

Abstract
Revealing detailed structural and dynamic information of membrane embedded or associated proteins is challenging due to their hydrophobic nature which makes NMR and X-ray crystallographic studies challenging or impossible. Electron paramagnetic resonance (EPR) has emerged as a powerful technique to provide essential structural and dynamic information for membrane proteins with no size limitations in membrane systems which mimic their natural lipid bilayer environment. Therefore, tremendous efforts have been devoted toward the development and application of EPR spectroscopic techniques to study the structure of biological systems such as membrane proteins and peptides.
This chapter introduces a novel approach established and developed in the Lorigan lab to investigate membrane protein and peptide local secondary structures utilizing the pulsed EPR technique electron spin echo envelope modulation (ESEEM) spectroscopy. Detailed sample preparation strategies in model membrane protein systems and the experimental setup are described. Also, the ability of this approach to identify local secondary structure of membrane proteins and peptides with unprecedented efficiency is demonstrated in model systems. Finally, applications and further developments of this ESEEM approach for probing larger size membrane proteins produced by over-expression systems are discussed.
1. INTRODUCTION
Membrane-associated and embedded proteins comprise 30% of sequenced genes (Landreh & Robinson, 2015; Moraes, Evans, Sanchez-Weatherby, Newstead, & Stewart, 2014). They are responsible for the exchange of signals and physical materials across the membranes and play vital roles in different aspects of cellular activities (Baker, 2010b; Congreve & Marshall, 2010). Mutations or misfolding of membrane proteins are associated with numerous human dysfunctions, disorders, and diseases (Cheung & Deber, 2008; Conn, Ulloa-Aguirre, Ito, & Janovick, 2007). Currently, half of all the FDA approved drugs target membrane proteins (von Heijne, 2007). Detailed structural and dynamic information for membrane proteins are vital for elucidating protein functions, intermolecular interactions, and regulations. Better structural knowledge of membrane protein systems is also crucial to our understanding of the basic mechanisms of disease pathways and benefit novel clinical therapy development (Rask-Andersen, Almén, & Schioth, 2011; Shukla, Vaitiekunas, & Cotter, 2012). Despite the abundance and importance of membrane proteins, there is very limited knowledge about structure, function, and dynamics of these complicated biological systems (Das, Park, & Opella, 2015; Kang, Lee, & Drew, 2013).
1.1 Membrane Protein Secondary Structure
The majority of membrane proteins structural motifs fall into two categories: membrane-spanning or surface-associated α-helix or α-helical bundles and β-barrels (Chothia, Levitt, & Richardson, 1977; McLuskey, Roszak, Zhu, & Isaacs, 2010; White & Wimley, 1999). It has been shown previously that the local secondary structure affects membrane proteins packing and interactions with its lipid environment (Kurochkina, 2010). Generally, better knowledge about secondary structure, particularly site-specific secondary structure, is useful toward the understanding of the function, dynamics, and interactions of membrane proteins (Kubota, Lacroix, Bezanilla, & Correa, 2014; Yu & Lorigan, 2014). Also, the formation and transition of secondary structural components are crucial for a variety of cellular processes ranging from protein folding and refolding to the amyloid deposits in various neu-rodegenerative disorders such as Alzheimer’s disease, Huntington’s disease, and Parkinson’s syndrome (Gross, 2000).
While enormous efforts have been placed on accessing membrane protein structural information over the past two decades, membrane proteins are inherently difficult to study (Baker, 2010a; Kang et al., 2013). Traditional structural biology techniques such as NMR and X-ray crystallography have revealed an increasing number of atomic level 3D structures of proteins. However, only a small portion of those are membrane proteins (Garman, 2014; Harris, 2014; Wang & Ladizhansky, 2014). In addition to these traditional biophysical techniques, the structural biology community has also benefited greatly from other structural approaches to tackle challenging biological systems (Bahar, Lezon, Bakan, & Shrivastava, 2010; Cowieson, Kobe, & Martin, 2008; Feng, Pan, & Zhang, 2011). Biophysical and biochemical techniques such as mass spectrometry, IR, Raman spectroscopy, fluorescence resonance energy transfer spectroscopy, chemical cross-linking, and computational modeling have all been utilized successfully to provide valuable information about structure, dynamics, and interactions of membrane proteins (Chattopadhyay & Haldar, 2014; King et al., 2008; Ladokhin, 2014; Tang & Clore, 2006).
There are several established biophysical techniques that are used to study the secondary structure of membrane proteins. Circular dichroism (CD) is an excellent tool for rapid determination of the secondary structure and folding properties of proteins (Greenfield, 2006; Whitmore & Wallace, 2008). CD spectroscopy detects the differential absorption of left- and right-handed circular polarized light that can be used to determine the global secondary structure of a protein. CD has the advantage that it can measure samples containing 20 μg or less of proteins in physiological buffers in a short period of time. However, it only yields the overall secondary structure of the entire complex and does not provide the specific secondary structure of different segments of the protein. Solid-state NMR spectroscopy can be utilized to determine local secondary structures based on the backbone chemical shift assignment and dipolar couplings (Fritzsching, Yang, Schmidt-Rohr, & Hong, 2013). However, it requires milligram scales of isotope-labeled protein or peptide samples and days to weeks of data-acquisition time while still suffering from low sensitivity. Other methods such as FT-Raman spectroscopy, ATR FT-IR, and continuous-wave EPR dipolar wave analysis also can provide secondary structure information (Carbonaro & Nucara, 2010; Roach, Simpson, & JiJi, 2012). Data obtained by these methods are sometimes ambiguous and often require extensive data analysis.
Electron paramagnetic resonance (EPR) is a powerful and sensitive biophysical technique for studying chemical and biological systems with unpaired electron spins. It was first observed over a half century ago and has been particularly useful in characterizing organic radicals, metal complexes, and biomolecules with paramagnetic centers (Brückner, 2010; Goldfarb, 2006). However, with the development of site-directed spin-labeling (SDSL) techniques to target biological systems, there has been a significant increase in the application of EPR spectroscopy to study protein structure and dynamics (Alexander, Bortolus, Al-Mestarihi, Mchaourab, & Meiler, 2008; Altenbach, Flitsch, Khorana, & Hubbell, 1989; Fanucci & Cafiso, 2006; Hirst, Alexander, McHaourab, & Meiler, 2011; Hubbell, Gross, Langen, & Lietzow, 1998; Hubbell, López, Altenbach, & Yang, 2013; Sahu, McCarrick, & Lorigan, 2013; Sahu, McCarrick, Troxel, et al., 2013). SDSL EPR is sensitive to dynamics on the picoseconds to microsecond timescales, which cover a wide range of motions in biological and molecular systems (Barnes, Liang, Mchaourab, Freed, & Hubbell, 1999; Casey et al., 2014; Nesmelov, 2014). Also, the topology of a membrane protein can be explored with respect to the lipid bilayer with SDSL coupled with CW-EPR spectroscopy. Adding relaxation enhancers such as chelated nickel and oxygen can alter the electron spin–lattice and spin–spin relaxation rates and distinguish between solvent-exposed regions and residues buried in the membrane (Altenbach, Greenhalgh, Khorana, & Hubbell, 1994; Huang et al., 2015; van Wonderen et al., 2014). Utilizing different experimental approaches, EPR spectroscopy can also access distance information between different spin labels from several angstroms up to 10 nm (Baber, Louis, & Clore, 2015; Edwards et al., 2013; Sahu, Hustedt, et al., 2014; Sahu, McCarrick, Troxel, et al., 2013). Pulsed EPR techniques such as double electron–electron resonance provide important structural information on membrane proteins (Baber et al., 2015; Sahu, Kroncke, et al., 2014).
