability. Huh7 HCV replicon cells (1.5 × 105 cells) were incubated with SAM 1 mmol/L, some cells were treated with actinomycin D (4 μg/mL) or CHX (50 μg/mL) for 2 h, then SAM was added. Cells were harvested 16 h later and HCV-RNA levels were quantified by real-time RT-PCR and normalized with GAPDH using ΔΔCt method. Mean results from three experiments are shown; B: CHX partially reverts the downregulated NS5A protein expression induced by SAM. Huh7 HCV replicon cells (1.5 × 105 cells) were incubated with SAM 1 mmol/L, some cells were treated CHX (50 μg/mL) after 4 h of SAM treatment. Cells were harvested at different time points (0-36 h), then total protein was extracted and western blot for NS5A and GAPDH was performed. CHX: Cycloheximide; SAM: S-adenosyl-L-methionine; HCV: Hepatitis C virus.
Moreover, in the cells treated with CHX for the indicated times, we observed that HCV-RNA levels were not affected by blocking translational machinery but upon SAM addition, a slight increase of viral RNA was observed (Figure 3A). Interestingly, when we analyzed the NS5A protein levels upon CHX alone or combined with SAM treatment, we observed that protein levels were slightly decreased at 16, 24 and 36 h upon CHX stimulation compared with control cells (non-treated cells) (Figure 3B). However, when cells were exposed to CHX + SAM, the NS5A protein levels were greater than those observed in cells incubated only with SAM at the same times (Figure 3B). We found that CHX partially reversed the negative effect of SAM on HCV expression (Figure 3B; lane 4, 8 and 12). After a cell is infected by virus, numerous cellular mechanisms are triggered to prevent the virus from completing its viral cycle. Many of these mechanisms depend on the activation and increased synthesis of proteins with specific antiviral activity (as PKR, Mx1, OAS), so when cellular translation is inhibited, the synthesis of these proteins also decreases, then the levels of viral RNA could increase during this period due to the absence of inhibiting replication proteins. Together, these results suggest that cellular translation machinery and protein stability could play a role in the modulation of HCV protein synthesis by SAM in cultured cells.
Effect of SAM exposition on oxidative stress markers
Cells are protected against oxidative damage by natural antioxidant molecules, notably glutathione, and several antioxidant enzymes. It is reported that one of the biological activities of SAM is as antioxidative agent[23]. However, there is not enough data on whether its antioxidant properties could be implicated in the downregulation of HCV expression. We studied the modulation of oxidative stress markers in the negative regulation of HCV-RNA induced by SAM. First, we were interested in determining whether SAM could influence cellular enzymatic antioxidant systems expression, such as SOD 1 and 2 enzymes, CAT, and TRX1 in order to decrease viral expression. To evaluate this effect, mRNAs levels of these enzymes were examined by quantitative real-time RT-PCR in cells expressing HCV-proteins exposed to 1 mmol/L SAM for 0 to 72 h, and then compared to non-treated cells. We observed that transcriptional expression of SOD-1, SOD-2 and TRX1 was upregulated (0.5-fold, 2.5-fold and 2 fold, respectively compared to control) at different times of exposition (24-72 h) in the same cell system (Figure 4A).
Figure 4.
