

Abstract
Background
Understanding the cause of therapeutic resistance and identifying new biomarkers in breast cancer to predict therapeutic responses will help optimise patient care. Calcium (Ca2+)-signalling is important in a variety of processes associated with tumour progression, including breast cancer cell migration and proliferation. Ca2+-signalling is also linked to the acquisition of multidrug resistance. This study aimed to assess the expression level of proteins involved in Ca2+-signalling in an in vitro model of trastuzumab-resistance and to assess the ability of identified targets to reverse resistance and/or act as potential biomarkers for prognosis or therapy outcome.
Methods
Expression levels of a panel of Ca2+-pumps, channels and channel regulators were assessed using RT-qPCR in resistant and sensitive age-matched SKBR3 breast cancer cells, established through continuous culture in the absence or presence of trastuzumab. The role of Cav3.2 in the acquisition of trastuzumab-resistance was assessed through pharmacological inhibition and induced overexpression. Levels of Cav3.2 were assessed in a panel of non-malignant and malignant breast cell lines using RT-qPCR and in patient samples representing different molecular subtypes (PAM50 cohort). Patient survival was also assessed in samples stratified by Cav3.2 expression (METABRIC and KM-Plotter cohort).
Results
Increased mRNA of Cav3.2 was a feature of both acquired and intrinsic trastuzumab-resistant SKBR3 cells. However, pharmacological inhibition of Cav3.2 did not restore trastuzumab-sensitivity nor did Cav3.2 overexpression induce the expression of markers associated with resistance, suggesting that Cav3.2 is not a driver of trastuzumab-resistance. Cav3.2 levels were significantly higher in luminal A, luminal B and HER2-enriched subtypes compared to the basal subtype. High levels of Cav3.2 were associated with poor outcome in patients with oestrogen receptor positive (ER+) breast cancers, whereas Cav3.2 levels were correlated positively with patient survival after chemotherapy in patients with HER2-positive breast cancers.
Conclusion
Our study identified elevated levels of Cav3.2 in trastuzumab-resistant SKBR3 cell lines. Although not a regulator of trastuzumab-resistance in HER2-positive breast cancer cells, Cav3.2 may be a potential differential biomarker for survival and treatment response in specific breast cancer subtypes. These studies add to the complex and diverse role of Ca2+-signalling in breast cancer progression and treatment.
Electronic supplementary material
The online version of this article (doi:10.1186/s12935-016-0299-0) contains supplementary material, which is available to authorized users.
Keywords: Breast cancer, Trastuzumab-resistance, Calcium-signalling, Cav3.2 (CACNA1H), Therapeutic response, Biomarker
Background
Approximately 20–25 % of breast tumours display overexpression of human epidermal growth factor receptor 2 (HER2, ERBB2) [1]. The monoclonal antibody trastuzumab (Herceptin®) represents a major advancement in the treatment of HER2-positive breast cancers [2, 3]. However, some patients can develop resistance to trastuzumab therapy [4, 5] while others exhibit initial therapeutic insensitivity, despite their tumours being identified as HER2-positive and the patients being naïve to trastuzumab therapy (intrinsic resistance) [6, 7].
Overcoming therapeutic resistance is a major challenge and an understanding of the underlying mechanism will inform the development of second generation treatments [8]. For example, resistance to tyrosine kinase inhibitors in lung cancer is attributed to the acquisition of an EGFR T790M mutation, and evidence suggests that a combination of irreversible tyrosine kinase inhibitors and an anti-EGFR antibody is effective in overcoming this particular resistance mechanism [9].
Different mechanisms may be responsible for acquired and intrinsic trastuzumab-resistance. These mechanisms include, but are not limited to, mutations in ERBB2 resulting in the expression of a modified HER2 receptor altering trastuzumab binding [10, 11] and upregulation of proteins that sterically hinder trastuzumab binding [12, 13]. Increased signalling through HER1, HER2, HER3 (receptors of the EGFR-family) and IGF-1R [14–17] as well as downstream signalling such as activation of the PTEN/PI3K/Akt pathway also represent potential pathways for trastuzumab-resistance [7, 18–20]. Many of the aforementioned studies have been evaluated in breast cancer cell lines established from HER2-positive breast cancer cells cultured in the presence of trastuzumab, including the HER2-positive SKBR3 cell line [15, 16, 21].
A remodelling of Ca2+-signalling occurs in some breast cancers and is thought to be an important contributor or biomarker of breast tumourigenesis [22]. For example, enhanced expression of the Ca2+-channel TRPV6 is a feature of oestrogen receptor negative breast cancers [23] and alteration in the relative levels of the store operated Ca2+-influx pathway regulators STIM1 and STIM2 are a feature of the basal molecular breast cancer subtype and is associated with poor survival [24]. Ca2+ is a critical regulator of many processes important in cancer [25], including proliferation and migration [26, 27]. Indeed, inhibition of the Orai1 Ca2+-channel reduces the metastatic potential of breast cancer cells [28].
Ca2+-signalling is also implicated in some therapeutic resistance pathways in breast cancer. For example, the Ca2+-permeable ion channel TRPC5 plays a role in p-glycoprotein-mediated resistance to adriamycin in MCF-7 breast cancer cells [29]. However, the potential contribution of remodelling of Ca2+-signalling in trastuzumab-resistance has not yet been explored. Herein we sought to determine alterations of Ca2+-signalling proteins in the context of trastuzumab-resistance using HER2-positive SKBR3 breast cancer cell lines as models of intrinsic (no previous trastuzumab exposure) and acquired resistance. This work had the goal of identifying calcium channels and pumps that when inhibited could restore sensitivity to therapy and/or serve as biomarkers for prognosis or response to therapy.
Methods
Cell culture and development of resistant cell lines
Human breast cell lines were purchased from ATCC, provided by UQCCR or were a gift from the late Professor Rob Sutherland (Garvan Institute, Sydney, Australia). SKBR3 cells were subcultured in McCoy’s 5A media (Invitrogen) supplemented with 10 % foetal bovine serum and 1 % penicillin/streptomycin mixture (100 U/mL/100 µg/mL, Invitrogen) at 37 °C and 5 % CO2. Cells were routinely tested for mycoplasma infection and the SKBR3 parental cell line was STR profiled as previously described [30]. Trastuzumab-resistant cell lines were developed as follows, adapted from [31]. Briefly, cells were cultured in the presence of trastuzumab (10 µg/mL, Herceptin®, Roche Products, Dee Why, Australia) over a 7 month period. Trastuzumab treatment was initiated 24 h after seeding. Age-matched controls (no trastuzumab) were produced over a similar time period. Media (±trastuzumab) was replaced every 3 days.
Cell viability, MTS assay
Cell viability was assessed using the CellTiter 96® aqueous non-radioactive cell proliferation assay (Promega) using the manufacturer’s instructions. Cell lines were treated with trastuzumab or control media without antibiotics.
Real time RT-qPCR (RT-qPCR)
Real time RT-qPCR (RT-qPCR) was used to assess mRNA levels of target genes as previously described [32]. Briefly, total RNA (Qiagen RNeasy™ Plus Mini Kit (Qiagen, Hilden, Germany) was reverse transcribed (Ominiscript RT Kit Qiagen) and amplified using TaqMan Universal or TaqMan Fast Universal PCR master mix (Life technologies, Australia), with TaqMan Gene expression Assays (Additional file 1: Data S1). Experiments were performed using a StepOnePlus Real Time RT-qPCR instrument (Applied Biosystems, Carlsbad, USA) with universal cycling conditions. Results are expressed normalised to 18S rRNA and analysed using the comparative CT method. A CT value of 35 was assigned to samples where amplification did not occur within 40 cycles for characterisation of trastuzumab-resistant and aged-matched control SKBR3 cell lines and assessment of Cav3.2 levels in breast cancer cell lines (Taqman Universal PCR master mix). A CT of 40 was assigned to samples where amplification did not occur within 40 cycles for studies assessing potential mRNA changes induced by Cav3.2 overexpression (TaqMan Fast PCR master mix).
Immunoblotting
Whole cell lysates were prepared using lysis buffer containing protease and phosphatase inhibitors (Roche, Applied Science, Penzberg, Germany). Proteins were separated using 4–12 % NuPAGE Bis–Tris gels with MOPS SDS running buffer (Invitrogen) and transferred to a PVDF membrane. Membranes were blocked in phosphate buffered saline (PBS)/1 %Tween20 or in PBS/5 % skim milk powder for 1 h. Antibodies included: HER2 polyclonal rabbit (Tyr 1222, Cell Signaling), EGFR polyclonal rabbit (Tyr 992, Cell Signaling), β-actin loading control (AC-15, Sigma Aldrich) and horseradish peroxidase conjugated goat anti-rabbit IgG (170-6516, Biorad). Membranes were incubated with SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) and images analysed using a Versadoc MP400 Imaging system (BioRad). Protein density was normalised to β-actin.