This work describes a novel approach established and developed in our lab to investigate membrane protein and peptide secondary structure utilizing the pulsed EPR technique electron spin echo envelope modulation (ESEEM). ESEEM spectroscopy coupled with SDSL can provide valuable local secondary structural information (α-helix and β-strand) of membrane proteins and peptides in a lipid bilayer with short data-acquisition times and straightforward data analysis.
1.2 ESEEM Spectroscopy
ESEEM spectroscopy has been widely utilized to study the electronic environment of paramagnetic metal centers and metalloproteins and provide valuable information on metalloenzyme mechanisms, metal binding, substrate binding, and the ligand coordination sphere (Cieslak, Focia, & Gross, 2010; Deligiannakis, Boussac, & Rutherford, 1995; Hernández-Guzmán et al., 2013; Warncke, 2005). SDSL and ESEEM spectroscopy have been used to study the supermolecular structure of biological systems, the penetration depth of water into the membrane and in KcsA K+ channels, localization of proteins or lipids in membranes, and protein folding (Carmieli et al., 2006; Cieslak et al., 2010) (Bartucci, Guzzi, Esmann, & Marsh, 2014; Dzuba & Raap, 2013; Matalon, Faingold, Eisenstein, Shai, & Goldfarb, 2013).
In this chapter, a novel ESEEM approach developed in the Lorigan lab is discussed. By using SDSL coupled with ESEEM spectroscopy, the secondary structure of membrane peptides and proteins can be determined by detecting 2H modulation between a 2H-labeled amino acid and a nearby spin-labeled cysteine residue (Liu et al., 2012; Mayo et al., 2011; Zhou et al., 2012). A cysteine-mutated nitroxide spin label (MTSL) is positioned strategically at 1, 2, 3, and 4 residues away from an amino acid (i) with a deuterated side chain (denoted as i+1 to i+4). The characteristic periodicity of the α-helix or β-strand structure has unique patterns in the individual ESEEM spectra. A typical α-helical periodicity consists of 3.6 amino acids per turn. The distance from the beginning to the end of the turn in the α-helix is 5.4 Å. Taking this into account every three or four residues in an α-helical segment should have a minimum distance between the side chain residues, assuming that the helix is straight. The second predominant secondary structure, β-strand, is an extended stretch of polypeptides chain with every other two amino acid side chains approximately 6 Å apart. ESEEM spectroscopy can detect dipolar interactions between a nitroxide spin label and a 2H nucleus out to a maximum of approximately 8 Å. For an α-helical structure, one set of ESEEM data from different samples should show a pattern in which 2H modulation can be detected for i+3 and i+4 samples, but not for the i+2 samples because they are outside the 8 Å detection limit. However, for peptides or protein segments adopting an extended structure such as a β-strand, the ESEEM spectra should show exactly the opposite results. In this case, 2H modulation would be detected for the i +2 sample, but not for the i+3 and i+4 samples.
This novel pulsed EPR ESEEM secondary structure approach is advantageous because it has no protein or protein-complex size limitations and is very sensitive when compared to NMR spectroscopy. Moreover, this approach can provide direct local secondary structural information qualitatively without complicated data analysis. Generally, each set of ESEEM experiments requires small amounts and concentrations of labeled protein sample (~25 μL and ~100 μM). Also, the ESEEM data acquisition is fairly fast when compared to NMR and only takes about an hour. With selective isotopic labeling, this approach can be adopted in an overexpression system and, therefore, can be applied to larger proteins and protein complexes. In Section 2, detailed experimental procedures for sample preparation, spectrometer setup, and data analysis are described.
2. INTEGRATION OF MEMBRANE PEPTIDES INTO LIPID BILAYER
In order to demonstrate the ability of this novel pulsed EPR approach for determining secondary structural components, well-characterized model peptides with known α-helical or β-sheet secondary structures were chosen to prove the concept (Opella et al., 1999; Zerella, Chen, Evans, Raine, & Williams, 2000). The nicotinic acetylcholine receptor M2δ segment, which is a well-studied 23 amino acid residue transmembrane peptide was selected to represent a model α-helix in a lipid bilayer (PDB entry: 1EQ8). The 17-amino acid residue of Ubiquitin was chosen to represent a model β-strand (PDB entry: 1E0Q). In this section, sample preparation with model peptides in a membrane mimetic system is described.
Solid-phase peptide synthesis (SPPS) : SPPS is the standard method for synthesizing peptides and small proteins in the lab. It allows for the synthesis of natural peptides and small proteins which are difficult to express, as well as the incorporation of unnatural or isotope-labeled amino acids and the synthesis of D-amino acid proteins. Also, unlike ribosomal protein synthesis, SPPS can proceed in both C-terminal and N-terminal fashions. SPPS has the ability to synthesize peptides with up to 70 amino acids and can potentially make proteins and peptides with 150+ amino acids with the help of chemical ligation (Chandrudu, Simerska, & Toth, 2013; Raibaut, El Mahdi, & Melnyk, 2015). The two most commonly used forms of SPPS are Fmoc and Boc, which have different protecting groups used on the C-terminal or N-terminal residues of each amino acid block. Table 1 shows the wild-type and ESEEM experimental construct sequences for the two model peptides AChR M2δ (α-helix) and Ubiquitin 17 (β-sheet). Four different peptides were designed by positioning the 2H-labeled amino acid at position i and the cysteine (X) at four successive positions (i+1 to i+4). Both the M2δ and Ubiquitin peptide constructs were synthesized on a CEM microwave-assisted peptide synthesizer using Fmoc protection chemistry. Low loading (0.2 mmol/g) and high swelling rate solid supports were chosen to increase the yield of these relatively hydrophobic peptide sequences. A solution of 2H-labeled amino acid such as d10 Leu or d8 Val (Isotec) dissolved in N-methyl-2-pyrrolidone was used as the 2H probe and incorporated into peptides at a designated position (i). The peptides were cleaved, deprotected, and isolated from their resin support in an acidic environment. The cleavage and deprotection cocktail was designed and optimized according to the sequence, length, and protection groups used on the amino acid side chain (Góngora-Benítez, Tulla-Puche, & Albericio, 2013; Mäde, Els-Heindl, & Beck-Sickinger, 2014). Most cocktails are TFA-based and the amino acid composition of the peptide dictates the final concentration of TFA, type of scavengers used, and reaction times.
Peptide purification and validation: After the peptides were cleaved from their solid support, the cleavage cocktail was evaporated by N2 gas flow or via rotary evaporation until the peptide precipitation started to appear. Methyl tert-butyl ether was added to assist the precipitation of peptide and wash off residual TFA. The crude peptide was dried under a vacuum overnight followed by purification via reverse-phase HPLC with a C4 preparation column using a linear gradient of 5–95% solvent B (90% ace-tonitrile). This gradient is usually sufficient to purify typical peptides from SPPS. However, high impurity and multiple major truncations from a bad synthesis can increase the difficulty of the separation. In those cases, a mobile-phase gradient and component can be adjusted to achieve a better separation. The HPLC fraction of the target peptide was collected and lyophilized to a solid powder for further steps and storage. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectroscopy was used to confirm the molecular weight and purity of the peptides after HPLC purification.
