

Abstract
Src homology 2 domains are interaction modules dedicated to the recognition of phosphotyrosine sites incorporated in numerous proteins found in intracellular signaling pathways. Here we provide for the first time structural insight into the dimerization of Fyn SH2 both in solution and in crystalline conditions, providing novel crystal structures of both the dimer and peptide‐bound structures of Fyn SH2. Using nuclear magnetic resonance chemical shift analysis, we show how the peptide is able to eradicate the dimerization, leading to monomeric SH2 in its bound state. Furthermore, we show that Fyn SH2's dimer form differs from other SH2 dimers reported earlier. Interestingly, the Fyn dimer can be used to construct a completed dimer model of Fyn without any steric clashes. Together these results extend our understanding of SH2 dimerization, giving structural details, on one hand, and suggesting a possible physiological relevance of such behavior, on the other hand.
Keywords: SH2, homodimer, crystal structures, NMR analysis, Fyn
Abbreviations
- CD
far‐UV circular dichroism
- ITC
isothermal titration calorimetry
- SH2
Src homology 2
- NMR
nuclear magnetic resonance
- PDB
protein database
- PLC‐γ1C
phospholipase Cγ1
- pTyr
phosphotyrosine
- SH3
Src homology 3
Introduction
In the complex cellular environment, effective signaling is often mediated through the association between peptides and modular binding domains. The Src homology 2 (SH2) domain is a classic example of such domains. Belonging to more than 100 different human proteins, it is considered to be the largest domain family associated with phosphotyrosine‐based recognition processes.1, 2 While it is well documented that SH2 domains perform their binding function in a monomeric configuration (reviewed in3, 4), multiple reports have also highlighted the tendency of the isolated forms of these domains to dimerize.5, 6, 7, 8 In a recent systematic analysis9 it was shown that such SH2 intertwined homomers belong to the biggest category of intertwined of homo‐oligomeric proteins currently available in the PDB: ∼72% of the structures used in that study belong to the so‐called S‐type swaps where either terminal chain segments or nonterminal ones are exchanged. This group also includes 3D domain swapped structures,10, 11, 12, 13, 14, 15 wherein hinge regions change conformation, replacing intra‐molecular interactions within the monomer by inter‐molecular interactions in the dimer.
As the intertwining process alters the SH2 ligand interaction surface, interfering as a consequence with target binding,5, 7 the SH2 homodimers belong to more general group of intertwined dimers and not to the category of 3D domain swapped constructs. For instance, NMR backbone dynamics of the C‐terminal SH2 domain of phospholipase C‐γ1 (PLC‐γ1C) suggested that the free domain is in a monomer‐dimer equilibrium.16, 17 This domain is characterized by an aggregation propensity starting at concentrations of 2–4 mg/mL. Similar NMR dynamical experiments, analytical ultracentrifugation, and dynamic light scattering revealed this associative phenomenon in case of SH2 domain of human tyrosine kinase Fyn at 50 µM (corresponding to ± 0.7 mg/mL).18 Self‐aggregation tendencies were also observed in SH2 domains of Hck,19 SAP,20 and the p85α subunit of PI3K.21 In the case of the Grb2 adaptor protein, the SH2 dimerization modifies the affinity of ligand binding.7 Interestingly, the reported SH2 dimers5, 6, 8 exhibit different structural features in their aggregated forms, which may have distinct biological consequences.
All these findings, together with the pronounced aggregation tendency observed for the Fyn SH2 domain,18, 22 lead us to further investigate the potential significance of its dimerization behavior and its implication in modifying the binding capabilities of these widely known phosphotyrosine‐binding modules. By solving the crystal structures of both the intertwined dimer and the monomer in complex with a high affinity phosphotyrosine‐containing peptide, we characterize in detail the association process. By comparing these structural data and making use of solution NMR we provide detailed insight into the interaction between the Fyn SH2 dimer and the high‐affinity phosphotyrosine‐containing peptide. Size‐exclusion chromatography, circular dichroism, and NMR experiments demonstrate the presence in solution of Fyn SH2 dimer, while adding an excess of high affinity peptide results in the depletion of the dimer in favor of the monomer. Our results broaden the understanding on the ligand binding capabilities of Fyn SH2 domain and its dimerization tendency.