Pharmacological inhibition of Cav3.2
Cells were treated with mibefradil (0.01–1 µM; Sigma Aldrich) [33] or ML218 (0.1–10 µM; Sigma Aldrich) [34] either alone or in combination with trastuzumab (10 µg/mL), 24 h after seeding (2000 cells/well in a 96 well plate). All treatments and media changes were repeated every 2 days in antibiotic-free media. Sensitivity to trastuzumab was assessed using a MTS assay 192 h after treatment start.
Measurement of cytosolic free calcium levels
Cytosolic free Ca2+ ([Ca2+]CYT) levels were measured using a fluorometric imaging plate reader (FLIPRTETRA, Molecular Devices, Sunnyvale, USA). Cells (2000 cells/well in a 96-well black-walled imaging plate; Corning) were assessed 216 h after seeding in antibiotic-free media. Cells were treated with Fluo-4 AM (4 µM) for 30 min at 37 °C, washed three times in physiological salt solution (PSS) buffer [NaCl (140 mM)], glucose (11.5 mM), CaCl2 (1.8 mM), HEPES (10 mM), KCl (5.9 mM), MgCl2 (1.4 mM), NaH2PO4 (1.2 mM), NaHCO3 (5 mM) pH 7.3] and incubated in PSS at room temperature (15 min). Measurements were performed at 470–495 nm excitation and 515–575 nm emission and analysed using ScreenWorks Software (v2.0.0.27, Molecular Devices).
Overexpression of Cav3.2 (CACNA1H)
SKBR3 cells were transfected in 6-well plates (375 000 cells/well) 24 h after seeding using Lipofectamine®3000 and PC3000 enhancer (Life Technologies) with 2.5 µg of total DNA [pCDNA3.1 (Invitrogen) + pEGFP-N1 (Clontech)] (EGFP MOCK) or (α1Ha (Adgene, 45809, Cambridge, USA) + pEGFP-N1) (EGFP Cav3.2) at equimolar ratio of 10:1. After transfection (24 h) cells were sorted into EGFP enriched (>90 % purity) and EGFP depleted populations using a MoFlo Astrios FACS, RNA was isolated and the effect on expression of selected markers was analysed using RT-qPCR.
Relative CACNA1H (Cav3.2) expression levels in breast cancer subtypes
The relative expression levels of CACNA1H were analysed in the cancer genome atlas network (TCGA) [35] breast cancer data set. Expression levels were defined as log2 transformed mean-centred transcript quantification produced by the TCGA consortium through RSEM (RNA-seq by expectation–maximization) [36]. TCGA tumours were divided into quartiles of CACNA1H expression. ERBB2, ESR1 and PGR levels were compared in each quartile to the quartile with the lowest level of CACNA1H expression.
Assessment of patient survival and response to chemotherapy (CT) in oestrogen receptor positive (ER+), HER2-positive (HER+) and triple negative (TNBC) tumours based on CACNA1H expression
Receiver operator curve (ROC) analysis was performed in each clinical subgroup and within the METABRIC [37] cohort that received chemotherapy (CT). The variable for ROC analysis was CACNA1H expression while the classification variable was patient survival (1 = death from breast cancer, 0 = non-event). The optimal criterion, as determined by MedCalc (https://www.medcalc.org/) factoring in disease prevalence as death caused by breast cancer, was used to stratify tumours into high and low expressing groups. Groups were used for stratifying patients by overall survival using Kaplan–Meier curves. The online tool KM-Plotter [38] was used to generate a validation set of Kaplan–Meier curves based on CACNA1H expression. Survival of patients was stratified using the “Auto select best cutoff” feature, which determines the optimal patient stratification based on median, tertile and quartile groupings. Logrank P values and hazard ratios were determined using MedCalc. Hazard ratios with corresponding 95 % confidence interval and P values are as indicated.
Statistical analyses
GraphPad Prism (v6.05 for Windows; GraphPad Software, Inc., La Jolla, CA, USA) was used to determine statistical differences.
Results
Development and characterisation of trastuzumab-resistant SKBR3 cell lines
Trastuzumab-resistant cell lines and sensitive age-matched control cell lines were established from trastuzumab-sensitive parental SKBR3 cells by continuous culture in trastuzumab (10 µg/mL) for 7 months. Cell lines were assigned as “T” for continuous culture in trastuzumab or “V” for vehicle, “S” for sensitive and “R” for resistant. Cell line status after the 7 months of culture was analysed in the presence of trastuzumab (10 µg/mL) over 192 h (Fig. 1a). Most cell lines (6 of 8) cultured in the absence of trastuzumab retained sensitivity e.g. [SV1, SV2, Fig. 1a (i)]. Two cell lines cultured in the presence of trastuzumab were selected for further study based on the acquisition of trastuzumab-resistance. In these two cell lines the relative cell viability was not decreased by trastuzumab treatment compared to vehicle controls [RT1, RT2, Fig. 1a (ii)]. Two of eight age-matched control cell lines cultured continuously for 7 months in the absence of trastuzumab spontaneously developed resistance to trastuzumab and were defined as exhibiting intrinsic resistance to trastuzumab [RV1, RV2, Fig. 1a (iii)]. Trastuzumab-sensitive cell lines (SV1 and SV2) showed a significant decrease in cell viability after trastuzumab treatment, while acquired (RT1 and RT2) and intrinsic (RV1 and RV2) resistant cells showed no significant difference compared to vehicle controls (**p ≤ 0.1) (Fig. 1b).
Fig. 1.
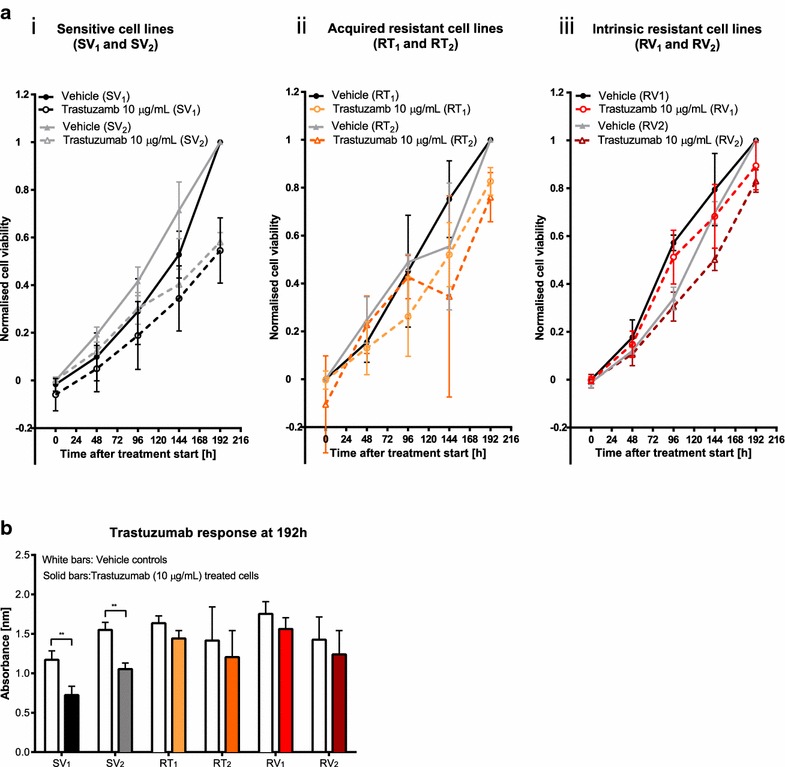
Development of SKBR3-derived cell lines as in vitro models for acquired and intrinsic drug-resistance. SKBR3 cells were continuously cultured for 7 months in the absence or the presence of 10 µg/mL trastuzumab. a Cells were seeded at a density of 2000 cells/well and treated with 10 µg/mL trastuzumab or equal volume of H2O (vehicle), 24 h after seeding and relative cell viability was assessed over 192 h with a MTS assay. Two cell lines continuously cultured in the absence of trastuzumab (SV1, SV2) retained trastuzumab-sensitivity (i), two cell lines cultured in the presence of trastuzumab (RT1, RT2) acquired trastuzumab-resistance (ii) and two cell lines continuously cultured in the absence of trastuzumab (RV1, RV2) developed intrinsic resistance to trastuzumab (iii). Viable cell number of all samples are expressed relative to control cells (no trastuzumab) at 192 h. Normalised cell viability = [Absorbance of sample (t) − Average absorbance of control (t = 0)]/[Average absorbance control (t = 192 h) − Average absorbance control (t = 0)], (n = 3 ± SD). b Relative viable cell numbers of all cell lines were compared at the end of the protocol (192 h after start of treatment). Trastuzumab sensitive cell lines (SV1, SV2) showed a significant decreased cell viability in the presence of trastuzumab compared to vehicle control cells, whereas the relative viable cell number of acquired resistant cell lines (RT1, RT2) and intrinsic resistant cell lines (RV1, RV2) was not significantly altered by trastuzumab (n = 3 ± SD). Statistical analyses were performed using multiple t tests with Holm Sidak correction (**p ≤ 0.01). White bars present vehicle controls and solid coloured bars represent trastuzumab (10 µg/mL) treated cells
Assessment of HER2 and EGFR expression in trastuzumab-resistant SKBR3 cell lines
To confirm that trastuzumab-resistance amongst the model cell lines was not due to loss of HER2 expression, or alterations in EGFR expression, mRNA and protein levels of HER2 and EGFR were quantified in all cell lines (Fig. 2). Resistant cell lines showed similar levels of HER2 and EGFR mRNA compared to trastuzumab-sensitive cell lines (p > 0.5) (Fig. 2a, b). HER2 protein (185 kDa) was seen in all samples, with no truncated receptor detected. HER2 protein levels were similar in all SKBR3-derived cell lines compared to parental SKBR3 cells and no differences in protein levels were observed between resistant and sensitive cell lines (p > 0.5) (Fig. 2c). EGFR protein levels were also similar in sensitive and resistant cell lines (Fig. 2d). Collectively these data suggested that the mechanism of resistance in this model was not related solely to changes in the expression of HER2 or EGFR.