Attachment of spin label: Purified peptides were labeled with S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate (MTSL) at 10× molar excess in DMSO for 12 h. Reversed-phase HPLC was used to remove the excess MTSL (Gorka et al., 2012; Zhao et al., 2012). For small proteins and peptides, chromatography is more efficient when compared to dialysis for the removal of excess MTSL. HPLC fractions of the targeted peptides were lyophilized and stored in −20 °C for further processing. MALDI-TOF was utilized to confirm the molecular weight, purity, and labeling efficiency of the target peptides qualitatively. A series of tempo solutions with standardized concentrations were prepared and a spin concentration calibration curve was generated. The concentrations of the spin-labeled peptide samples were directly calculated from the CW-EPR spectra. Spin-labeling efficiency was determined by comparing the spin concentration obtained from CW-EPR data with the protein or peptide concentration.
Integration of synthetic peptide into lipid bilayers: MTSL-labeled M2δ peptides were integrated into DMPC/DHPC (3.5/1) bicelles at a 1:1000 peptide to lipid molar ratio. Both spin-labeled peptide and lipids were dissolved in chloroform in a pear-shape flask. N2 gas was applied to evaporate the solvent, while the flask was slowly rotated to form a uniform film of lipid and peptide mixture along the wall of the flask. This lipid/ peptide film was dried under vacuum overnight to remove any remaining solvent. 200 μL HEPES buffer at pH 7.4 was added to rehydrate the lipid/peptide film followed by a combination of vortex, freeze–thaw, and sonication steps until the bicelle sample turned clear. For these experiments, bicelles were used as a membrane mimic system and yielded high-quality ESEEM data. The final spin label concentrations of the peptides were −100 μM. Comparable ESEEM data could be obtained with micelles, vesicles, and lipodisq nanoparticles (data not published). The Ubiquitin peptide was dissolved in an aqueous buffer using a previously published protocol (Zerella et al., 2000). CW-EPR spectra of bicelle samples were taken to verify the incorporation of peptide and successful removal of free spin label.
Peptide secondary structure validation: M2δ bicelles and aqueous Ubiquitin peptide samples with concentrations ranging from 0.01 to 0.1 mg/mL were analyzed using a Jasco J-810 spectrometer over a wavelength range of 190–250 nm. The CD spectrum of i+3 Ubiquitin construct in Fig. 1 shows a large and broad negative band centered at 218 nm indicating a β-sheet secondary structure. The M2δ CD spectrum indicates α-helical secondary structure through the two negative bands at 222 nm and 208 nm. After baseline subtraction, CD data were analyzed with DichroWeb and showed pure α-helical content for M2δ and β-sheet for the Ubiquitin peptide after spin label incorporation.
Table 1.
Peptide Constructs for AChR M2δ and Ubiquitin Peptides
| AChR M2δ | Ubiquitin Peptide | |
|---|---|---|
| Wild type | NH2-EKMSTAISVLLAQAVFLLLTSQR-COOH | NH2-MQIFVKTLDGKTITLEV-COOH |
| i+1 | NH2-EKMSTAISVXiAQAVFLLLTSQR-COOH | NH2-MQIFVKXiDGKTITLEV-COOH |
| i+2 | NH2-EKMSTAISXLiAQAVFLLLTSQR-COOH | NH2-MQIFVXTiDGKTITLEV-COOH |
| i+3 | NH2-EKMSTAIXVLiAQAVFLLLTSQR-COOH | NH2-MQIFXKTiDGKTITLEV-COOH |
| i+4 | NH2-EKMSTAXSVLiAQAVFLLLTSQR-COOH | NH2-MQIXVKTiDGKTITLEV-COOH |
Wild-type and experimental constructs of AChR M2δ (α-helix) and Ubiquitin peptide (β-sheet) were listed in this table. i stands for positions where 2H-labeled d10 Leu was placed. X makes positions where amino acid is replaced by Cys for MTSL incorporation.
Figure 1.

CD spectra of i+ 3 AChR M2δ and Ubiquitin peptide constructs in DMPC/DHPC bicelles with a lipid protein ratio of 200:1 at 298 K and pH 7. AChR M2δ spectrum (red line (dark gray in the print version)) shows two negative bands at 208 nm and 222 nm which indicate α-helical structure. The Ubiquitin peptide spectrum (black line) shows a large and broad negative band centered at 218 nm indicating a β-sheet secondary structure.
The strategies described above can be applied to other synthetic proteins and peptides with minor modifications. Utilizing site-directed mutagenesis and selective isotope labeling, this ESEEM approach has been demonstrated that it can be adapted to overexpressed and reconstituted proteins and peptides (data not shown).
3. ESEEM SPECTROSCOPY ON MODEL PEPTIDES IN A LIPID BILAYER
3.1 ESEEM Principles
Pulsed EPR techniques such as ESEEM involve the application of a series of short time-dependent microwave pulses at the appropriate frequency to an electron spin system in a constant external magnetic field. The corresponding magnetization of the electron spins can be measured in the form of an emitted microwave signal, which provides information about the local environment of the electron spin system. The standard two-pulse spin echo or “Hahn echo” sequence is shown in Fig. 2A. In the two-pulse experiment, two microwave pulses separated by a time interval τ are applied to the electron spin system at a microwave frequency and magnetic field, which satisfies the magnetic resonance condition of the electron spin system. The first π/2 pulse rotates the electron spin magnetization by 90°, thus creating a short free induction signal. However, this signal rapidly decays due to the rapid spin dephasing resulting from inherent inhomogeneous line broadening. Thus, instead of directly observing the free induction decay as for NMR, a second π pulse is applied, which flips the magnetization of the spin system 180° in such a way that the spins refocus at the moment τ after the second pulse and create an electron spin echo that can be easily measured. Generally, in a two-pulse EPR experiment, the intensity of the echo is measured after the instrumental dead time as a function of increasing τ which generates the original time domain signal (Schweiger & Jeschke, 2001). The intensity of this echo decreases with increasing τ due to transverse spin relaxation. Nuclear spins that are coupled to the electron spin modulate the amplitude of the electron spin echo periodically as a function of τ. This phenomenon is called ESEEM. This periodic modulation results from the nuclear spin precession of nuclei in close vicinity of the unpaired electron spin. Basically, ESEEM spectroscopy indirectly observes NMR transitions through an electron spin coupled to a nearby NMR active nucleus (Deligiannakis & Rutherford, 2001; Hoffman, 2003).
Figure 2.
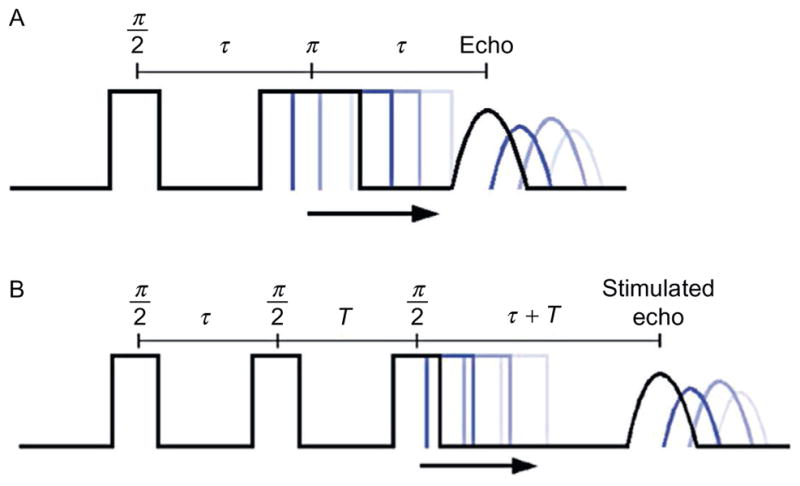
Two-pulse and three-pulse ESEEM pulse sequence.