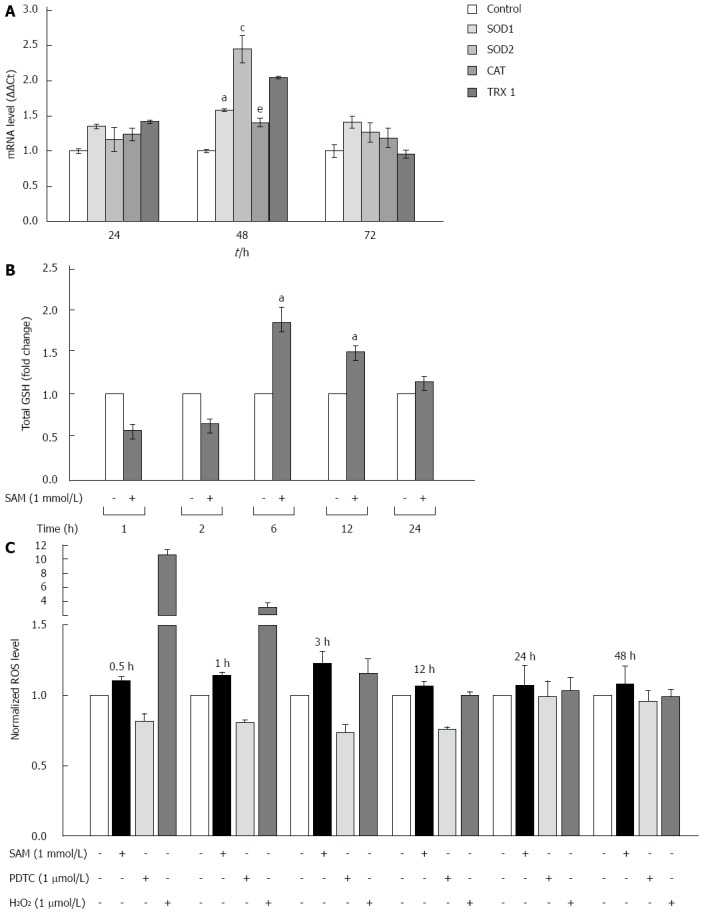
Effect of S-adenosyl-L-methionine exposition on oxidative stress markers. A: Effect of SAM on antioxidant enzyme expression. Huh7 HCV replicon cells (2 × 105 cells) were incubated with 1 mmol/L SAM and then cells were harvested at 24, 48 and 72 h. SOD1, SOD2, catalase and thioredoxin 1 mRNA levels were quantified by real-time RT-PCR and normalized with GAPDH and RPS18 using the delta delta Ct method. Mean results from three independent experiments are shown. aP ≤ 0.05 comparison of SOD1 expression in cells treated with SAM vs untreated cells; cP ≤ 0.05, SOD2 expression of SAM treated cells vs untreated; and eP ≤ 0.05, TRX1 expression of SAM treated cells vs untreated; B: Total glutathione amount in HCV-replicon cells treated with SAM. Huh7 HCV-replicon cells (2 × 105 cells) were treated with 1 mmol/L SAM. Total glutathione level was evaluated at different times by Ellman’s recycling method (0-24 h). Mean results from three independent experiments are shown; C: Effect of SAM in ROS levels. HCV replicon-containing cells (2 × 104 cells) were incubated with 1 mmol/L SAM at different time points (0.5, 1, 3, 12, 24 and 48 h). ROS levels were assessed by DCFH-DA assay. Fluorescence was measured at 503 nm and 530 nm, excitation and emission wavelengths, respectively. Hydrogen peroxide (1 μmol/L H2O2) was used as a positive damage control and pyrrolidine dithiocarbamate (5 μmol/L PDTC) as antioxidant control. SAM: S-adenosyl-L-methionine; HCV: Hepatitis C virus; SOD: Superoxide dismutase; ROS: Reactive oxidative species; PDTC: Pyrrolidin dithiocarbamate.
SAM is the main precursor of glutathione synthesis. Next, we measured the total glutathione levels in HCV-replicon cells treated with 1 mmol/L SAM from 0 to 24 h by using Ellman’s recycling method. As we expected, we found that total glutathione levels were increased in treated cells upon 6 h post-treatment (about 80%-60%) compared to control cells without SAM, and then levels began to decrease (Figure 4B).
Finally, we measured if these antioxidant mechanisms up-regulated by SAM are enough to counteract the oxidative stress induced in the cells by HCV proteins and then decrease viral expression. Huh7 replicon-cells were incubated with 1 mmol/L SAM at different time points (0 to 48 h) and then ROS levels were assessed by DCFH-DA assay. A potent antioxidant, pyrrolidine dithiocarbamate (PDTC), was used as a control to decrease ROS[24] and H2O2 was used as a positive damage control. Interestingly, we observed that there was no significant variation in ROS levels in both cell types after SAM exposition at all times evaluated, in contrast to the observed with PDTC exposition where an average of 40% reduction was observed (0.5-24 h) (Figure 4C). Our results suggest that the antiviral activity of SAM could be mediated at least partially by the induction of enzymatic antioxidant systems and total GSH turnover, triggering unknown signaling pathways but with minimal effect on ROS levels.