Fig. 2.
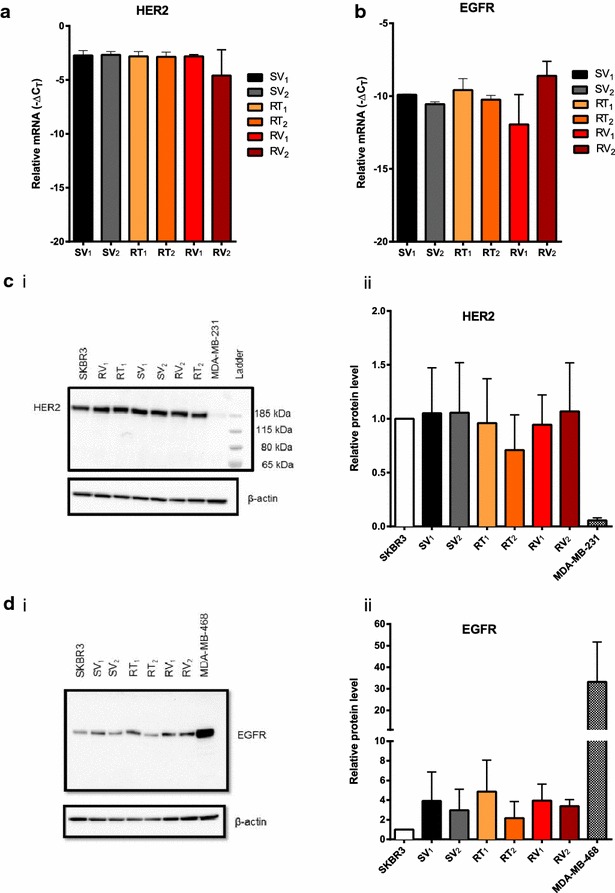
Sensitive and resistant SKBR3-derived cell lines retained their HER2 and EGFR expression levels. Assessment of mRNA levels in SKBR3-derived cell lines showed no differences in the mRNA levels of HER2 (a) and EGFR (b) in trastuzumab-resistant cell lines (RT1, RT2, RV1, RV2) compared to sensitive age-matched control cell lines (SV1, SV2). All mRNA levels were normalised to 18S rRNA and values expressed as −ΔCT (n = 3 ± SD). The protein level of HER2 (c) and EGFR (d) were analysed in all SKBR3-derived cell lines and representative immunoblots are shown in (c i) and d i). All protein levels were normalised to β-actin protein expression, parental SKBR3 cells were used to assess changes in protein levels through continuous culture, MDA-MB-231 cells were used as a HER2-negative control (c) and MDA-MB-468 as an EGFR positive control (d). HER2 (c ii) and EGFR (d ii) protein expression was normalised to the SKBR3 parental cell line in biological replicates (n = 3 ± SD). Sensitive and resistant SKBR3-derived cell lines showed similar levels of EGFR and HER2 protein expression. Statistical analyses were performed using one-way ANOVA with Bonferroni post-tests (p > 0.05)
Assessment of mRNA levels of calcium channels, pumps and regulating proteins identifies elevated Cav3.2 in trastuzumab-resistant cell lines
Assessment of over 40 targets including purinergic receptors (P2RX2/4/5, P2RY2/6), calcium pumps (PMCAs, SPCAs, SERCAs) and calcium permeable ion channels (Orai, TRPs, IP3Rs) demonstrated similar mRNA levels of most targets between trastuzumab-sensitive and resistant cell lines, except for the voltage gated calcium channel Cav3.2 (Fig. 3a). Further analysis confirmed the elevation of Cav3.2 mRNA in the RV1, RV2 and RT1 resistant cell lines in comparison to both sensitive cell lines SV1 and SV2 (Fig. 3b).
Fig. 3.
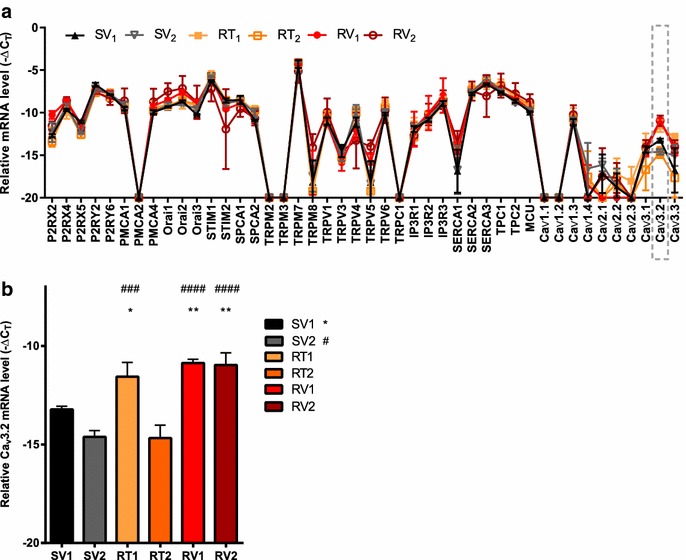
Cav3.2 upregulation in trastuzumab-resistant cell lines. a Assessment of mRNA levels of 44 calcium pumps, channels and calcium regulating proteins identified increased expression of Cav3.2 in three of four trastuzumab resistant cell lines. All values were measured relative to 18 S rRNA and expressed as −ΔCT. Data points at Y = −20 represent no mRNA detection (n = 3 ± SD). b Acquired resistant cell line RT1 and both intrinsic resistant cell lines (RV1, RV2) showed significantly increased expression of Cav3.2 compared to trastuzumab sensitive age-matched control (SV1) (n = 3 ± SD). Statistical analyses were performed using one-way ANOVA with Bonferroni post-tests (*p ≤ 0.05, **p ≤ 0.01)
Pharmacological inhibition of Cav3.2 does not restore trastuzumab-sensitivity in trastuzumab-resistant SKBR3 cells
Cav3.2 inhibition was assessed in the trastuzumab-resistant cell line (RV1), which had the most pronounced upregulation of Cav3.2 relative to trastuzumab-sensitive SKBR3 cell lines. Cells were treated with mibefradil, a calcium channel blocker, which inhibits both T-type and L-type voltage-gated calcium channels, but with greater effectiveness for T-type calcium channel inhibition [33]. Treatment with mibefradil (0.01–1 µM) did not enhance the trastuzumab (10 µg/mL) response in the RV1 resistant cell line (Fig. 4a). Treatment with ML218, a pharmacological inhibitor of Cav3.1, Cav3.2 and Cav3.3 channels with higher selectivity for Cav3.2 [34] at 0.1–10 µM also did not enhance the trastuzumab (10 µg/mL) response in the RV1 cell line (Fig. 4b). Thus pharmacological inhibition of the Cav3.2 calcium channel with the inhibitors used in this study did not restore sensitivity to trastuzumab.
Fig. 4.