In the three-pulse ESEEM sequence (Fig. 2B), three π/2 pulses are used to monitor the modulated echo as function of time. Instead of refocusing the spin, it creates a polarization grading with the first two pulses. The third π/2 pulse flips the stored polarization back to the transverse plane for measurement. All three pulses together generated a stimulated echo at the time of τ after the third pulse. Since only the longitudinal magnetization created by the second pulse contributes to the formation of the stimulated echo signal, the signal decay depends on the corresponding spin–lattice relaxation time T1. By utilizing the three-pulse technique, ESEEM data can be obtained much further out in time and dramatically increases the resolution of the frequency domain data following Fourier transformation (FT) (Kevan & Schwartz, 1979).
For three-pulse ESEEM, the intensity of the stimulated echo is measured as a function of the evolution time T. However, the value of the fixed τ leads to artifacts within the ESEEM data, which are known as “τ-dependent blind spots” (Stoll, Calle, Mitrikas, & Schweiger, 2005). A nuclear spin with a Larmor frequency ωn is suppressed at the appropriate τ value (τ = 2πn/ωn). Therefore, for ESEEM experiments with multiple NMR active nuclei involved, different τ values should be examined to optimize the spectra. Proton modulation can be suppressed and deuterium modulation can be maximized with this approach.
Another shortcoming of the three-pulse ESEEM is that it generates more than the stimulated echo: it also generates one refocused echo and three primary echoes. For certain values of T and τ, those echoes can overlap with the stimulated echo and cause spectrum distortion. However, four-step phase cycling can be utilized to remove those unwanted echoes and prevent distortion of the spectrum.
The modulation depths of both two- and three-pulse ESEEM experiments have been discussed thoroughly in previous publications and reviews (Schweiger & Jeschke, 2001). It is affected by the static field B0, g tensor of the electron spin and gyromagnetic ratio of the nuclei. For the same type of nucleus in a constant magnetic field, the modulation depth is proportional to r−6, where r is the distance between the NMR active nucleus and the center of the unpaired electron spin density. Thus, it suffers from a rapid decay as the distance between the electron spin and nuclei spins increases. Typically, ESEEM can only detect dipolar interactions between a NMR active nucleus and a spin label through a dipolar coupling within a short of distance. For deuterium coupled to a nitroxide spin label, the detection limitation is about 8 Å. In the case of several nuclei, the ESEEM signal can be calculated as a product of the expression for each individual nuclear spin. In the next session, the analysis and interpretation of our ESEEM data will be discussed in detail.
3.2 ESEEM Experimental Setup and Data Analysis
In order to determine local secondary structural information from this ESEEM approach, a set of ESEEM spectra from different spin-labeled position need to be compared. Therefore, it requires consistency with sample preparation, experimental setup, data collection, and analysis.
Experimental setup: ESEEM data were collected at X-band on a Bruker ELEXSYS E580 spectrometer equipped with an MS3 split ring resonator. The three-pulse ESEEM sequence was chosen to maximize the low-frequency modulation of 2H. The measurements were conducted at 80 K at a microwave frequency of 9.269 GHz with 16 ns π/2 pulse widths. A starting T of 368 ns and 512 points in 12 ns increments were used for all samples. A τ value of 200 ns was chosen to suppress proton modulation. 30% glycerol was added to rehydrolyzed bicelle sample as a cryoprotectant to prevent water crystallization during the freezing process. 40 μL of sample with a final concentration of 100 μmol was pipetted into a 3 mm ESEEM tube and fast frozen in liquid N2. 30 scans with four-step phase cycling were used to obtain the required signal-to-noise ratio.
Data analysis and interpretation: The original ESEEM time domain data consists of two components: the unmodulated decay and the modulation of nuclei at the corresponding Larmor frequency. The time domain data were fit to a two-component exponential decay. The maximum value of the exponential fit was scaled to 1 and the same factor was applied to the time domain data. The exponential fit was then subtracted from the time domain data and yielded a scaled ESEEM spectrum with modulation about 0. A cross-term averaged FT was performed to the resulting spectrum to generate the corresponding frequency domain with minimized dead time artifacts (Tarabek, Bonifacić, & Beckert, 2006). The analysis can be performed use Matlab and our ESEEM data-processing package at http://epr.muohio.edu/user-resources/oaeprl-plotting-package. Any modulation presented from every weakly coupled nucleus will show a peak at its corresponding Larmor frequency. The maximum intensity of the deuterium peak at 2.3 MHz was measured in an arbitrary unit and peak intensity was recorded for further analysis.
4. DEVELOPMENT OF ESEEM SECONDARY STRUCTURE DETERMINATION APPROACH
Here, we demonstrate the ability and efficiency of this ESEEM approach to probe the secondary structure of membrane proteins and peptides in a model lipid bilayer system.
4.1 Determine α-Helical Secondary Structure of Membrane Peptides
In order to map out the α-helical content of a segment of the M2δ peptide, 2H-labeled d8 Val was positioned at Val9 (i) and the MTSL spin label (X) was strategically placed at four successive positions (i+1 to i+4). Figure 3 shows the normalized ESEEM spectra for 2H-labeled d8 Val9 AChR M2δ (EKMSTAISiLLXQAVFLLLTSQR) including both the time domain and frequency domain data (Fig. 3, red (light gray in the print version) and blue (dark gray in the print version)). A control sample was prepared such that Val9 was not 2H-labeled (Fig. 3, black) at position i+3 (EKMSTAISVLLXQAVFLLLTSQR). The spectra compared the three-pulse ESEEM data of the M2δ peptide with 2H-labeled d8 Val and a SL three residues away (i+3) at two different τ values with a non-deuterated sample as a control. Figure 3A reveals an obvious low-frequency 2H modulation in the time domain data for both τ values in the 2H-labeled Val sample when compared to the control sample, which had normal protonated Val instead of 2H-labeled Val. The corresponding Fourier transform frequency domain data in Fig. 3B revealed a large well-resolved peak centered at the 2H-Larmor frequency of 2.3 MHz originating from weakly coupled 2H nuclei. 2H modulation is not detected in the control sample at any τ values. Also, the ESEEM data showed that both τ values provided high-quality time domain and FT ESEEM data. However, it was obvious that when 2H modulation was optimized, the proton modulation was also effectively suppressed with a τ equal to 200 ns. The optimal τ values can vary depending on the field and frequency under which the experiment was performed, as well as the type of isotopic label used in the experiment (Kevan & Schwartz, 1979).
Figure 3.