Results and Discussion
Human fyn SH2 domain is prone to aggregate
While there is an increasing body of literature on SH2 domains, only a fraction of the studies require high concentrations and purity levels of these protein domains. A global profiling study using all the known SH2 domains showed that about half of these protein modules are highly soluble.23 However, when produced in elevated concentrations, typically required for crystallographic and NMR studies, SH2 domains show tendency of self‐association.16, 18, 19, 20, 21
To investigate the aggregation tendency of the Fyn SH2 domain, in a first stage, analytical gel filtration, and far‐UV circular dichroism (CD) were employed. The protein was purified by a combination of affinity and size exclusion chromatography techniques;24 the last separation step produced a mixture of dimer and monomer forms, in a ratio of 1 : 3, respectively. Figure 1(a) illustrates the final gel‐filtration along with the molecular‐weight standards. As can be seen from the gel‐filtration profile, the dimer and the monomer fractions can be efficiently separated. To further assess the Fyn SH2 association tendency, the monomer form was collected and concentrated to 1 mM. In this initial stage [Fig. 1(c)], the monomeric state was confirmed, but after 48‐h incubation at 25°C, the dimer‐monomer mixture with the same 1 : 3 ratio was observed in the sample [Fig. 1(d)]. Our results indicate a high self‐association tendency of this SH2 domain, a behavior that was also observed previously using analytical ultracentrifugation and dynamic light scattering experiments.18
Figure 1.
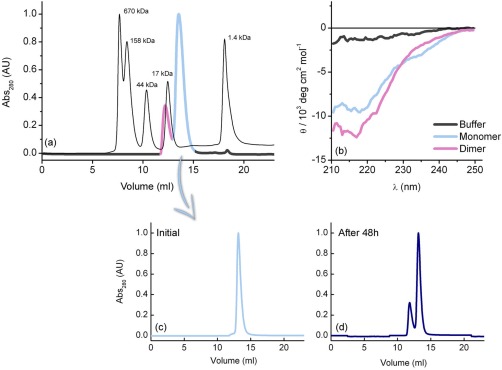
Solution studies of human Fyn SH2 domain. (a) Analytical gel‐filtration profile on a Superdex 75 HR column as the last purification step of the non‐His‐tagged Fyn SH2 domain. The molecular‐weight standards are shown in black, while the protein chromatogram is represented in purple (dimer fraction) and light blue (monomer fraction). (b) Far‐UV circular dichroism spectra of the monomer and the dimer fractions. The CD spectra are shown only for the 210–250 nm wavelength intervals, where there is no buffer interference. (c, d) Stability test of the Fyn SH2 monomer fraction performed using size exclusion chromatography. The monomer sample from (a) was concentrated to 1 mM and incubated at 25°C for different time intervals. Analytical gel‐filtration profiles were obtained by loading SH2 at 1 mM concentration on a Superdex 75 HR column.
Additionally, the far‐UV circular dichroism (CD) experiments for both Fyn SH2 fractions were recorded to scrutinize the possible differences in their secondary structures [Fig. 1(b)]. The observed spectra are typical for folded proteins and the profiles exhibited a minimum around 215–220 nm, which indicates the predominance of β‐sheets. Interestingly, the data show differences in the secondary structures of the two Fyn SH2 fractions. When compared with the monomer, the dimer state shows a more pronounced β‐sheet shape, highlighting that the Fyn SH2 monomer is folded and the dimerization process coincides with an increase in β‐strand content in the SH2 dimer.