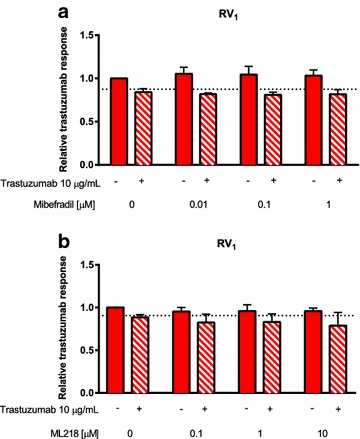
The effect of pharmacological inhibition of Cav3.2 on trastuzumab-resistance. a Cells were treated with mibefradil (0.01–1 µM) alone or with trastuzumab (10 µg/mL) 24 h after seeding for 192 h (n = 3 ± SD). Mibefradil did not promote the response to trastuzumab (p > 0.05). b Cells were treated with ML218 (0.1–10 µM) alone or with trastuzumab (10 µg/mL) 24 h after seeding for 192 h (n = 3 ± SD). ML218 did not promote the response to trastuzumab (p > 0.05). Statistical analyses were performed using two-way ANOVA with Bonferroni post-tests
Assessment of ATP-mediated alterations in [Ca2+]CYT in trastuzumab-sensitive and resistant SKBR3 derived cell lines
To assess if Ca2+-signalling alterations were associated with trastuzumab-resistance, [Ca2+]CYT in response to purinergic receptor activation with ATP (1 mM) was measured. Some resistant cell lines appeared to have a delayed recovery in [Ca2+]CYT after ATP stimulation (Fig. 5a). ATP concentration–response curves (1 nM to 1 mM) were used to characterise changes in intracellular Ca2+-signalling. The relative [Ca2+]CYT at 800 s, a measure of recovery after ATP stimulation, was higher in acquired trastuzumab-resistant SKBR3 cell lines (RT1 and RT2) compared to trastuzumab-sensitive age-matched controls (Fig. 5b).
Fig. 5.
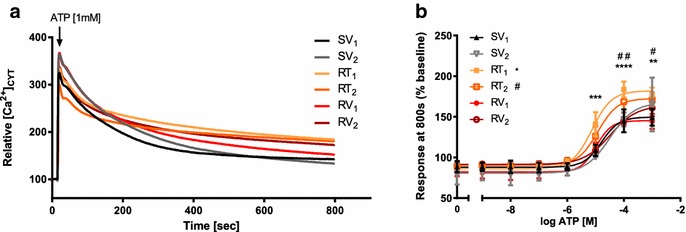
Intracellular calcium measurements with ATP stimulation in trastuzumab-sensitive and resistant cell lines. a Example of relative cytosolic calcium [Ca2+]CYT responses after ATP [1 mM] stimulation in trastuzumab-sensitive age-matched controls (SV1, SV2), acquired resistant cell lines (RT1, RT2) and intrinsic resistant cell lines (RV1, RV2). b Relative calcium [Ca2+]CYT responses at 800 s with ATP stimulation [1 nM to 1 mM] in aged-matched control and resistant cell lines (n = 3 ± SD). Acquired resistant cell lines (RT1, RT2) showed a slower recovery after ATP stimulation with a significant higher relative calcium [Ca2+]CYT level at 800 s while intrinsic resistant cell lines (RV1, RV2,) showed similar recovery rate and similar relative calcium [Ca2+]CYT levels compared to aged matched control SV1. Statistical analysis was performed using two-way ANOVA with Bonferroni post-tests, compared to SV1 (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001)
Overexpression of Cav3.2 in parental SKBR3 cells does not increase expression of selected mRNA markers associated with drug-resistance
Since Cav3.2 is upregulated in trastuzumab-resistant SKBR3 cells and given the role of Ca2+-signalling in the activation of transcription factors [39] we hypothesised that Cav3.2 may induce the expression of genes related to therapeutic resistance and/or with cellular phenotypes associated with therapeutic resistance [40–44]. SKBR3 parental cells were either co-transfected with pEGFP-N1 + α1Ha (EGFP Cav3.2) or with pEGFP-N1 + the empty plasmid backbone (EGFP MOCK) as a control (Fig. 6). Overexpression of Cav3.2 did not produce any pronounced increase of the mRNA levels of vimentin, snail, KRT5, KRT6A, CXCR4, FOXM1 or HSP90AA1 (Fig. 6).
Fig. 6.
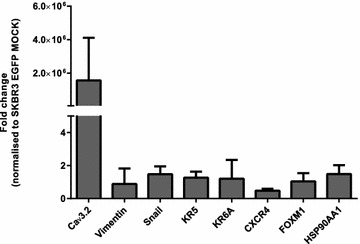
Effect of CACNA1H (Cav3.2) overexpression on expression of mRNA markers associated with tumour progression and drug-resistance. SKBR3 parental cells were co-transfected with α1Ha + pEGFP-N1 (EGFP Cav3.2) or pCDNA3.1 + pEGFP-N1 (EGFP MOCK) and sorted into EGFP enriched and depleted populations 24 h after transfection. Results are expressed as fold change normalised to EGFP MOCK. EGFP Cav3.2 cells showed an approximate greater than 2 106 fold increase in expression compared to EGFP MOCK cells (n = 3 ±SD). Overexpression of Cav3.2 did not increase levels of markers associated with EMT (vimentin, snail), basal markers (KR5, KR6A ) and markers associated with drug-resistance (CXCR4, FOXM1, HSP90AA1) (n = 3 ±SD)
Assessment of Cav3.2 expression levels in cell lines and clinical breast cancer molecular subtypes
Levels of Cav3.2 mRNA were then assessed in a panel of non-malignant breast and breast cancer cell lines representing different molecular subtypes (Fig. 7). Cav3.2 mRNA levels in three of the trastuzumab-resistant cell lines (RT1, RV1 and RV2) were similar to the basal-like HER2 overexpressing trastuzumab-resistant cell line HCC1569 [45]. Basal-like breast cancer cell lines (MDA-MB-231, MDA-MB-468) had undetectable levels of Cav3.2 mRNA, except for HCC1569 cells. In non-malignant breast cell lines (184A1, 184B5, MCF10A, Bret-80-Tert) Cav3.2 mRNA was not detected. Highest levels of Cav3.2 were seen in the luminal-like breast cancer lines MCF-7 and T47D. However, Cav3.2 mRNA levels were dramatically different between luminal-like breast cancer cell lines, with very low levels in ZR-75-1 and parental SKBR3 cell lines and very high levels in MCF-7 and T47D (Fig. 7).
Fig. 7.
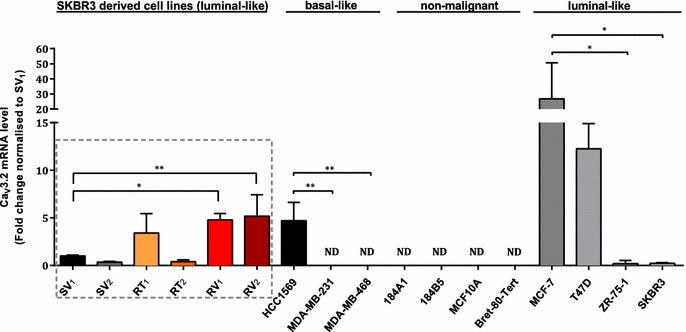
Assessment of the expression level of CACNA1H (Cav3.2) in SKBR3 derived trastuzumab-resistant cell lines, different breast cancer cell lines and non-malignant breast cell lines. Cav3.2 was not detected (ND) in non-malignant breast cell lines and the basal breast cancer cell lines MDA-MB-231 and MDA-MB-468. The luminal cell lines MCF-7 and T47D showed the highest expression of Cav3.2. Statistical analysis was performed within each subtype and trastuzumab-resistant cell lines were compared to the SV1 trastuzumab sensitive SKBR3 cell line. For all amplified targets (n = 3 ±SD), statistical analysis was performed using one-way ANOVA with Bonferroni post-test (*p ≤ 0.05, **p ≤ 0.01)
Cav3.2 expression was also assessed in clinical breast cancer samples stratified into the intrinsic molecular subtypes using the cancer genome atlas expression data (TCGA) [35] (Fig. 8a). The luminal A and B subtypes showed the highest expression level of CACNA1H (Cav3.2), with the basal subtype expressing significantly lower levels of Cav3.2 compared to the luminal A, B and HER2 subtypes. Consistent with the cell line data, luminal breast cancers were associated with a wide range of Cav3.2 levels.
Fig. 8.