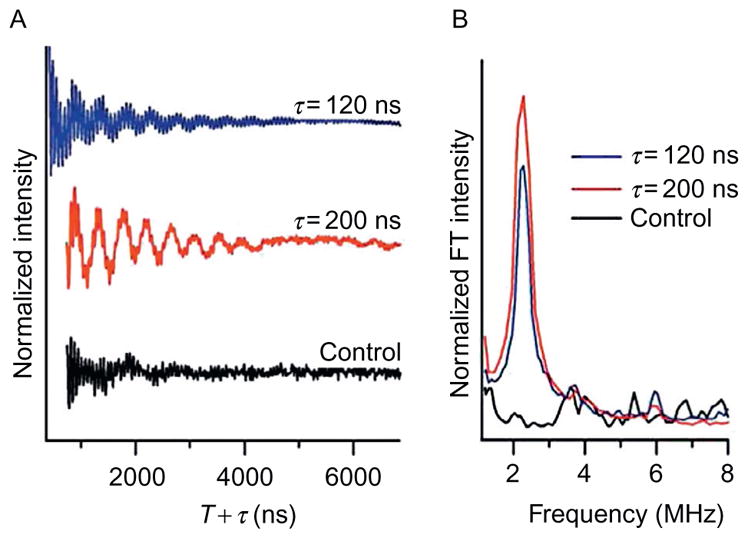
Three-pulse ESEEM spectra of AChR M2δ with 2H-labeled d8 Val9 in DMPC/DHPC bicelles. (A) Time-domain data for i+ 3 AChR M2δ with 2H-labeled d8 Val9 at τ = 120 ns (blue (dark gray in the print version)), τ= 200 ns (red (light gray in the print version)) and non-deuterated control sample at τ = 200 ns (black). (B) Corresponding frequency domain data for i + 3 AChR M2δ with 2H-labeled d8 Val9 at τ = 120 ns (blue (dark gray in the print version)), τ = 200 ns (red (light gray in the print version)) and nondeuterated control sample at τ= 200 ns (black).
Figure 4 shows the three-pulse ESEEM data for 2H-labeled d8 Val9 M2δ peptides at all four successive positions (i+1 to i+4) in bicelles. 2H modulation was observed in the time domain spectrum and a corresponding peak centered at the 2H-Larmor frequency in the frequency domain data for the i+3 and i+4 2H-labled d8 Val9 M2δ samples. However, no 2H modulation was detected for the i+1 or i+2 positions. These ESEEM spectral pattern of large 2H peaks observed at i+3 and i+4 positions were consistent with the structural characteristic that there are 3.6 amino acids per turn for an α-helix. The 2H-labeled Val side chain and the spin labels are located on the same side of the helix when they are three or four amino acid residues away. These data clearly showed the utility of this technique for determining the α-helical secondary structure of membrane peptides and proteins.
Figure 4.

Three-pulse ESEEM experimental data of AChR M2δ with 2H-labeled d8 Val9 in DMPC/DHPC bicelles at τ = 200 ns for the i+ 1 to i +4 in (A) time domain and (B) frequency domain.
This approach was further explored and expanded to utilize 2H-labeled d10 Leu as a 2H-labeled probe for this novel ESEEM approach. The three-pulse ESEEM data for the 2H-labeled d10 Leu11 (i+1 through i+4) M2δ peptides are shown in Fig. 5. For 2H-labeled d10 Leu11 M2δ peptides, the 2H modulation was observed in the time domain for i+1, i+3, and i+4 samples. Also, a 2H peak was clearly observed at the 2H-Larmor frequency in the frequency domain data. However, there was no 2H modulation for the 2H-labeled d10 Leu11 i+2 M2δ sample. Despite the longer side chain of Leu when compared to Val, ESEEM spectra still revealed a similar pattern for this α-helix. Moreover, Fig. 6A shows the comparison of the ESEEM frequency domain data between 2H-labeled d10 Leu10 and 2H-labeled d8 Val9 peptides at the i+4 position. This frequency domain datarevealed a dramatic signal enhancement when 2H-labeled d10 Leu was usedinstead of 2H-labeled d8 Val. Distances and conformations provided by molecular dynamic simulations also supported this result (Liu et al., 2012). Figure 6B indicates that the additional C–C bonds in the Leu side chain brought the deuterons closer to the N–O nitroxide bond of the spin label when compared to the Val side chain. Therefore, the MTSL had a higher probability of being able to detect the 2H nuclei at a closer distance resulting in a significant increase in 2H peak intensity for Leu at this position, when compared to Val.
Figure 5.

Three-pulse ESEEM experimental data of AChR M2δ with 2H-labeled d10 Leu11 in DMPC/DHPC bicelles at τ = 200 ns for the i+ 1 to i+ 4 in (A) time domain and (B) frequency domain.
Figure 6.

Comparison between 2H-labeled d8 Val9 and 2H-labeled d10Leu10 M2δ bicelle samples. (A) Normalized FT frequency domain modulation data for 2H-labeled d10Leu10i+ 4 (black) and 2H-labeled d8 Val9 i+ 4 (red (dark gray in the print version)). (B) Likely conformation of M2δ with and 2H-labeled d10Leu10 (left) and M2δ with 2H-labeled d8Val9 (right) both with MTSL at i+ 4 from MD simulation.
4.2 Distinguishing α-Helices from β-Strands
In addition to demonstrating this novel ESEEM approach’s ability to identify an α-helix, this exact labeling paradigm can be applied to an ideal β-sheet peptide to distinguish these two most predominant secondary structures.
Figure 7 compares three-pulse ESEEM data for samples with the spin label in the i +2 and i +3 positions for both 2H-labeled d10 Leu17 M2δ and 2H-labeled d10 Leu8 Ubiquitin peptide constructs. In Fig. 7A, the presence of 2H modulation in the i+2 Ubiquitin time domain spectrum indicated a weak dipolar coupling between deuterium nuclei and the spin label. Also, a corresponding FT peak at the 2H-Larmor frequency was revealed in the frequency domain which indicated that the distance between the 2H nuclei on Leu and the spin labels must be within the ~8 Å detection limit. The absence of 2H modulation in the i+3 Ubiquitin sample implied that the 2H-SL distances are greater than 8 Å. This ESEEM spectra pattern was caused by the extended structure of the β-sheet in which the 2H nucleus is closer to the i+2 position than the i +3 position. Also, the MTSL and the d10 Leu side chain pointed toward the same side of the β-sheet in i+2 constructs, while they pointed to opposite sides of the β-sheet in i+3 constructs. Figure 7B shows the corresponding ESEEM data for the M2δ peptide in a bicelle. Conversely, the M2δ i +2 spectrum did not contain any 2H modulation, whereas the M2δ i+3 spectrum clearly shows 2H modulation and a 2H FT peak. The MTSL and d10 Leu side chain pointed toward opposite sides of the helix in i+2 constructs and wrap around to point toward the same side of the helix in the i+3 construct which was similar to the illustrations shown in Fig. 6B. The complementary results of the i+2/i+3 spectra for an α-helices and β-sheets obtained utilizing this novel ESEEM approach demonstrated the establishment of a simple qualitative method for determining site-specific secondary structure of any given protein system.
Figure 7.

Three-pulse ESEEM experimental data. (A) Ubiquitin peptide with 2H-labeled d10 Leu8 in buffer at τ = 200 ns for the i+ 2 and i+ 3 in time domain (left) and frequency domain (right). (B) AChR M2δ with 2H-labeled d10 Leu11 in DMPC/DHPC bicelles at τ= 200 ns for the i+ 2 and i+ 3 in time domain (left) and frequency domain (right).