Structural view into the intertwined Fyn SH2 dimer
To gain more insights into the structural organization of the self‐associated SH2 products, the crystal structure of the Fyn SH2 dimer is solved here (see Table 1). Expression, purification, and crystallization screens have been previously described.24 The Fyn SH2 crystal reveals two SH2 domains positioned in an intertwined arrangement. Each of the domains shows a partially conserved SH2 fold with most of the typical secondary structure elements, yet significant changes are localized within the βE‐ and βF‐strands and the EF loop [Fig. 2(a)]. Both SH2 domains belonging to the dimer display the extensive β‐sheet with a central position and the αA‐helix as known from the classical fold. The βE‐strand, EF loop and βF‐strand adopt an extended conformation [denoted βE‐EF‐βF in Fig. 2(a)], which results in an altered position of the αB‐helix and the BG loop. The canonical SH2 fold is restored by the αB‐helix of the symmetry‐related dimer [Fig. 2(b)]. Consequently, the classical SH2 fold is now defined by two chains: a core containing the central β‐sheet (residues Trp 149 ‐ Thr 215) from one chain and residues Thr 216 ‐ Lys 248 from the second polypeptide chain. In the new configuration of each chain, the N‐ and C‐terminal regions are located far apart (50 Å), in contrast with the classical fold where the two termini are separated by a distance of less than 10 Å. The buried surface area is 2200 Å2, 29% of the total surface per open monomer. This large interaction surface is assured by hydrophobic contacts and stabilized by an extensive hydrogen bond network. These bonds are located not only between the two extended βE‐EF‐βF regions, but also between the central β‐sheet, from one chain, and the C‐terminal region, of the complementary chain. The latter contacts assure the symmetry of the domain and the almost complete recovery of the canonical SH2 fold. In addition, the new extended β‐sheet formed by the dimerization process is in agreement with the CD data, suggesting an increase in this secondary structural content of the dimer fraction with respect to the monomer [Fig. 1(b)].
Table 1.
Data Collection and Refinement Statistics
| Fyn SH2:specific‐pY complex | Dimer of the Fyn‐SH2 domain | |
|---|---|---|
| Wavelength (Å) | 0.9334 | 0.9801 |
| Resolution range (Å) | 23.34 ‐ 1.45 (1.45 ‐ 1.40) | 34.92 ‐ 1.99 (2.06 ‐ 1.99) |
| Space group | P 43 21 2 | C 2 |
| Unit cell | 39.25 39.25 145.20 90.00 90.00 90.00 | 88.96 57.87 101.19 90.00 90.60 90.00 |
| Total reflections | 343996 | 126420 (17997) |
| Unique reflections | 23359 (2286) | 35049 (3365) |
| Multiplicity | 14.7 (13.3) | 3.6 (3.6) |
| Completeness (%) | 99.93 (99.96) | 98.66 (95.57) |
| Mean I/sigma(I) | 24.43 (7.73) | 7.7 (3.2) |
| Wilson B‐factor | 14.34 | 36.29 |
| R‐merge | 0.076 (0.361) | 0.081 (0.303) |
| R‐work | 0.1534 (0.2206) | 0.2064 (0.3069) |
| R‐free | 0.1876 (0.1946) | 0.2554 (0.3369) |
| Number of H atoms | 1234 | 2642 |
| Macromolecules | 1039 | 2440 |
| Ligands | 12 | 51 |
| Water | 183 | 151 |
| Protein residues | 120 | 308 |
| Average B‐factor | 23.10 | 55.40 |
| Macromolecules | 19.40 | 55.00 |
| Ligands | 46.50 | 64.90 |
| Solvent | 43.00 | 59.00 |
| RMS (Bonds) | 0.028 | 0.012 |
| RMS (Angles) | 1.50 | 1.41 |
| Ramachandran favored (%) | 99.0 | 97.7 |
| Ramachandran allowed (%) | 1.0 | 2.3 |
| Ramachandran outliers (%) | 0.0 | 0.0 |
| PDB ID | 4U1P | 4U17 |
Statistics for the highest‐resolution shell are shown in parentheses.
Figure 2.
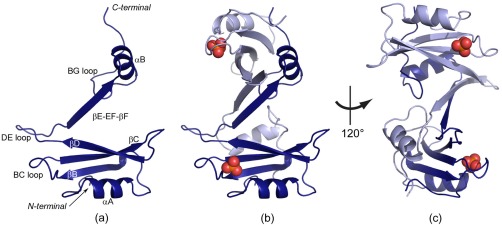
The Fyn SH2 dimer. (a) The monomer unit of the SH2‐dimer crystal structure with the main secondary structure elements labelled. The extended βE‐EF‐βF region and the αB‐helix adopt a different conformation than in the canonical SH2 fold. (b,c) The structure of the dimer in two orientations. The two monomer units are colored in light and dark blue, respectively. The cocrystallized phosphate is shown as spheres (phosphorus atoms are colored orange, oxygen atoms in red).