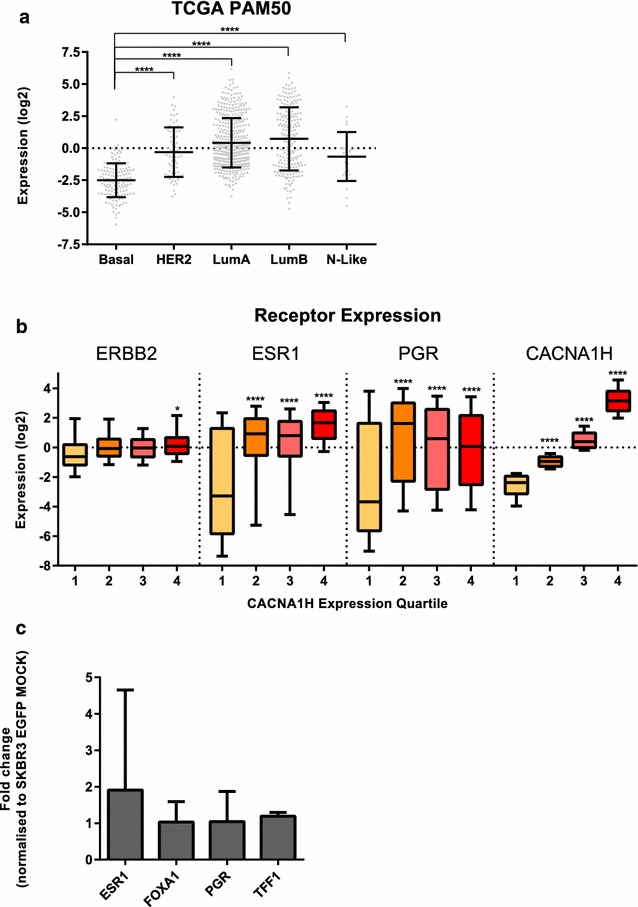
a Analysis of CACNA1H (Cav3.2) expression across the intrinsic molecular subtypes of breast cancer in clinical patient samples. The relative CACNA1H expression level (log2 normalised) was produced by the TCGA consortium through RSEM [36]. The TCGA tumour cohort consists of 845 tumours with 140 basal-like (Basal), 67 HER2-enriched (HER2), 420 luminal A (LumA), 194 luminal B (LumB) and 24 normal-like (N-Like) as determined by RNA-Seq based PAM50 allocations by the TCGA consortium. The PAM50 intrinsic molecular subtypes are based on the classifications of gene expression patterns previously described [66, 67]. This classification was performed by the TCGA consortium [35]. Horizontal lines represent data means with standard deviation and data points in grey. Statistical analysis was performed on expression levels of CACNA1H in basal-like compared to HER2-enriched and luminal subtypes using a one-way ANOVA with Sidak corrected multiple comparisons,(****p ≤ 0.0001). b Relative gene expression for breast cancer receptors ERBB2 (HER2), ESR1 (oestrogen receptor) and PGR (progesterone receptor) within each quartile of CACNA1H expression. Statistical analysis was performed using a one-way ANOVA with Sidak corrected multiple comparisons, comparing expression levels between the highest and the lowest quartile for each gene (*p ≤ 0.05, ****p ≤ 0.0001). c Cav3.2 overexpression in SKBR cells (SKBR3 EGFP) did not increase expression of hormone receptors or proteins involved in oestrogen-receptor mediated signalling TFF1, FOXA1, quantified using RT-qPCR. Results are expressed as fold change normalised to EGFP MOCK (n = 3 ±SD)
High levels of Cav3.2 are associated with increased levels of ESR1 and PGR receptor but Cav3.2 itself does not induce hormone receptor expression or expression of luminal markers in SKBR3 cells
Given the strong association between Cav3.2 and the luminal A and B molecular subtypes (Fig. 8a), we also assessed hormone receptor levels in different CACNA1H (Cav3.2) expression quartiles using the TCGA database, to address the hypothesis that this may reflect a direct regulatory effect. Breast cancers with high levels of CACNA1H (Cav3.2) (2nd, 3rd, 4th quartile) showed a significant elevation in levels of ESR1 and PGR compared to breast cancers with significantly low levels of Cav3.2 (1st quartile) (Fig. 8b). A significant correlation between ERBB2 was also observed, however, only minor compared to ESR1 and PGR (Fig. 8b). To define a potential role for Cav3.2 in the expression of ESR1 and PGR, we analysed the expression levels of ESR1, FOXA1, PGR and TFF1 in SKBR3 cells with induced overexpression of Cav3.2 (EGFP Cav3.2 SKBR3 cells). These results indicated that Cav3.2 is not a regulator of the expression of ESR1 and PGR or the luminal markers FOXA1 and TFF1 (Fig. 8c).
High CACNA1H (Cav3.2) expression levels in ER-positive (ER+) breast cancer are associated with poor prognosis, whereas in HER2-positive (HER2+) breast cancer patients it is associated with better responses to chemotherapy (CT)
Given the association between Cav3.2 and trastuzumab-resistance in HER2-positive breast cancer cell lines in vitro and increased expression within the luminal subtype, we explored the potential of Cav3.2 as a biomarker in predicting patient survival and/or outcomes with chemotherapy. The overall survival of patients from both METABRIC [37] and KM-Plotter [38] cohorts were analysed based on their expression level of CACNA1H (Cav3.2) (high/low) in each subtype and within each group for patients receiving chemotherapy (CT) (Fig. 9a). Our analysis identified that high levels of Cav3.2 are associated with poor survival in ER + tumours in METABRIC and KM-Plotter cohorts [Fig. 9a, b (i, ii)]. Note that beyond 20 years, survival of ER+ patients appear to be better for those with high expression, however, due to a small sample size remaining in the study at this time (9 patients) we were unable to explore this further. In all other subtypes, expression levels of Cav3.2 were not consistently stratified with overall survival in both cohorts (Fig. 9a). We then analysed the treatment response from the METABRIC and KM-Plotter cohorts towards chemotherapy in each subgroup. Interestingly, this analysis identified that HER2-positive patients receiving chemotherapy with tumours expressing high levels of CACNA1H demonstrated a better overall survival after chemotherapy, compared to patients with low levels of CACNA1H in both, METABRIC and KM-Plotter cohorts [Fig. 9a, b (iii, iv)]. In all other subtypes, Cav3.2 did not significantly affect survival with therapy in both cohorts (Fig. 9a).
Fig. 9.
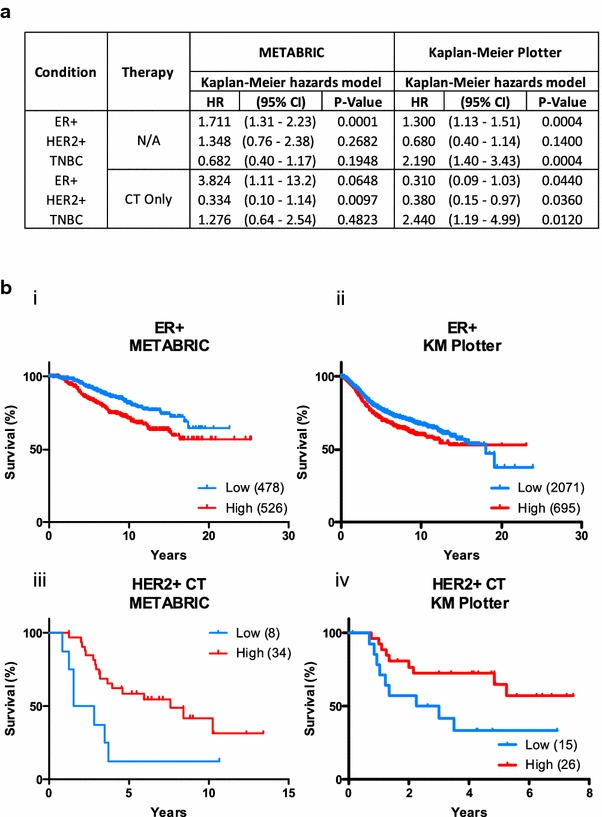
Cav3.2 expression stratifies the survival of breast cancer patients. The survival of patients from both the METABRIC and Kaplan–Meier Plotter cohorts were stratified based on the expression of Cav3.2 for each of the clinical subgroups and within those groups, in patients treated with chemotherapy (CT). METABRIC patients were stratified on overall survival (OS) and the KM-Plotter cohort on relapse free survival (RFS). Log-rank hazard ratios (HR) and corresponding P values are shown (a). Kaplan–Meier curves from data shown in panel A for ER+ patients (b i, ii) and those with HER2-positive tumours (HER+) treated with CT (b iii, iv). Tumours were stratified on the basis of their CACNA1H expression into low and high expressing groups, numbers indicated in brackets
Discussion
Our work explores alterations in the expression of calcium channels in the context of trastuzumab-resistance and therapeutic response. The work presented here demonstrated that levels of Cav3.2, a voltage gated T-type calcium channel, are elevated in acquired and intrinsic SKBR3 breast cancer cell line models of trastuzumab-resistance. Whilst we found no evidence for a direct role of Cav3.2 in driving or reversing trastuzumab-resistance, our data suggest that Cav3.2 may be an informative prognosis marker in ER+ breast cancer patients for overall survival and in HER2-positive breast cancer patients with chemotherapy.