Turnover of MAT1A/MAT2A is induced by SAM treatment in Huh7 replicon-cells
In normal liver MAT1A is responsible for SAM biosynthesis, but upon several liver insults it is replaced by MAT2A in liver regeneration and HCC. Huh7 cells are derived from a hepatocarcinoma cell line, then in our experimental setting it was expected that MAT1A levels were downexpressed and MAT2A were major expressed. To further address the mechanisms involved in the SAM-induced downregulation of HCV expression, we evaluated the role of MAT1A and MAT2A enzymes in our cell system expressing HCV proteins. HCV replicon-cells were treated with SAM as mentioned before and then qPCR and western blot assays were performed to evaluate enzyme expression. From our results we observed two complementary phenomena (Figure 5). First, a transcriptional downregulation was observed in both enzymes at 24 h because cells were plenty of SAM, thus they do not need to synthesize it more; but later on we observed a major upregulation of MAT1A-mRNA at 48 h upon treatment (2.8 fold-times), while MAT2A-mRNA synthesis was less induced (1.4 fold) (Figure 5A). This turnover from MAT1A /MAT2A at 48 h upon SAM exposition is better represented in Figure 5B.
Figure 5.
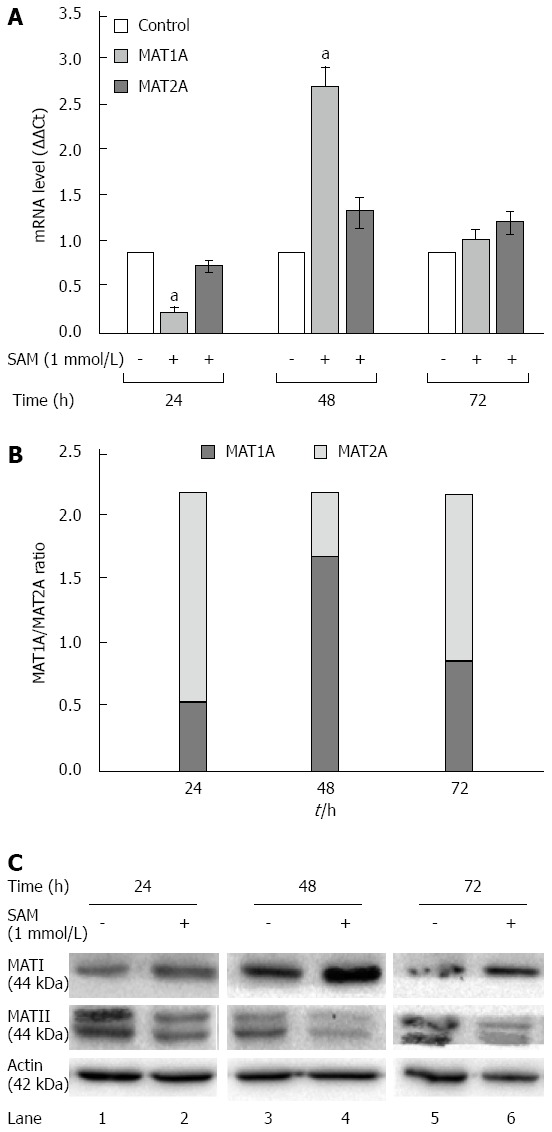
Effect of S-adenosyl-L-methionine on methionine adenosyltransferase 1/2 turnover. A: Assesment of MAT-mRNA: Huh7 HCV replicon cells (2 × 105 cells) were incubated with 1 mmol/L SAM and then cells were harvested at 24, 48 and 72 h and MAT1 and MAT2 mRNA levels were quantified by real-time RT-PCR and normalized with GAPDH and RPS18 using delta delta Ct method. Mean results from three independent experiments are shown. aP ≤ 0.05 comparison of SAM vs untreated cells; B: MAT1A/MAT2A Turnover: Measured MAT1A/MAT2A expression represented as a total; C: MAT1 and MAT2 protein expression: Huh7 HCV replicon cells (2 × 105 cells) were incubated with SAM 1 mmol/L. Cells were harvested at 24, 48 and 72 h and total proteins were obtained. Cell lysates were prepared, and equal amounts of protein extracts (40 μg) were subjected to immunoblot analysis to detect MAT1, MAT2 and actin levels. SAM: S-adenosyl-L-methionine; HCV: Hepatitis C virus; MAT: Methionine adenosyltransferase.
Second, there was a translational upregulation of MAT1A protein since 24 to 72 h upon SAM exposition. Contrary to these, MAT2A protein levels were decreased upon SAM exposition since 24 to 72 h compared with untreated cells (Figure 5C).