5. SUMMARY AND FUTURE DIRECTION
Here, we established and developed a novel approach to probe the secondary structure of membrane proteins and peptides in lipid bilayers. Results showed that this approach can be used to identify α-helical and β-sheet secondary structures with multiple isotopically labeled amino acids. The modulation depth with different probes varied according to the side chain length, side chain flexibility, backbone motion, and the local environment. However, the i+x pattern for each secondary structure is similar regardless of which amino acid probe was used. Thus, this ESEEM approach should be valuable for identifying an α-helical region, as well as distinguishing between α-helical and a β-strand in a peptide or protein. This efficient ESEEM spectroscopic technique does not provide the same high-resolution structural information obtained from NMR spectroscopy or X-ray crystallography, but does provide very important qualitative secondary structural information for membrane protein systems or other biological systems where those techniques are not applicable. For SDSL EPR researchers, this approach will provide additional spectroscopic tools to probe the structures of biological systems.
In order to fully establish and expand the application of this very effective secondary structure approach, other amino acids with different side chain lengths and flexibility need to be examined. Also, the potential of utilizing other isotope-labeled amino acids (such as 13C, 15N, or 19F) could be investigated with this novel technique. This powerful method has the potential to be extended to detecting random coils and less predominant secondary structure such as 310 and π helices. Also, it can be adapted to detect secondary structural transitions and local conformational changes.
Further studies will be conducted to apply this ESEEM SDSL approach to larger integral membrane proteins, which are overexpressed in bacteria and then reconstituted into lipid bilayers. Cys residues can be introduced to desired positions through site-directed mutagenesis. Also, selective amino acid isotopic labeling can be used to 2H label certain amino acids such as Leu or Val. However, overexpression conditions need to be optimized to prevent scrambling of the isotope-labeled amino acids.
Acknowledgments
This work was generously supported by National Institutes of Health Grant R01 GM108026 and by the National Science Foundation Grant CHE-1011909. The pulsed EPR spectrometer was purchased through the NSF and the Ohio Board of Regents Grants (MRI-0722403).
References
- Alexander N, Bortolus M, Al-Mestarihi A, Mchaourab H, Meiler J. De novo high-resolution protein structure determination from sparse spin-labeling EPR data. Structure. 2008;16(2):181–195. doi: 10.1016/j.str.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C, Flitsch SL, Khorana HG, Hubbell WL. Structural studies on transmembrane proteins. 2. Spin labeling of bacteriorhodopsin mutants at unique cysteines. Biochemistry. 1989;28(19):7806–7812. doi: 10.1021/bi00445a042. [DOI] [PubMed] [Google Scholar]
- Altenbach C, Greenhalgh DA, Khorana HG, Hubbell WL. A collision gradient method to determine the immersion depth of nitroxides in lipid bilayers: Application to spin-labeled mutants of bacteriorhodopsin. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(5):1667–1671. doi: 10.1073/pnas.91.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baber JL, Louis JM, Clore GM. Dependence of distance distributions derived from double electron–electron resonance pulsed EPR spectroscopy on pulse-sequence time. Angewandte Chemie (International Ed in English) 2015;54:5336–5339. doi: 10.1002/anie.201500640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar I, Lezon TR, Bakan A, Shrivastava IH. Normal mode analysis of biomolecular structures: Functional mechanisms of membrane proteins. Chemical Reviews. 2010;110(3):1463–1497. doi: 10.1021/cr900095e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M. Making membrane proteins for structures: A trillion tiny tweaks. Nature Methods. 2010a;7(6):429–434. doi: 10.1038/nmeth0610-429. [DOI] [PubMed] [Google Scholar]
- Baker M. Structural biology: The gatekeepers revealed. Nature. 2010b;465(7299):823–826. doi: 10.1038/465823a. [DOI] [PubMed] [Google Scholar]
- Barnes JP, Liang Z, Mchaourab HS, Freed JH, Hubbell WL. A multifrequency electron spin resonance study of T4 lysozyme dynamics. Biophysical Journal. 1999;76(6):3298–3306. doi: 10.1016/S0006-3495(99)77482-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartucci R, Guzzi R, Esmann M, Marsh D. Water penetration profile at the protein-lipid interface in Na, K-ATPase membranes. Biophysical Journal. 2014;107(6):1375–1382. doi: 10.1016/j.bpj.2014.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brückner A. In situ electron paramagnetic resonance: A unique tool for analyzing structure-reactivity relationships in heterogeneous catalysis. Chemical Society Reviews. 2010;39(12):4673–4684. doi: 10.1039/b919541f. [DOI] [PubMed] [Google Scholar]
- Carbonaro M, Nucara A. Secondary structure of food proteins by Fourier transform spectroscopy in the mid-infrared region. Amino Acids. 2010;38(3):679–690. doi: 10.1007/s00726-009-0274-3. [DOI] [PubMed] [Google Scholar]
- Carmieli R, Papo N, Zimmermann H, Potapov A, Shai Y, Goldfarb D. Utilizing ESEEM spectroscopy to locate the position of specific regions of membrane-active peptides within model membranes. Biophysical Journal. 2006;90(2):492–505. doi: 10.1529/biophysj.105.062992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey TM, Liu Z, Esquiaqui JM, Pirman NL, Milshteyn E, Fanucci GE. Continuous wave W- and D-band EPR spectroscopy offer “sweet-spots” for characterizing conformational changes and dynamics in intrinsically disordered proteins. Biochemical and Biophysical Research Communications. 2014;450(1):723–728. doi: 10.1016/j.bbrc.2014.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrudu S, Simerska P, Toth I. Chemical methods for peptide and protein production. Molecules. 2013;18(4):4373–4388. doi: 10.3390/molecules18044373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay A, Haldar S. Dynamic insight into protein structure utilizing red edge excitation shift. Accounts of Chemical Research. 2014;47(1):12–19. doi: 10.1021/ar400006z. [DOI] [PubMed] [Google Scholar]
- Cheung JC, Deber CM. Misfolding of the cystic fibrosis transmembrane conductance regulator and disease. Biochemistry. 2008;47(6):1465–1473. doi: 10.1021/bi702209s. [DOI] [PubMed] [Google Scholar]
- Chothia C, Levitt M, Richardson D. Structure of proteins: Packing of alpha-helices and pleated sheets. Proceedings of the National Academy of Sciences of the United States of America. 1977;74(10):4130–4134. doi: 10.1073/pnas.74.10.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieslak JA, Focia PJ, Gross A. Electron spin-echo envelope modulation (ESEEM) reveals water and phosphate interactions with the KcsA potassium channel. Biochemistry. 2010;49(7):1486–1494. doi: 10.1021/bi9016523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve M, Marshall F. The impact of GPCR structures on pharmacology and structure-based drug design. British Journal of Pharmacology. 2010;159(5):986–996. doi: 10.1111/j.1476-5381.2009.00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PM, Ulloa-Aguirre A, Ito J, Janovick JA. G protein-coupled receptor trafficking in health and disease: Lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacological Reviews. 2007;59(3):225–250. doi: 10.1124/pr.59.3.2. [DOI] [PubMed] [Google Scholar]