The phosphotyrosine (pTyr) binding pocket is located between the αA‐helix and the main β‐sheet. The structure of this binding pocket is not perturbed by the dimerization process. Even more, the residues forming the positively charged groove are accommodating a phosphate ion from the buffer. The second binding pocket, commonly named specificity pocket, is normally located between the EF and BG loops. In the intertwined dimer, the EF loop becomes a secondary structure element involved in the interactions with the symmetric unit and the BG loop is located far from its initial position. Even if the SH2 fold is almost reconstituted through the symmetric unit, the specificity pocket is completely altered by the dimerization. The new extended element βE‐EF‐βF is obstructing the formation of this second binding cavity, which will be analyzed in more detail in the following sections.
The crystal structure of Fyn SH2 in complex with the high‐affinity peptide
Next to the homodimer structure, the crystal structure of the Fyn SH2:specific‐pY complex is also solved (see Table 1). As previously described,24 the Fyn SH2:specific‐pY complex was crystallized at 20°C, using the Morpheus Screen Kit (Molecular Dimensions25). Visual inspection of the peptide−protein complex structure reveals the canonical SH2 fold [Fig. 3(a)] and the specific‐pY peptide binds according to the classical “two‐pronged‐plug” mechanism.26 A detailed view of the residues in the pTyr and specificity pockets that are involved in the interaction with the peptide is provided in Figures 3(b,c). The peptide pTyr side‐chain interacts with the SH2 pocket‐forming residues through electrostatic interactions at the phosphate moiety level and engages also in van der Waals contacts with the aromatic group.27 Figure 3(b) shows only the main interactions involving the phosphate moiety for reasons of clarity. The highly conserved Arg 176 (βB6) is located at the bottom of the pocket and is the main residue involved in the interaction with the phosphate moiety of the peptide. Besides the amino‐aromatic interactions with the pTyr ring, residues in the pTyr pocket also interact with the peptide residues located at pTyr‐1 and pTyr‐2.
Figure 3.
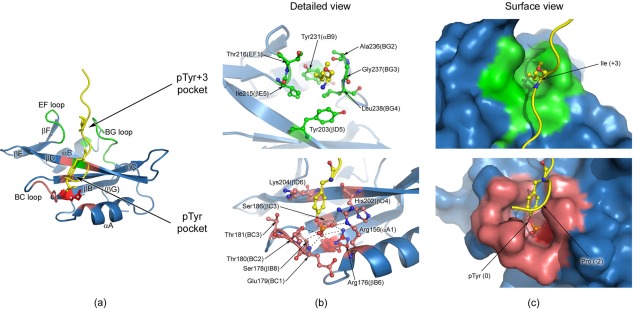
Human Fyn SH2:specific‐pY complex and detailed views of the two binding pockets. (a) The structure of the complex, with the regions forming the pTyr binding pocket colored in light red and the specificity pocket shown in green. The highly conserved Arg 176 (βB6) is depicted in red and represented in sticks. The specific‐pY peptide is colored in yellow and only the pTyr (0) and Ile (+3) residues are shown in sticks. (b) Detailed view of the two peptide binding pockets, where the main residues involved in interactions are represented in sticks. For the phosphate moiety the schematic directions of interaction are indicated by the dashed lines (bottom figure). Nitrogen atoms are coloured blue, oxygen atoms red, phosphorus atoms orange, and carbon atoms from the specific‐pY peptide yellow. The carbon atoms of the residues involved in the specificity pocket are shown in green (upper figure), while the ones from the pTyr pocket are depicted in light red (bottom figure). All the residues not directly involved in binding are coloured blue. The highly conserved Arg 176 (βB6) is colored red (bottom figure). (c) Surface view of the two pockets, where SH2 domain is shown in surface representation and the peptide is depicted as cartoon. The peptide interaction residues (pTyr and Ile+3) are illustrated as sticks. The structures in (b) and (c) have the same orientation.
The specificity pocket is neutral in charge, being formed by hydrophobic residues that contact the peptide at the position pTyr+3, which is the case for all SH2 domains in Src‐like kinases.28 The EF and BG loops, the main constituents of this pocket, play a major role in modifying the binding pocket surface, as well as in engaging the ligand. In case of the specific‐pY peptide, the Ile (+3) side‐chain is almost completely buried in the specificity pocket [Fig. 3(b), top]. The main residues forming this second pocket are shown in Figure 3(b) (top panel). The central position of Gly 237 (BG3) in the BG loop is an important factor for determining the loop shape and consequently of the specificity pocket architecture.29
Next to the interactions in both binding pockets, the specific‐pY peptide is also involved in weak contacts with the Fyn SH2 surface, as was previously reported the for Src SH2 complex.29 The two glutamate residues [Glu (+1), Glu (+2)] located between the main anchoring points of the two pockets, play an important role in these surface interactions.