DISCUSSION
Recently it has been reported that SAM treatment improves early virological response in CHC patients with previous nonresponse treated with PEG-IFN + RBV[25]. In this study we demonstrated that SAM alone has the capacity to downregulate transcriptional and translational HCV expression in a hepatoma cell line expressing HCV viral proteins. In addition, there are in vivo and in vitro experiments reporting that SAM could enhance the antiviral effect of IFN, based on that SAM might be related to an early viral response. Our findings reinforce partially the data presented by Feld et al[26] showing that SAM possesses antiviral action against HCV. In Feld’s report the effect in vitro of the addition of SAM on viral clearance was not apparent until 72 h after interferon treatment, while in our experiments we found a clear decline of HCV expression since 24 h upon SAM alone or combined (SAM + IFN + RBV) exposition (Figures 1B and 2) and that could be due to the higher dose used in our experiments (1 mmol/L SAM). Contrary to results found by Feld et al[26], our results reveal that SAM possesses an early synergic antiviral effect when it is added to IFN + RBV treatment in HCV replicon cell culture system.
On the other hand, in our study the addition of SAM to PEG-IFNα and RBV does not improve the expression of ISGs genes. We observed that PKR and STAT1 protein levels were stimulated in the presence of IFNα + RBV but not upon SAM exposition, suggesting that HCV downregulation mediated by SAM is independent of IFNα stimulated pathways. These findings could have clinical application in terms of understanding the pathophysiology of the disease.
SAM is a pleiotropic molecule involved in multiple cellular reactions, participating directly in the following three types of reactions: transmethylation, trans-sulfuration and aminopropylation[27]. SAM is also involved in many other biochemical reactions in the human body, serving as a key metabolite that regulates hepatocyte growth, death and differentiation[28,29]. Based on this, we explored if alteration of RNA stability or basal levels are triggered by SAM or its metabolites. Based on our finding, we can suggest that the decrease in viral RNA mediated by SAM is due to an independent mechanism that does not involve alterations in its half-life and reduction of HCV-RNA stability (Figure 3A). However, SAM needs a cellular protein translation process to subsequently decrease the expression of viral proteins (Figure 3B).
It has been well established that SAM is the main methyl donor in methyltransferase reactions and that SAM supplementation restores hepatic glutathione deposits, which is a well-known anti-oxidant important for prevention of liver injury. To investigate the possible mechanism(s) responsible for the SAM anti-HCV observed effect we examined antioxidant enzyme RNA levels and GSH/GSSG ratio. Previously we have reported that SOD-1, CAT and GPX enzymes expression were already induced in Huh7 replicon cells while it was almost undetectable in parental cells[30]. In this study we found that SAM up-regulated SOD1, SOD2, and thioredoxin-1 expression; induced total glutathione levels, and decreased HCV-RNA levels (Figure 4A and B) but without modification of ROS levels in Huh7 HCV replicon cells. These findings are difficult to explain because we expected ROS levels could decrease upon antioxidants enzyme and GSH/GSSG were upregulated. There are some explanations for the divergent results. One explanation could be that the site of major expression of these enzymes and turnover ratio is the cytoplasm; and as we previously reported, ROS induced by HCV proteins are mainly produced in the mitochondria[30]. Another plausible explanation is that antioxidant enzymes are triggering different signaling pathways not affecting ROS amount but decreasing viral proteins. It is possible that additional immediate-early genes are also directly affected by SOD1, SOD2 or TRX and then also affects downstream events. Furthermore, oxidants/antioxidants may have different effects on other steps of the HCV life cycle. The possibility of considering SAM as a modulator of oxidative stress levels is not new. Recent evidence from Brown et al[23] have demonstrated that an overdose of acetaminophen induces oxidative stress in the whole liver and mitochondrial subcellular fractions which in turn increases cellular levels of ROS and reactive nitrogen species and induces GSH depletion as a critical early events in acetaminophen hepatotoxicity. In cells treated with SAM, they found that acetaminophen hepatotoxicity was blocked by SAM treatment[23]. These data suggest that the antioxidant status was compromised with several important components of the antioxidant defense mechanism being significantly decreased or increased in HCV-infected cells.
These findings provide new clues to further explain the mechanism(s) that indirectly suppressed HCV replication; however, the mechanism(s) still are poorly defined and additional approaches should be designed.