- Cowieson NP, Kobe B, Martin JL. United we stand: Combining structural methods. Current Opinion in Structural Biology. 2008;18(5):617–622. doi: 10.1016/j.sbi.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Das BB, Park SH, Opella SJ. Membrane protein structure from rotational diffusion. Biochimica et Biophysica Acta. 2015;1848(1 Pt B):229–245. doi: 10.1016/j.bbamem.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deligiannakis Y, Boussac A, Rutherford AW. ESEEM study of the plastoquinone anion radical (QA--) in 14N- and 15N-labeled photosystem II treated with CN. Biochemistry. 1995;34(49):16030–16038. doi: 10.1021/bi00049a017. [DOI] [PubMed] [Google Scholar]
- Deligiannakis Y, Rutherford AW. Electron spin echo envelope modulation spectroscopy in photosystem I. Biochimica et Biophysica Acta. 2001;1507(1–3):226–246. doi: 10.1016/s0005-2728(01)00201-8. [DOI] [PubMed] [Google Scholar]
- Dzuba SA, Raap J. Spin-echo electron paramagnetic resonance (EPR) spectroscopy of a pore-forming (Lipo)peptaibol in model and bacterial membranes. Chemistry & Biodiversity. 2013;10(5):864–875. doi: 10.1002/cbdv.201200387. [DOI] [PubMed] [Google Scholar]
- Edwards DT, Ma Z, Meade TJ, Goldfarb D, Han S, Sherwin MS. Extending the distance range accessed with continuous wave EPR with Gd3+ spin probes at high magnetic fields. Physical Chemistry Chemical Physics. 2013;15(27):11313–11326. doi: 10.1039/c3cp43787f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanucci GE, Cafiso DS. Recent advances and applications of site-directed spin labeling. Current Opinion in Structural Biology. 2006;16(5):644–653. doi: 10.1016/j.sbi.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Feng W, Pan L, Zhang M. Combination of NMR spectroscopy and X-ray crystallography offers unique advantages for elucidation of the structural basis of protein complex assembly. Science China. Life Sciences. 2011;54(2):101–111. doi: 10.1007/s11427-011-4137-2. [DOI] [PubMed] [Google Scholar]
- Fritzsching KJ, Yang Y, Schmidt-Rohr K, Hong M. Practical use of chemical shift databases for protein solid-state NMR: 2D chemical shift maps and amino-acid assignment with secondary-structure information. Journal of Biomolecular NMR. 2013;56(2):155–167. doi: 10.1007/s10858-013-9732-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garman EF. Developments in x-ray crystallographic structure determination of biological macromolecules. Science. 2014;343(6175):1102–1108. doi: 10.1126/science.1247829. [DOI] [PubMed] [Google Scholar]
- Goldfarb D. High field ENDOR as a characterization tool for functional sites in microporous materials. Physical Chemistry Chemical Physics. 2006;8(20):2325–2343. doi: 10.1039/b601513c. [DOI] [PubMed] [Google Scholar]
- Góngora-Benítez M, Tulla-Puche J, Albericio F. Handles for Fmoc solid-phase synthesis of protected peptides. ACS Combinatorial Science. 2013;15(5):217–228. doi: 10.1021/co300153c. [DOI] [PubMed] [Google Scholar]
- Gorka J, Rohmer M, Bornemann S, Papasotiriou DG, Baeumlisberger D, Arrey TN, et al. Perfusion reversed-phase high-performance liquid chromatography for protein separation from detergent-containing solutions: An alternative to gel-based approaches. Analytical Biochemistry. 2012;424(2):97–107. doi: 10.1016/j.ab.2012.02.021. [DOI] [PubMed] [Google Scholar]
- Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nature Protocols. 2006;1(6):2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross M. Proteins that convert from alpha helix to beta sheet: Implications for folding and disease. Current Protein & Peptide Science. 2000;1(4):339–347. doi: 10.2174/1389203003381289. [DOI] [PubMed] [Google Scholar]
- Harris JR. Transmission electron microscopy in molecular structural biology: A historical survey. Archives of Biochemistry and Biophysics. 2014 doi: 10.1016/j.abb.2014.11.011. In press. [DOI] [PubMed] [Google Scholar]
- Hernández-Guzmán J, Sun L, Mehta AK, Dong J, Lynn DG, Warncke K. Copper(II)-bis-histidine coordination structure in a fibrillar amyloid β-peptide fragment and model complexes revealed by electron spin echo envelope modulation spectroscopy. Chembiochem. 2013;14(14):1762–1771. doi: 10.1002/cbic.201300236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst SJ, Alexander N, McHaourab HS, Meiler J. RosettaEPR: An integrated tool for protein structure determination from sparse EPR data. Journal of Structural Biology. 2011;173(3):506–514. doi: 10.1016/j.jsb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman BM. Electron-nuclear double resonance spectroscopy (and electron spin-echo envelope modulation spectroscopy) in bioinorganic chemistry. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(7):3575–3578. doi: 10.1073/pnas.0636464100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Green B, Thompson M, Chen R, Thomaston J, DeGrado WF, et al. C-terminal juxtamembrane region of full-length M2 protein forms a membrane surface associated amphipathic helix. Protein Science. 2015;24(3):426–429. doi: 10.1002/pro.2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell WL, Gross A, Langen R, Lietzow MA. Recent advances in site-directed spin labeling of proteins. Current Opinion in Structural Biology. 1998;8(5):649–656. doi: 10.1016/s0959-440x(98)80158-9. [DOI] [PubMed] [Google Scholar]
- Hubbell WL, López CJ, Altenbach C, Yang Z. Technological advances in site-directed spin labeling of proteins. Current Opinion in Structural Biology. 2013;23(5):725–733. doi: 10.1016/j.sbi.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Lee C, Drew D. Breaking the barriers in membrane protein crystallography. The International Journal of Biochemistry & Cell Biology. 2013;45(3):636–644. doi: 10.1016/j.biocel.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Kevan L, Schwartz RN, editors. Time domain electron spin resonance. New York: Wiley-Interscience; 1979. [Google Scholar]
- King GJ, Jones A, Kobe B, Huber T, Mouradov D, Hume DA, et al. Identification of disulfide-containing chemical cross-links in proteins using MALDI-TOF/ TOF-mass spectrometry. Analytical Chemistry. 2008;80(13):5036–5043. doi: 10.1021/ac702277q. [DOI] [PubMed] [Google Scholar]
- Kubota T, Lacroix JJ, Bezanilla F, Correa AM. Probing α-3(10) transitions in a voltage-sensing S4 helix. Biophysical Journal. 2014;107(5):1117–1128. doi: 10.1016/j.bpj.2014.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurochkina N. Helix-helix interactions and their impact on protein motifs and assemblies. Journal of Theoretical Biology. 2010;264(2):585–592. doi: 10.1016/j.jtbi.2010.02.026. [DOI] [PubMed] [Google Scholar]