Although a large number of SH2 domain structures have been solved,1 the Fyn SH2:specific‐pY complex determined here is the first X‐ray structure of the human Fyn kinase in its peptide bound state. The complex is characterized by the same overall structure found in a typical SH2 domain.4 Additionally, the protein‐peptide interactions identified in our high‐resolution crystal structure are preserved in SH2 domains from closely related proteins.22, 29, 30 Of particular interest is the published NMR solution structure of the same complex (PDB code 1AOU22). A comparison of the Fyn:specific‐pY complexes obtained with X‐ray and NMR techniques, reveal a similar orientation of the main residues involved in key contacts of the two binding pockets.
Detailed analysis of the interaction between the Fyn SH2 domain and the specific‐pY peptide
Making use of NMR chemical shifts data,31 the perturbations determined by the specific‐pY peptide binding at the Fyn SH2 domain were scrutinized at the atomic level. At the protein level the peptide‐binding event promotes a series of changes or shifting's of conformations. Any modification in the local environment of an atom is translated in a chemical shift change. By analyzing the chemical shift values of the free and bound state, alterations in the domain structure can be detected. The chemical shift differences are illustrated in Figure 4 along with the threshold that allows one to distinguish between significant and insignificant changes.
Figure 4.
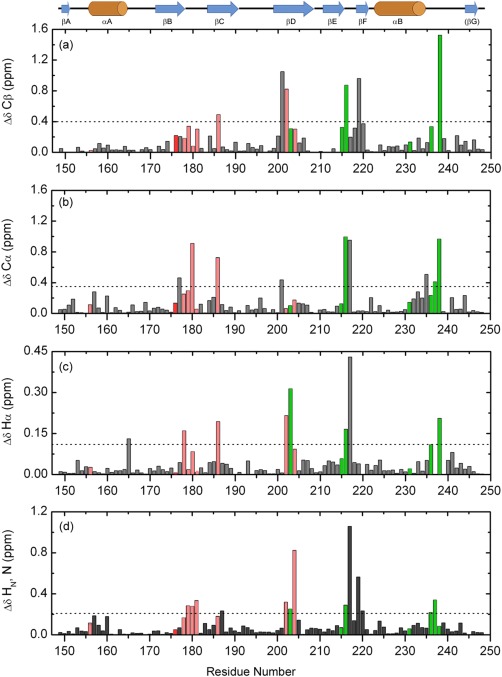
Chemical shift perturbations in Fyn SH2 domain upon specific‐pY peptide binding, in function of the residue number. The figure shows the absolute differences calculated for Cβ (a), Cα (b), Hα (c), and the combined backbone amides (d). The significance criteria are represented by the average chemical shift difference plus one standard deviation for each atom type, indicated by the dashed lines. The secondary structure elements of Fyn SH2 domain are depicted above. The residues involved in the specificity pocket are shown in green, while the ones from the pTyr pocket are depicted in light red. All the residues not directly involved in binding are colored gray. The highly conserved Arg 176 (βB6) is colored red.
The specific‐pY peptide binding induces significant chemical shift changes confined to three regions: the BC loop, the βD secondary structure element and the EF/BG loops. As observed in the crystal structure [Fig. 3(b,c), bottom panels], the residues from the BC loop are in close contact with the pTyr residue from the peptide and are characterized by important perturbations [see Fig. 3(b)]. Some amino acids from the central βD‐sheet are also part of the pTyr binding pocket and are involved in the interactions with the aromatic ring from the pTyr residue.
The clusters of chemical shift perturbations observed in the EF and BG loops confirm a binding event at the specificity site. Almost all the residues forming this second binding pocket experience significant changes upon peptide binding. Among these residues, Leu 238 (BG4) exhibits one of the largest chemical shift changes. Moreover, in our high‐resolution crystal structure of the complex and also in the existing NMR ensemble (PDB code 1AOU22), the side‐chain of Leu 238 is one of the main contact points with the Ile (+3) from the specific‐pY peptide. Our results and observations are consistent with the existing structures and confirm the binding of the specific‐pY peptide at both pockets.