In vitro models for trastuzumab-resistance include cell lines representing intrinsic resistance such as those established from the breast cancers of patients that do not respond to trastuzumab therapy despite HER2 overexpression [46], and those that represent acquired resistance, established through continuous culture of HER2-positive breast cancer cell lines in the presence of trastuzumab [31, 47]. Intrinsic resistance and acquired resistance may be mediated through different mechanisms [10]. In this study we developed a model system derived from the same cell line, suitable for assessing the mechanistic pathways that may be responsible for spontaneous (intrinsic) and acquired resistance derived from the same cell line. Intrinsic trastuzumab-resistant SKBR3 cell lines were established during the production of age-matched controls, which was not unexpected as continuous culturing can have a major impact on cellular phenotypes [48, 49].
The dysregulation of calcium homeostasis and the expression levels of specific calcium channels and pumps can be a feature of tumour progression [26, 50, 51] and is associated with the acquisition of resistance to adriamycin, the IGF-1 receptor inhibitor ganitumab and other agents [29, 52]. Our assessment of a wide panel of calcium pumps, channels and channel modulators identified an upregulation of the voltage gated Ca2+ channel Cav3.2 in three of four trastuzumab-resistant SKBR3 cell lines. Consistent with the variety of pathways associated with trastuzumab resistance [10, 16, 18], Cav3.2 was not elevated in all of the resistant cell lines, with elevated Cav3.2 absent from RT2 cells. Cav3.2 is a T-type voltage-gated calcium channel [53] generally expressed in cells of the heart, brain and liver and some tumours [53, 54]. High expression levels of T-type channels are associated with increased proliferation in breast cancer cells [55] and tumour progression in prostate cancer cells [56]. Cav3.2 is also proposed as an oncogene candidate in T cell leukaemia [57]. A higher level of Cav3.2 was also identified in the basal-like, HER2-positive, intrinsically trastuzumab-resistant cell line HCC1569. Intrinsic resistance to trastuzumab is associated with expression of basal markers [58]. Thus Cav3.2 expression might be a feature of breast cancers that are trastuzumab-resistant and also express basal markers. Our studies also demonstrated that although Cav3.2 may be a marker for activation of some trastuzumab-resistance pathways, inhibition of Cav3.2 is unable to restore trastuzumab-sensitivity in SKBR3 cell lines with acquired or intrinsic trastuzumab-resistance. Cav3.2 inhibitors therefore do not appear to represent an effective way to overcome trastuzumab-resistance in HER2-positive breast tumours. Although induced overexpression did not promote expression of resistance markers, future studies using T-type Ca2+ channel activators when available should assess the effect of these agents on the expression of resistance markers and trastuzumab resistance, analogous to studies which have evaluated the effects of pharmacological activation of L-type Ca2+ channels with (S)-(-)-Bay K 8644 [59, 60]. Only the acquired trastuzumab-resistant cell lines RT1 and RT2 showed a different calcium response after ATP stimulation compared to trastuzumab-sensitive controls, resulting in a delayed recovery. Although the response to ATP showed only minor alterations between sensitive and resistant cell lines and was not observed in all resistant cell lines, Cav3.2 might be involved in activation of transcription factors as previously shown for other voltage gated calcium channels [39]. However, our studies identified that Cav3.2 is not a driver of basal, epithelial to mesenchymal (EMT) transition or markers associated with resistance in SKBR3 cells. Cav3.2 mRNA was highly expressed in some luminal-like breast cancer cell lines (MCF-7 and T47D) compared to basal breast cancer cell lines that lack HER2 amplification, and was undetectable in cell lines derived from non-malignant breast tissue. The relationship between Cav3.2 and the luminal subtype was also observed in clinical breast cancer samples, with significantly higher Cav3.2 levels in luminal A and B subtypes compared to basal breast cancers. Although not a driver for ESR1 and PGR levels in SKBR3 cells, there was a strong correlation between Cav3.2 and levels of these hormone receptors in breast cancers. Analyses of clinical data showed that high levels of Cav3.2 were associated with poor prognosis in ER+ breast cancer patients. Surprisingly, our studies identified that high levels of Cav3.2 was associated with a better response to chemotherapy in patients with HER2-positive breast cancers.
The apparent contradiction of Cav3.2 mRNA levels being negatively associated with survival in ER+ cancers and positively associated with survival with chemotherapy in HER-positive breast cancers, may be reflective not only of potential differential contribution of calcium signalling in different breast cancer subtypes but also in the diversity of the Ca2+-signal. Some aspects of calcium signalling could be quite different between HER2+ and ER+ breast cancers and this could contribute to differences in the association between Cav3.2 levels and survival, for example Orai3-mediated Ca2+-influx is Ca2+-store dependent in ER positive but not ER negative breast cancer cells lines [61]. Ca2+-influx can promote cellular proliferation or be an inducer of cell death [50]. In the context of the association between high Cav3.2 levels and poor prognosis in ER+ breast cancers, Cav3.2-mediated constitutive Ca2+-influx contributes to enhanced proliferation in the LNCaP prostate cancer cell line [62]. Whereas the association between high levels of Cav3.2 and better outcomes with chemotherapy in HER2-positive breast cancer may be related to important role of Ca2+-increases during cell death [63]. Although not yet studied in HER2-positive breast cancer cell lines, the T-type Ca2+-channel Cav3.1 is a key mechanism by which cyclophosphamide induces apoptosis in luminal MCF-7 breast cancer cells [64] and Cav3.2 is essential in the inhibitory effects of epigallocatechin-3-gallate on the viability of MCF-7 breast cancer cells [65].
Conclusion
Our data suggest that at the clinical level, Cav3.2 may be similarly important in the responses to chemotherapy in HER2-positive breast cancers, however, further studies are required. In summary, these studies have identified enhanced expression of Cav3.2 as a feature of trastuzumab-resistant breast cancer cells, however, Cav3.2 does not seem to be a driver of trastuzumab-resistance. Further studies are now required to elucidate the potential differential roles of Cav3.2 in different breast cancer subtypes and its utility as a potential biomarker of prognosis and therapeutic responsiveness in patients with ER + and HER2-positive breast cancers respectively.
Authors’ contributions
EP contributed to study design, performed experiments, conducted data analysis and contributed to the writing of the manuscript. EK performed experiments, contributed to experimental design and wrote the manuscript. MJGM, KTDSY, JSO performed experiments and contributed to data analysis. MAB, FS, AAP and SRT contributed to study design, data analysis and the writing of the manuscript. GM designed experiments, contributed to the data analysis and the writing of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This study was supported by the National Health and Medical Research Council (NHMRC, Grant: 1079672) of Australia and The University of Queensland. EP was funded by a UQ Centennial Scholarship and International Postgraduate Research Scholarship. GM was supported by the Mater Foundation. The Translational Research Institute is supported by a grant from the Australian Government. This study makes use of data generated by the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC). Funding for the project was provided by Cancer Research UK and the British Columbia Cancer Agency Branch [37].
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- CT
chemotherapy
- EMT
epithelial to mesenchymal transition
- ER+
oestrogen receptor positive
- HER2
human epidermal growth factor 2
- ROC
receiver operator curve
- RSEM
RNA seq by expectation maximization
- RT-qPCR
real time quantitative polymerase chain reaction
- TCGA
the cancer genome atlas network
- TNBC
triple negative breast cancer
Additional file
10.1186/s12935-016-0299-0 Gene expression assays used in these studies to assess relative mRNA levels of target genes.
Footnotes
Elena Pera and Elke Kaemmerer contributed equally to this work
Contributor Information
Elena Pera, Email: epera@me.com.
Elke Kaemmerer, Email: e.kaemmerer@uq.edu.au.
Michael J. G. Milevskiy, Email: michael.milevskiy@uqconnect.edu.au
Kunsala T. D. S. Yapa, Email: k.yapa@uq.edu.au
Jake S. O’Donnell, Email: jake.odonnell@uq.edu.au
Melissa A. Brown, Email: melissa.brown@uq.edu.au
Fiona Simpson, Email: f.simpson@uq.edu.au.