The molecular mechanism of HCV pathogenesis remains unclear but it is well known that both viral and host factors contribute to disease outcome. MAT is an essential cellular enzyme that catalyzes the formation of SAM. In mammals, two genes (MAT1A and MAT2A) encode homologous MAT catalytic subunits which are differentially expressed among different tissue[16]. MAT1A encodes for α1 that forms dimer (MATIII) and tetramer (MATI) that are predominantly present in liver parenchymal cells; while MAT2A encoding the α2 catalytic subunit of the MATII isoenzyme that is expressed in all other tissues. In patients with chronic liver disease there is a decrease of SAM biosynthesis exacerbating liver injury, and then exogenous supplementation might represent a useful therapy[17,31,32]. However, the effectiveness of SAM treatment in chronic liver disease has not been adequately studied.
Results found in this study suggest that a possible mechanism by which SAM decreases HCV expression could involve an additive effect between modulation of antioxidant enzyme systems, glutathione restore levels (GSH/GSSG) and switching MAT2A/MAT1A turnover in treated cells, because all these molecules participate in cell surviving pathways. In addition, Feld et al[26] found that STAT1, a transcription factor responsible for antiviral/IFN signaling activation, was methylated in cells treated with SAM, so, in that way, SAM could be available to translocate to the nucleus and do its function. Together these results provide new clues to explain the mechanism(s) involved in the antiviral effect of SAM against HCV. In addition, a fine balance of the MAT1A/MAT2A ratio induced by SAM should play a role in HCV replication and in the modulation of antioxidant enzymes. Therefore, if SAM can modify oxidative stress systems, gluthatione restoring and MAT1A/2A cell signaling pathways, these activities can provide hepatocytes with the capacity to counteract the damage induced by HCV infection.
ACKNOWLEDGMENTS
We thank Sergio Lozano-Rodriguez, MD (UANL) for his careful language assistance in the manuscript.
COMMENTS
Background
S-adenosyl-L-methionine (SAM) is a pleiotropic molecule that is involved in multiple cellular reactions, participating directly in the following three types of reactions: transmethylation, trans-sulfuration and aminopropylation. SAM is also involved in many other biochemical reactions in the human body, serving as a key metabolite that regulates hepatocyte growth, death and differentiation.
Research frontiers
It has been reported that SAM treatment improves early virological response in chronic hepatitis C subjects with previous nonresponse treated with PEG-IFN + RBV, while in vitro decreases the levels of hepatitis C virus (HCV) RNA, however the implicated mechanisms are unknown.
Innovations and breakthroughs
Results from this study suggest that SAM is able to diminish HCV expression at least in part through modulation of antioxidant enzymes, biosynthesis of glutathione and switching methionine adenosyltransferase 1A (MAT1A)/MAT2A turnover in HCV expressing cells.
Applications
These findings suggest the possibility that SAM modulation could help to counteract the HCV-induced damage.
Terminology
HCV infection is a major global health issue, with an estimated 3% of the world’s populations being chronically infected. HCV is a noncytophatic hepatotropic member of the Flaviviridae that causes acute and chronic hepatitis, and hepatocellular carcinoma. It has been shown that HCV modifies antioxidant defense mechanisms yielding a cellular oxidative imbalance and further cell death.
Peer-review
The authors investigated the antiviral effect of SAM with a HCV subgenomic replicon cell culture system. It’s a well written paper with clear hypothesis, the data is solid and well presented.
Footnotes
Supported by CONACYT-Mexico, grant register CB2010-01-155082 to Rivas-Estilla AM.
Institutional review board statement: In this study, we performed all experiments in vitro using cell lines and did not include humans or animals; therefore this requirement does not apply.
Conflict-of-interest statement: All listed authors in this manuscript do not have financial relationships to disclose.