- Ladokhin AS. Measuring membrane penetration with depth-dependent fluorescence quenching: Distribution analysis is coming of age. Biochimica et Biophysica Acta. 2014;1838(9):2289–2295. doi: 10.1016/j.bbamem.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreh M, Robinson CV. A new window into the molecular physiology of membrane proteins. The Journal of Physiology. 2015;593(2):355–362. doi: 10.1113/jphysiol.2014.283150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Sahu ID, Mayo DJ, McCarrick RM, Troxel K, Zhou A, et al. Enhancement of electron spin echo envelope modulation spectroscopic methods to investigate the secondary structure of membrane proteins. The Journal of Physical Chemistry B. 2012;116(36):11041–11045. doi: 10.1021/jp304669b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäde V, Els-Heindl S, Beck-Sickinger AG. Automated solid-phase peptide synthesis to obtain therapeutic peptides. Beilstein Journal of Organic Chemistry. 2014;10:1197–1212. doi: 10.3762/bjoc.10.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matalon E, Faingold O, Eisenstein M, Shai Y, Goldfarb D. The topology, in model membranes, of the core peptide derived from the T-cell receptor transmembrane domain. Chembiochem. 2013;14(14):1867–1875. doi: 10.1002/cbic.201300191. [DOI] [PubMed] [Google Scholar]
- Mayo D, Zhou A, Sahu I, McCarrick R, Walton P, Ring A, et al. Probing the structure of membrane proteins with electron spin echo envelope modulation spectroscopy. Protein Science. 2011;20(7):1100–1104. doi: 10.1002/pro.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLuskey K, Roszak AW, Zhu Y, Isaacs NW. Crystal structures of all-alpha type membrane proteins. European Biophysics Journal. 2010;39(5):723–755. doi: 10.1007/s00249-009-0546-6. [DOI] [PubMed] [Google Scholar]
- Moraes I, Evans G, Sanchez-Weatherby J, Newstead S, Stewart PD. Membrane protein structure determination—The next generation. Biochimica et Biophysica Acta. 2014;1838(1 Pt A):78–87. doi: 10.1016/j.bbamem.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesmelov YE. Protein structural dynamics revealed by site-directed spin labeling and multifrequency EPR. Methods in Molecular Biology. 2014;1084:63–79. doi: 10.1007/978-1-62703-658-0_4. [DOI] [PubMed] [Google Scholar]
- Opella SJ, Marassi FM, Gesell JJ, Valente AP, Kim Y, Oblatt-Montal M, et al. Structures of the M2 channel-lining segments from nicotinic acetylcholine and NMDA receptors by NMR spectroscopy. Nature Structural Biology. 1999;6(4):374–379. doi: 10.1038/7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raibaut L, El Mahdi O, Melnyk O. Solid phase protein chemical synthesis. Topics in Current Chemistry. 2015;363:103–154. doi: 10.1007/128_2014_609. [DOI] [PubMed] [Google Scholar]
- Rask-Andersen M, Almén MS, Schioth HB. Trends in the exploitation of novel drug targets. Nature Reviews Drug Discovery. 2011;10(8):579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- Roach CA, Simpson JV, JiJi RD. Evolution of quantitative methods in protein secondary structure determination via deep-ultraviolet resonance Raman spectroscopy. Analyst. 2012;137(3):555–562. doi: 10.1039/c1an15755h. [DOI] [PubMed] [Google Scholar]
- Sahu ID, Hustedt EJ, Ghimire H, Inbaraj JJ, McCarrick RM, Lorigan GA. CW dipolar broadening EPR spectroscopy and mechanically aligned bilayers used to measure distance and relative orientation between two TOAC spin labels on an antimicrobial peptide. Journal of Magnetic Resonance. 2014;249C:72–79. doi: 10.1016/j.jmr.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu ID, Kroncke BM, Zhang R, Dunagan MM, Smith HJ, Craig A, et al. Structural investigation of the transmembrane domain of KCNE1 in proteoliposomes. Biochemistry. 2014;53(40):6392–6401. doi: 10.1021/bi500943p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu ID, McCarrick RM, Lorigan GA. Use of electron paramagnetic resonance to solve biochemical problems. Biochemistry. 2013;52(35):5967–5984. doi: 10.1021/bi400834a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu ID, McCarrick RM, Troxel KR, Zhang R, Smith HJ, Dunagan MM, et al. DEER EPR measurements for membrane protein structures via bifunctional spin labels and lipodisq nanoparticles. Biochemistry. 2013;52(38):6627–6632. doi: 10.1021/bi4009984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger A, Jeschke G. Principles of pulse electron paramagnetic resonance. Oxford: Oxford University Press; 2001. [Google Scholar]
- Shukla HD, Vaitiekunas P, Cotter RJ. Advances in membrane proteomics and cancer biomarker discovery: Current status and future perspective. Proteomics. 2012;12(19–20):3085–3104. doi: 10.1002/pmic.201100519. [DOI] [PubMed] [Google Scholar]
- Stoll S, Calle C, Mitrikas G, Schweiger A. Peak suppression in ESEEM spectra of multinuclear spin systems. Journal of Magnetic Resonance. 2005;177(1):93–101. doi: 10.1016/j.jmr.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Tang C, Clore GM. A simple and reliable approach to docking protein-protein complexes from very sparse NOE-derived intermolecular distance restraints. Journal of Biomolecular NMR. 2006;36(1):37–44. doi: 10.1007/s10858-006-9065-2. [DOI] [PubMed] [Google Scholar]
- Tarabek P, Bonifacić M, Beckert D. Time-resolved FT EPR and optical spectroscopy study on photooxidation of aliphatic alpha-amino acids in aqueous solutions; electron transfer from amino vs carboxylate functional group. The Journal of Physical Chemistry A. 2006;110(22):7293–7302. doi: 10.1021/jp060764m. [DOI] [PubMed] [Google Scholar]
- van Wonderen JH, McMahon RM, O’Mara ML, McDevitt CA, Thomson AJ, Kerr ID, et al. The central cavity of ABCB1 undergoes alternating access during ATP hydrolysis. The FEBS Journal. 2014;281(9):2190–2201. doi: 10.1111/febs.12773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. The membrane protein universe: What’s out there and why bother? Journal of Internal Medicine. 2007;261(6):543–557. doi: 10.1111/j.1365-2796.2007.01792.x. [DOI] [PubMed] [Google Scholar]
- Wang S, Ladizhansky V. Recent advances in magic angle spinning solid state NMR of membrane proteins. Progress in Nuclear Magnetic Resonance Spectroscopy. 2014;82:1–26. doi: 10.1016/j.pnmrs.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Warncke K. Characterization of the product radical structure in the Co(II)-product radical pair state of coenzyme B12-dependent ethanolamine deaminase by using three-pulse 2H ESEEM spectroscopy. Biochemistry. 2005;44(9):3184–3193. doi: 10.1021/bi048196t. [DOI] [PubMed] [Google Scholar]
- White SH, Wimley WC. Membrane protein folding and stability: Physical principles. Annual Review of Biophysics and Biomolecular Structure. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- Whitmore L, Wallace BA. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers. 2008;89(5):392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- Yu X, Lorigan GA. Secondary structure, backbone dynamics, and structural topology of phospholamban and its phosphorylated and Arg9Cys-mutated forms in phospholipid bilayers utilizing 13C and 15N solid-state NMR spectroscopy. The Journal of Physical Chemistry B. 2014;118(8):2124–2133. doi: 10.1021/jp500316s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerella R, Chen PY, Evans PA, Raine A, Williams DH. Structural characterization of a mutant peptide derived from ubiquitin: Implications for protein folding. Protein Science. 2000;9(11):2142–2150. doi: 10.1110/ps.9.11.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Guo L, Zeng H, Yang X, Yuan J, Shi H, et al. Purification and characterization of a novel antimicrobial peptide from Brevibacillus laterosporus strain A60. Peptides. 2012;33(2):206–211. doi: 10.1016/j.peptides.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Zhou A, Abu-Baker S, Sahu ID, Liu L, McCarrick RM, Dabney-Smith C, et al. Determining α-helical and β-sheet secondary structures via pulsed electron spin resonance spectroscopy. Biochemistry. 2012;51(38):7417–7419. doi: 10.1021/bi3010736. [DOI] [PMC free article] [PubMed] [Google Scholar]