Amelia A. Peters, Email: a.peters1@uq.edu.au
Sarah J. Roberts-Thomson, Email: sarahrt@uq.edu.au
Gregory R. Monteith, Email: gregm@uq.edu.au
References
- 1.Ross JS, Slodkowska EA, Symmans WF, Pusztai L, Ravdin PM, Hortobagyi GN. The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist. 2009;14:320–368. doi: 10.1634/theoncologist.2008-0230. [DOI] [PubMed] [Google Scholar]
- 2.Dawood S, Broglio K, Buzdar AU, Hortobagyi GN, Giordano SH. Prognosis of women with metastatic breast cancer by HER2 status and trastuzumab treatment: an institutional-based review. J Clin Oncol. 2010;28:92–98. doi: 10.1200/JCO.2008.19.9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nahta R, Esteva FJ. Trastuzumab: triumphs and tribulations. Oncogene. 2007;26:3637–3643. doi: 10.1038/sj.onc.1210379. [DOI] [PubMed] [Google Scholar]
- 4.Chung A, Cui X, Audeh W, Giuliano A. Current status of anti-human epidermal growth factor receptor 2 therapies: predicting and overcoming herceptin resistance. Clin Breast Cancer. 2013;13:223–232. doi: 10.1016/j.clbc.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nahta R, Esteva FJ. HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Res. 2006;8:215. doi: 10.1186/bcr1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolff AC, Hammond MEH, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, McShane LM, Paik S, Pegram MD, Perez EA, Press MF, Rhodes A, Sturgeon C, Taube SE, Tubbs R, Vance GH, van de Vijver M, Wheeler TM, Hayes DF. American society of clinical oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med. 2007;131:18–43. doi: 10.5858/2007-131-18-ASOCCO. [DOI] [PubMed] [Google Scholar]
- 7.Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–127. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 8.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 9.Oxnard GR, Arcila ME, Chmielecki J, Ladanyi M, Vincent A, Pao W. New strategies in overcoming acquired resistance to EGFR tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17:5530–5537. doi: 10.1158/1078-0432.CCR-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. 2012;17:1–16. doi: 10.1615/CritRevOncog.v17.i1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia W, Liu L-H, Ho P, Spector NL. Truncated ErbB2 receptor (p95ErbB2) is regulated by heregulin through heterodimer formation with ErbB3 yet remains sensitive to the dual EGFR/ErbB2 kinase inhibitor GW572016. Oncogene. 2004;23:646–653. doi: 10.1038/sj.onc.1207166. [DOI] [PubMed] [Google Scholar]
- 12.Nady P, Friedlander E, Tanner M, Kapanen AI, Carraway KL, Isola J, Jovin TM. Decreased accessibility and lack of activation of ErbB2 in JIMT-1; a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005;65:473–482. [PubMed] [Google Scholar]
- 13.Pályi-Krekk Z, Barok M, Isola J, Tammi M, Szöllosi J, Nagy P. Hyaluronan-induced masking of ErbB2 and CD44-enhanced trastuzumab internalisation in trastuzumab resistant breast cancer. Eur J Cancer. 2007;43:2423–2433. doi: 10.1016/j.ejca.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 14.Wehrman TS, Raab WJ, Casipit CL, Doyonnas R, Pomerantz JH, Blau HM. A system for quantifying dynamic protein interactions defines a role for Herceptin in modulating ErbB2 interactions. PNAS. 2006;103:19063–19068. doi: 10.1073/pnas.0605218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nahta R, Yuan LXH, Bing Z, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65:11118–11128. doi: 10.1158/0008-5472.CAN-04-3841. [DOI] [PubMed] [Google Scholar]
- 16.Browne BC, Crown J, Venkatesan N, Duffy MJ, Clynes M, Slamon D, O’Donovan N. Inhibition of IGF1R activity enhances response to trastuzumab in HER-2-positive breast cancer cells. Ann Oncol. 2011;22:68–73. doi: 10.1093/annonc/mdq349. [DOI] [PubMed] [Google Scholar]
- 17.Gallardo A, Lerma E, Escuin D, Tibau A, Muñoz J, Ojeda B, Barnadas A, Adrover E, Sánchez-Tejada L, Giner D, Ortiz-Martínez F, Peiró G. Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. Br J Cancer. 2012;106:1367–1373. doi: 10.1038/bjc.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 19.Clark AS, West K, Streicher S, Dennis PA. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther. 2002;1:707–717. [PubMed] [Google Scholar]
- 20.Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt is required for antibody-mediated Effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002;62:4132–4141. [PubMed] [Google Scholar]
- 21.Du C, Yi X, Liu W, Han T, Liu Z, Ding Z, Zheng Z, Piao Y, Yuan J, Han Y, Xie M, Xie X. MTDH mediates trastuzumab resistance in HER2 positive breast cancer by decreasing PTEN expression through an NFκB-dependent pathway. BMC Cancer. 2014;14:869. doi: 10.1186/1471-2407-14-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Azimi I, Roberts-Thomson SJ, Monteith GR. Calcium influx pathways in breast cancer: opportunities for pharmacological intervention. Br J Pharmacol. 2014;171:945–960. doi: 10.1111/bph.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peters AA, Simpson PT, Bassett JJ, Lee JM, DaSilva L, Reid LE, Song S, Parat M-O, Lakhani SR, Kenny PA, Roberts-Thomson SJ, Monteith GR. Calcium channel TRPV6 as a potential therapeutic target in estrogen receptor-negative breast cancer. Mol Cancer Ther. 2012;11:2158–2168. doi: 10.1158/1535-7163.MCT-11-0965. [DOI] [PubMed] [Google Scholar]
- 24.McAndrew D, Grice DM, Peters AA, Davis FM, Stewart T, Rice M, Smart CE, Brown MA, Kenny PA, Roberts-Thomson SJ, Monteith GR. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol Cancer Ther. 2011;10:448–460. doi: 10.1158/1535-7163.MCT-10-0923. [DOI] [PubMed] [Google Scholar]
- 25.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 26.Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer. 2011;11:609–618. doi: 10.1038/nrc3105. [DOI] [PubMed] [Google Scholar]
- 27.Monteith GR, Davis FM, Roberts-Thomson SJ. Calcium channels and pumps in cancer: changes and consequences. J Biol Chem. 2012;287:31666–31673. doi: 10.1074/jbc.R112.343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15:124–134. doi: 10.1016/j.ccr.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 29.Ma X, Cai Y, He D, Zou C, Zhang P, Lo CY, Xu Z, Chan FL, Yu S, Chen Y, Zhu R, Lei J, Jin J, Yao X. Transient receptor potential channel TRPC5 is essential for P-glycoprotein induction in drug-resistant cancer cells. PNAS. 2012;109:16282–16287. doi: 10.1073/pnas.1202989109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Azimi I, Beilby H, Davis FM, Marcial DL, Kenny PA, Thompson EW, Roberts-Thomson SJ, Monteith GR. Altered purinergic receptor-Ca2+ signaling associated with hypoxia-induced epithelial-mesenchymal transition in breast cancer cells. Mol Oncol. 2015:1–13. [DOI] [PMC free article] [PubMed]
- 31.Nahta R, Takahashi T, Ueno NT. P27 kip1 down-regulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res. 2004;64:3981–3986. doi: 10.1158/0008-5472.CAN-03-3900. [DOI] [PubMed] [Google Scholar]
- 32.Suchanek KM, May FJ, Robinson JA, Lee WJ, Holman NA, Monteith GR, Roberts-Thomson SJ. Peroxisome proliferator-activated receptor alpha in the human breast cancer cell lines MCF-7 and MDA-MB-231. Mol Carcinog. 2002;34:165–171. doi: 10.1002/mc.10061. [DOI] [PubMed] [Google Scholar]
- 33.Bertolesi GE, Shi C, Elbaum L, Jollimore C, Rozenberg G, Barnes S, Kelly MEM. The Ca2+ channel antagonists mibefradil and pimozide inhibit cell growth via Different cytotoxic mechanisms. Mol Pharmacol. 2002;62:210–219. doi: 10.1124/mol.62.2.210. [DOI] [PubMed] [Google Scholar]
- 34.Xiang Z, Thompson AD, Brogan JT, Schulte ML, Melancon BJ, Mi D, Lewis LM, Zou B, Yang L, Morrison R, Santomango T, Byers F, Brewer K, Aldrich JS, Yu H, Dawson ES, Li M, McManus O, Jones CK, Daniels JS, Hopkins CR, Xie XS, Conn PJ, Weaver CD, Lindsley CW. The discovery and characterization of ML218: a novel, centrally active T-type calcium channel inhibitor with robust effects in STN neurons and in a rodent model of Parkinson’s disease. ACS Chem Neurosci. 2011;2:730–742. doi: 10.1021/cn200090z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Network Cancer Genome Atlas. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li B, Dewey CN: RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12. [DOI] [PMC free article] [PubMed]