Data sharing statement: We did not include data sharing from other researchers.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: December 28, 2015
First decision: January 28, 2016
Article in press: March 2, 2016
P- Reviewer: Cao GW, Le MD S- Editor: Gong ZM L- Editor: A E- Editor: Ma S
References
- 1.Lavanchy D. The global burden of hepatitis C. Liver Int. 2009;29 Suppl 1:74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 2.Hepatitis C--global prevalence (update) Wkly Epidemiol Rec. 1999;74:425–427. [PubMed] [Google Scholar]
- 3.Burra P. Hepatitis C. Semin Liver Dis. 2009;29:53–65. doi: 10.1055/s-0029-1192055. [DOI] [PubMed] [Google Scholar]
- 4.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Gonçales FL, Häussinger D, Diago M, Carosi G, Dhumeaux D, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 5.Barnard RJ, Howe JA, Ogert RA, Zeuzem S, Poordad F, Gordon SC, Ralston R, Tong X, Sniukiene V, Strizki J, et al. Analysis of boceprevir resistance associated amino acid variants (RAVs) in two phase 3 boceprevir clinical studies. Virology. 2013;444:329–336. doi: 10.1016/j.virol.2013.06.029. [DOI] [PubMed] [Google Scholar]
- 6.Ivanov AV, Bartosch B, Smirnova OA, Isaguliants MG, Kochetkov SN. HCV and oxidative stress in the liver. Viruses. 2013;5:439–469. doi: 10.3390/v5020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358:958–965. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- 8.Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, Choi AM. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal. 2007;9:49–89. doi: 10.1089/ars.2007.9.49. [DOI] [PubMed] [Google Scholar]
- 9.Vendemiale G, Grattagliano I, Portincasa P, Serviddio G, Palasciamo G, Altomare E. Oxidative stress in symptom-free HCV carriers: relation with ALT flare-up. Eur J Clin Invest. 2001;31:54–63. doi: 10.1046/j.1365-2362.2001.00747.x. [DOI] [PubMed] [Google Scholar]
- 10.Yuan L, Kaplowitz N. Glutathione in liver diseases and hepatotoxicity. Mol Aspects Med. 2009;30:29–41. doi: 10.1016/j.mam.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Swietek K, Juszczyk J. Reduced glutathione concentration in erythrocytes of patients with acute and chronic viral hepatitis. J Viral Hepat. 1997;4:139–141. doi: 10.1111/j.1365-2893.1997.tb00217.x. [DOI] [PubMed] [Google Scholar]
- 12.Look MP, Gerard A, Rao GS, Sudhop T, Fischer HP, Sauerbruch T, Spengler U. Interferon/antioxidant combination therapy for chronic hepatitis C--a controlled pilot trial. Antiviral Res. 1999;43:113–122. doi: 10.1016/s0166-3542(99)00041-8. [DOI] [PubMed] [Google Scholar]
- 13.Venturini D, Simão AN, Barbosa DS, Lavado EL, Narciso VE, Dichi I, Dichi JB. Increased oxidative stress, decreased total antioxidant capacity, and iron overload in untreated patients with chronic hepatitis C. Dig Dis Sci. 2010;55:1120–1127. doi: 10.1007/s10620-009-0833-1. [DOI] [PubMed] [Google Scholar]
- 14.Shimoda-Matsubayashi S, Matsumine H, Kobayashi T, Nakagawa-Hattori Y, Shimizu Y, Mizuno Y. Structural dimorphism in the mitochondrial targeting sequence in the human manganese superoxide dismutase gene. A predictive evidence for conformational change to influence mitochondrial transport and a study of allelic association in Parkinson’s disease. Biochem Biophys Res Commun. 1996;226:561–565. doi: 10.1006/bbrc.1996.1394. [DOI] [PubMed] [Google Scholar]
- 15.Levent G, Ali A, Ahmet A, Polat EC, Aytaç C, Ayşe E, Ahmet S. Oxidative stress and antioxidant defense in patients with chronic hepatitis C patients before and after pegylated interferon alfa-2b plus ribavirin therapy. J Transl Med. 2006;4:25. doi: 10.1186/1479-5876-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kotb M, Mudd SH, Mato JM, Geller AM, Kredich NM, Chou JY, Cantoni GL. Consensus nomenclature for the mammalian methionine adenosyltransferase genes and gene products. Trends Genet. 1997;13:51–52. doi: 10.1016/s0168-9525(97)01013-5. [DOI] [PubMed] [Google Scholar]
- 17.Horikawa S, Tsukada K. Molecular cloning and developmental expression of a human kidney S-adenosylmethionine synthetase. FEBS Lett. 1992;312:37–41. doi: 10.1016/0014-5793(92)81405-b. [DOI] [PubMed] [Google Scholar]