Interestingly, no significant changes were detected for the two arginine residues directly involved in binding the pTyr. The highly conserved Arg 156 (αA1) and Arg 176 (βB6) are interacting with the peptide, but their backbone chemical environments are less affected by this interaction, as can be observed in Figure 4. This is probably due to the fact that the pTyr contacts determine changes at the level of the side‐chain atoms of the arginine residues and are not visible at the backbone level. This observation can also indicate a relatively rigid pTyr binding pocket.
Overall, the chemical shift perturbations upon specific‐pY peptide binding show changes at both interaction sites. A similar chemical shift perturbation pattern has been shown for the SH2 domain of the hematopoietic cellular kinase (Hck) in complex with the high affinity phosphorylated peptide.19 Additionally, the NMR results are completed by an isothermal titration calorimetry (ITC) study, which determined the binding affinity of the specific‐pY peptide for the Fyn SH2 domain (Fig. 5). The thermodynamic parameters obtained from the titration fit with a 1:1 stoichiometry characteristic of a single binding event. The data show a tight binding, characterized by a dissociation constant K d = 250 nM and a measured enthalpy <ΔH°> = −9.25 ± 0.1 kcal/mol. These results are in agreement with the previous experiments investigating the interaction of SH2 (originating in human Src kinase) with different binding peptides.26
Figure 5.
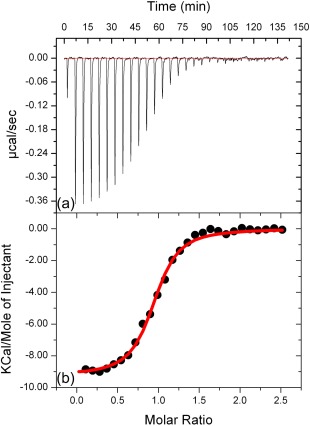
Isothermal calorimetric titrations for binding of the specific‐pY peptide to human Fyn SH2 domain. (a) The raw heat signal, the power output (microcalories per second) is represented as a function of time (minutes). (b) Integrated data obtained from the raw heat signal. The red line corresponds to the nonlinear least‐squares best fit to the data.
The dimerization distorts the Fyn SH2 peptide‐binding interface
To assess the impact of the Fyn SH2 dimerization on the binding site we compared the two crystal structures of the dimer/free state and monomer/specific‐pY peptide bound state (Fig. 6). While in the Fyn SH2:specific‐pY complex, the binding pockets are exposed and the peptide establishes interactions with both binding sites [Fig. 6(a)], in the Fyn SH2 dimer, the binding interface is partially distorted [Fig. 6(b,c)]. All the residues composing the pTyr binding pocket have similar orientations between the peptide‐free dimer and the pTyr‐bound Fyn SH2. The highly conserved Arg 176 (βB6), the main interaction point with the phosphate moiety of the peptide, is highlighted in red in Figure 6. On the other hand, the specificity‐binding pocket is altered. The SH2 domain from Fyn has the specificity pocket located at position pTyr+3, being formed by the EF and BG loops. In the dimer, the EF loop becomes structured and a new extended β‐strand is formed. Ile 215 (βE5), Thr 217 (EF2), and Ala 219 (βF1) are main interaction points in the intertwining, which makes them inaccessible for the ligand, and consequently unable to perform their normal function. Additionally, the new βE‐EF‐βF‐strand closes the space of the specificity pocket.
Figure 6.
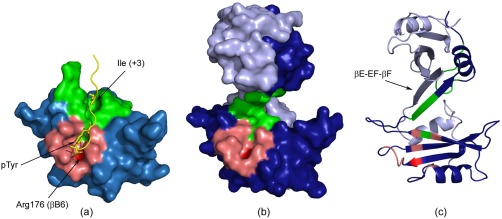
Human Fyn SH2 free and in complex with the specific‐pY peptide. (a) The crystal structure of the Fyn SH2:specific‐pY complex. The main regions forming the pTyr binding pocket are colored in light red, while the specificity pocket is shown in green. The highly conserved Arg 176 (βB6) is labelled and illustrated in red. The specific‐pY peptide is colored in yellow and only the pTyr (0) and Ile (+3) residues are shown in sticks. (b) Surface and (c) cartoon representations of the Fyn SH2 dimer. The two monomer units are illustrated in light and dark blue, while the two binding pockets are shown for only one monomer unit using the same color codes as in (a).