- 37.Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, Langerød A, Green A, Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Purushotham A, Børresen-Dale A-L, Brenton JD, Tavaré S, Caldas C, et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Györffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123:725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- 39.Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW. CaV1 and CaV2 channels engage distinct modes of Ca2+ signaling to control CREB-dependent gene expression. Cell. 2012;149:1112–1124. doi: 10.1016/j.cell.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin-Castillo B, Oliveras-Ferraros C, Vazquez-Martin A, Cufí S, Moreno JM, Corominas-Faja B, Urruticoechea A, Martín ÁG, López-Bonet E, Menendez JA. Basal/HER2 breast carcinomas integrating molecular taxonomy with cancer stem cell dynamics to predict primary resistance to trastuzumab (Herceptin) Cell Cycle. 2013;12:225–245. doi: 10.4161/cc.23274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oliveras-Ferraros C, Corominas-Faja B, Vazquez-Martin SA, Martin-Castillo B, Iglesias JM, López-Bonet E, Martin ÁG, Menendez JA. Epithelial-to-mesenchymal transition (EMT) confers primary resistance to trastuzumab (Herceptin) Cell Cycle. 2012;11:4020–4032. doi: 10.4161/cc.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carr JR, Park HJ, Wang Z, Kiefer MM, Raychaudhuri P. FoxM1 mediates resistance to herceptin and paclitaxel. Cancer Res. 2010;70:5054–5063. doi: 10.1158/0008-5472.CAN-10-0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chu S-H, Liu Y-W, Zhang L, Liu B, Li L, Shi J-Z. Regulation of survival and chemoresistance by HSP90AA1 in ovarian cancer SKOV3 cells. Mol Biol Rep. 2013;40:1–6. doi: 10.1007/s11033-012-1930-3. [DOI] [PubMed] [Google Scholar]
- 44.De Luca A, D’Alessio A, Gallo M, Maiello MR, Bode AM, Normanno N. Src and CXCR4 are involved in the invasiveness of breast cancer cells with acquired resistance to lapatinib. Cell Cycle. 2014;13:148–156. doi: 10.4161/cc.26899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Brien NA, Browne BC, Chow L, Wang Y, Ginther C, Arboleda J, Duffy MJ, Crown J, O’Donovan N, Slamon DJ. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol Cancer Ther. 2010;9:1489–1502. doi: 10.1158/1535-7163.MCT-09-1171. [DOI] [PubMed] [Google Scholar]
- 46.Tanner M, Kapanen AI, Junttila T, Raheem O, Grenman S, Elo J, Elenius K, Isola J. Characterization of a novel cell line established from a patient with herceptin-resistant breast cancer. Mol Cancer Ther. 2004;3:1585–1592. [PubMed] [Google Scholar]
- 47.Wang Y-C, Morrison G, Gillihan R, Guo J, Ward RM, Fu X, Botero MF, Healy N a, Hilsenbeck SG, Phillips G, Chamness GC, Rimawi MF, Osborne CK, Schiff R. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2- positive breast cancers – role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011, 13. [DOI] [PMC free article] [PubMed]
- 48.Masters JR, Stacey GN. Changing medium and passaging cell lines. Nat Protoc. 2007;2:2276–2284. doi: 10.1038/nprot.2007.319. [DOI] [PubMed] [Google Scholar]
- 49.Sambuy Y, De Angelis I, Ranaldi G, Scarino ML, Stammati A, Zucco F. The Caco-2 cell line as a model of the intestinal barrier: Influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol Toxicol. 2005;21:1–26. doi: 10.1007/s10565-005-0085-6. [DOI] [PubMed] [Google Scholar]
- 50.Monteith GR, McAndrew D, Faddy HM, Roberts-Thomson SJ. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer. 2007;7:519–530. doi: 10.1038/nrc2171. [DOI] [PubMed] [Google Scholar]
- 51.VanHouten J, Sullivan C, Bazinet C, Ryoo T, Camp R, Rimm DL, Chung G, Wysolmerski J. PMCA2 regulates apoptosis during mammary gland involution and predicts outcome in breast cancer. PNAS. 2010;107:11405–11410. doi: 10.1073/pnas.0911186107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fahrenholtz CD, Greene AM, Beltran PJ, Burnstein KL. A novel calcium-dependent mechanism of acquired resistance to IGF-1 receptor inhibition in prostate cancer cells. Oncotarget. 2014;5:9007–9021. doi: 10.18632/oncotarget.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Panner A, Wurster RD. T-type calcium channels and tumor proliferation. Cell Calcium. 2006;40:253–259. doi: 10.1016/j.ceca.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 54.Taylor JT, Zeng X-B, Pottle JE, Lee K, Wang AR, Yi SG, Scruggs JAS, Sikka SS, Li M. Calcium signaling and T-type calcium channels in cancer cell cycling. World J Gastroenterol. 2008;14:4984–4991. doi: 10.3748/wjg.14.4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taylor JT, Huang L, Pottle JE, Liu K, Yang Y, Zeng X, Keyser BM, Agrawal KC, Hansen JB, Li M. Selective blockade of T-type Ca2+ channels suppresses human breast cancer cell proliferation. Cancer Lett. 2008;267:116–124. doi: 10.1016/j.canlet.2008.03.032. [DOI] [PubMed] [Google Scholar]
- 56.Gackière F, Bidaux G, Delcourt P, Van Coppenolle F, Katsogiannou M, Dewailly E, Bavencoffe A, Van Chuoï-Mariot MT, Mauroy B, Prevarskaya N, Mariot P. CaV3.2 T-type calcium channels are involved in calcium-dependent secretion of neuroendocrine prostate cancer cells. J Biol Chem. 2008;283:10162–10173. doi: 10.1074/jbc.M707159200. [DOI] [PubMed] [Google Scholar]
- 57.Yoshida M, Nosaka K, Yasunaga J, Nishikata I, Kazuhiro M, Matsuoka M. Aberrant expression of the MEL1S gene identified in association with hypomethylation in adult T-cell leukemia cells. Blood. 2004;103:2753–2760. doi: 10.1182/blood-2003-07-2482. [DOI] [PubMed] [Google Scholar]
- 58.Oliveras-Ferraros C, Vazquez-Martin A, Martin-Castillo B, Martinez-Perez MC, Cufi S, del Barco S, Bernado L, Brunet J, Lopez-Bonet E, Menendez JA. Pathway-focused proteomic signatures in HER2-overexpressing breast cancer with a basal-like phenotyp: new insights into de novo resistance to trastuzumab (Herceptin) Int J Oncol. 2010;37:669–678. doi: 10.3892/ijo_00000716. [DOI] [PubMed] [Google Scholar]
- 59.Sekiguchi-Tonosaki M, Obata M, Haruki A, Himi T, Kosaka J. Acetylcholine induces Ca2+ signaling in chicken retinal pigmented epithelial cells during dedifferentiation. Am J Physiol Cell Physiol. 2009;296:C1195–C1206. doi: 10.1152/ajpcell.00423.2008. [DOI] [PubMed] [Google Scholar]
- 60.Viola HM, Davies SM, Filipovska A, Hool LC. L-type Ca(2+) channel contributes to alterations in mitochondrial calcium handling in the mdx ventricular myocyte. Am J Physiol Hear Circ Physiol. 2013;304:H767–H775. doi: 10.1152/ajpheart.00700.2012. [DOI] [PubMed] [Google Scholar]
- 61.Motiani RK, Abdullaev IF, Trebak M. A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J Biol Chem. 2010;285:19173–19183. doi: 10.1074/jbc.M110.102582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gackière F, Warnier M, Katsogiannou M, Derouiche S, Delcourt P, Dewailly E, Slomianny C, Humez S, Prevarskaya N, Roudbaraki M, Mariot P. Functional coupling between large-conductance potassium channels and Cav3.2 voltage-dependent calcium channels participates in prostate cancer cell growth. Biol Open. 2013;2:941–951. doi: 10.1242/bio.20135215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rizzuto R, Pinton P, Ferrari D, Chami M, Szabadkai G, Magalhães PJ, Di Virgilio F, Pozzan T. Calcium and apoptosis: facts and hypotheses. Oncogene. 2003;22:8619–8627. doi: 10.1038/sj.onc.1207105. [DOI] [PubMed] [Google Scholar]
- 64.Ohkubo T, Jun Y. T-type voltage-activated calcium channel Cav3.1, but not Cav3.2, is involved in the inhibition of proliferation and apoptosis in MCF-7 human breast cancer cells. Int J Oncol. 2012;41:267–275. doi: 10.3892/ijo.2012.1422. [DOI] [PubMed] [Google Scholar]
- 65.Ranzato E, Magnelli V, Martinotti S, Waheed Z, Cain SM, Snutch TP, Marchetti C, Burlando B. Epigallocatechin-3-gallate elicits Ca2+ spike in MCF-7 breast cancer cells: essential role of Cav3.2 channels. Cell Calcium. 2014;56:285–295. doi: 10.1016/j.ceca.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 66.Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein Lønning P, Børresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. PNAS. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lønning PE, Børresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]