- 18.Alvarez L, Corrales F, Martín-Duce A, Mato JM. Characterization of a full-length cDNA encoding human liver S-adenosylmethionine synthetase: tissue-specific gene expression and mRNA levels in hepatopathies. Biochem J. 1993;293(Pt 2):481–486. doi: 10.1042/bj2930481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lohmann V, Körner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 20.Trujillo-Murillo K, Rincón-Sánchez AR, Martínez-Rodríguez H, Bosques-Padilla F, Ramos-Jiménez J, Barrera-Saldaña HA, Rojkind M, Rivas-Estilla AM. Acetylsalicylic acid inhibits hepatitis C virus RNA and protein expression through cyclooxygenase 2 signaling pathways. Hepatology. 2008;47:1462–1472. doi: 10.1002/hep.22215. [DOI] [PubMed] [Google Scholar]
- 21.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 22.Rivas-Estilla AM, Svitkin Y, Lopez Lastra M, Hatzoglou M, Sherker A, Koromilas AE. PKR-dependent mechanisms of gene expression from a subgenomic hepatitis C virus clone. J Virol. 2002;76:10637–10653. doi: 10.1128/JVI.76.21.10637-10653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown JM, Kuhlman C, Terneus MV, Labenski MT, Lamyaithong AB, Ball JG, Lau SS, Valentovic MA. S-adenosyl-l-methionine protection of acetaminophen mediated oxidative stress and identification of hepatic 4-hydroxynonenal protein adducts by mass spectrometry. Toxicol Appl Pharmacol. 2014;281:174–184. doi: 10.1016/j.taap.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wild AC, Mulcahy RT. Pyrrolidine dithiocarbamate up-regulates the expression of the genes encoding the catalytic and regulatory subunits of gamma-glutamylcysteine synthetase and increases intracellular glutathione levels. Biochem J. 1999;338(Pt 3):659–665. [PMC free article] [PubMed] [Google Scholar]
- 25.Filipowicz M, Bernsmeier C, Terracciano L, Duong FH, Heim MH. S-adenosyl-methionine and betaine improve early virological response in chronic hepatitis C patients with previous nonresponse. PLoS One. 2010;5:e15492. doi: 10.1371/journal.pone.0015492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feld JJ, Modi AA, El-Diwany R, Rotman Y, Thomas E, Ahlenstiel G, Titerence R, Koh C, Cherepanov V, Heller T, et al. S-adenosyl methionine improves early viral responses and interferon-stimulated gene induction in hepatitis C nonresponders. Gastroenterology. 2011;140:830–839. doi: 10.1053/j.gastro.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu SC. S-Adenosylmethionine. Int J Biochem Cell Biol. 2000;32:391–395. doi: 10.1016/s1357-2725(99)00139-9. [DOI] [PubMed] [Google Scholar]
- 28.Anstee QM, Day CP. S-adenosylmethionine (SAMe) therapy in liver disease: a review of current evidence and clinical utility. J Hepatol. 2012;57:1097–1109. doi: 10.1016/j.jhep.2012.04.041. [DOI] [PubMed] [Google Scholar]
- 29.Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology. 2007;45:1306–1312. doi: 10.1002/hep.21650. [DOI] [PubMed] [Google Scholar]
- 30.Rivas-Estilla AM, Bryan-Marrugo OL, Trujillo-Murillo K, Pérez-Ibave D, Charles-Niño C, Pedroza-Roldan C, Ríos-Ibarra C, Ramírez-Valles E, Ortiz-López R, Islas-Carbajal MC, et al. Cu/Zn superoxide dismutase (SOD1) induction is implicated in the antioxidative and antiviral activity of acetylsalicylic acid in HCV-expressing cells. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1264–G1273. doi: 10.1152/ajpgi.00237.2011. [DOI] [PubMed] [Google Scholar]
- 31.Horikawa S, Ozasa H, Ito K, Katsuyama I, Tsukada K, Sugiyama T. Expression of S-adenosylmethionine synthetase isozyme genes in regenerating rat liver after partial hepatectomy. Biochem Mol Biol Int. 1996;40:807–814. doi: 10.1080/15216549600201413. [DOI] [PubMed] [Google Scholar]
- 32.Avila MA, Berasain C, Torres L, Martín-Duce A, Corrales FJ, Yang H, Prieto J, Lu SC, Caballería J, Rodés J, et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J Hepatol. 2000;33:907–914. doi: 10.1016/s0168-8278(00)80122-1. [DOI] [PubMed] [Google Scholar]