The ligand binding specificity of the Fyn SH2 domain is gained by the interactions at the second pocket, position pTyr+3.28 We hypothesized that it would be highly relevant to capture more insights about the dimerization and its consequences on the specificity pocket. We therefore used NMR and analyzed the dimer in presence and in absence of the high affinity (specific‐pY) peptide. Figure 7(a) shows the 1H‐15N HSQC spectra of both states, while the reported chemical shift assignments31 allow one to identify a significant number of additional signals in the free form. These signals, indicated by arrows in Figure 7(a), are doublings of an existing peak (in some cases) and are associated with the dimerization behavior of the SH2 domain. When adding an excess of specific‐pY peptide the doublings and additional NMR signals are not visible and bound state spectrum counts the expected number of signals as for a monomer state. These findings are in agreement with the gel filtration experiments, where a dimer‐monomer equilibrium was gained over time [Fig. 1(c,d)]. Additionally, we investigated the transverse relaxation (R 2) rates, which are informative for the size of the system.32 The overall reduction in the R 2 values is a good indication of the decrease in the mass of the system, while the R 2 values enhancement point to an increase in the mass. Monitoring the general trend of the R 2 values between the two Fyn SH2 states provided information on the system analyzed, as presented in Figure 7(b). The high transverse relaxation rates are shown for the free state, indicating the high mass of the system, in agreement with the dimer behavior. Note, the R 2 rates have been extracted only for the major peaks, attributed to the monomer state, but the results clearly indicate an equilibrium dimer‐monomer. Upon addition of the specific‐pY peptide to the solution containing the SH2 in the dimer form, a significant drop in the R 2 values is seen, suggesting a substantial decrease in mass.
Figure 7.
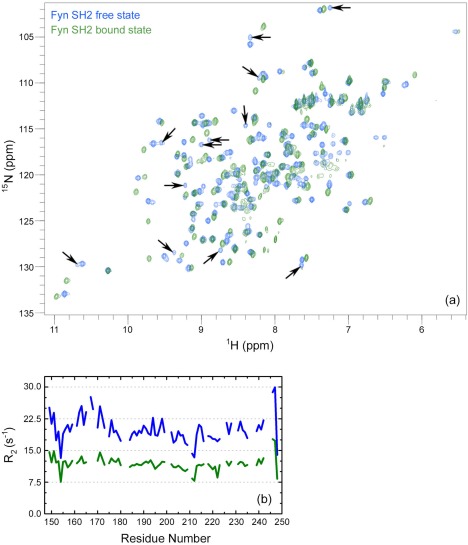
NMR studies of human Fyn SH2 domain (a) Overlay of the 800 MHz 1H‐15N HSQC spectra of Fyn SH2 in its free form (blue; dimeric state) and in complex with the specific‐pY peptide (green; monomeric state). The arrows indicate the doubling and/or additional peaks attributed to the dimeric state. (b) The Fyn SH2 transverse relaxation rates in function of the residue number for both states, maintaining the same coloring coding as in (a).
We interpret this important reduction in R 2 values as corresponding to the transition from the peptide‐free dimer to the specific‐pY complexed monomer population, indicating that the affinity of Fyn SH2 for the specific‐pY peptide is higher than the interactions driving the dimerization. The specific‐pY peptide binds Fyn SH2 domain with high affinity, engaging the SH2 domain at both binding sites, as clearly observed in our crystal structure of the Fyn SH2:specific‐pY complex. In case of the Fyn SH2 dimer only one pocket is available, as shown in Figure 6(b,c). We hypothesize that the peptide binds to the high affinity pocket via the phosphate moiety and also engages the residues in the specificity pocket. As the latter amino‐acids are the main interaction points of the extended βE‐EF‐βF‐strand which stabilize the structure, the dimer structure is disrupted and the specificity pocket is reconstituted. The EF and BG loops are now available to stabilize the Fyn SH2:specific‐pY complex by classical interactions at the pTyr+3 position.
Overall, the NMR experiments indicate, on one side, the presence of the Fyn SH2 dimer form in solution and, the ability of the high affinity peptide to efficiently reverse this aggregation tendency, on the other hand. Our findings are different from other SH2 dimer reported,5, 6, 7, 8 as it will be detailed in the next section.
Distinct features among different SH2 intertwined dimers
Several SH2 domains have been reported to form dimer structures. Besides the human Fyn SH2 domain described here [Fig. 8(a)], dimers were observed for the SH2 from Grb2,5 Nck1,6 and Itk.8
Figure 8.
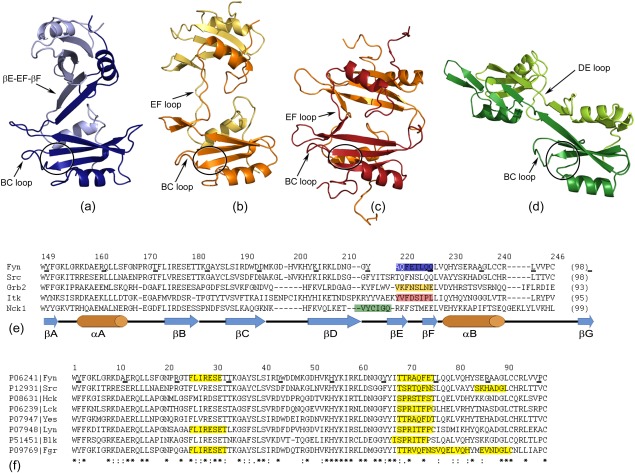
Different SH2 dimers originating in (a) Fyn (PDB code: 4U17); (b) Grb2 (PDB code: 1FYR; Schiering et al., 2000); (c) Itk (PDB code: 3S9K; Joseph et al., 2012) and (d) Nck1 (PDB code: 2CI8; Frese et al., 2006). The monomer units for each SH2 domain are illustrated in dark and light color and are shown in the same structure‐orientation. The BC loop and the extended hinge region are labeled in each dimer, while the pTyr binding pocket is indicated by a black circle. (e) A multiple sequence alignment of SH2 domains from human Fyn, human Src, human Grb2, mouse Itk, and human Nck1. The disposition of the hinge regions is highlighted in the corresponding color as in (a–d). The residue numbers of human Fyn SH2 and the secondary structure elements are indicated. (f) A multiple sequence alignment of Src‐related SH2 domains highlighting the regions having the potential to form amyloid aggregates.
Figure 9.
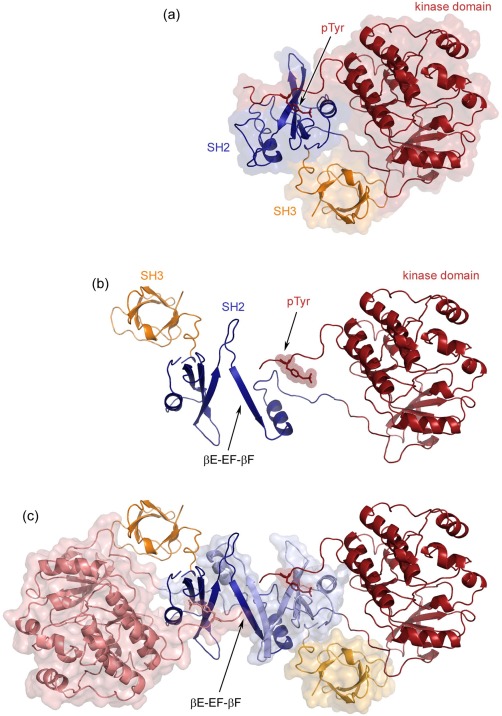
Model of the Fyn kinase dimer. (a) The crystal structure of the human Src kinase in its inactive form (PDB code: 2SRC). The SH2 domain (dark blue) is bound to the pTyr C‐terminal regulatory tail (labeled and shown as sticks), while the SH3 domain (dark yellow) stabilizes the closed confirmation. The kinase domain is represented in dark red. (b) The monomer unit of the hypothetical kinase dimer model, preserving the same coloring as in (a). (c) The complete kinase dimer highlighting the extended βE‐EF‐βF‐strand, the main interaction within the SH2 dimer. One monomer unit is colored as in (a) and the second one maintains the same colors in their light versions. Additionally, the last monomer unit is shown also in surface representation. The model was built with PyMOL, using our structure of the Fyn SH2 dimer and the human Src kinase (PDB code: 2SRC). The sequence identity between the full human Src and Fyn kinases is 76%.
The growth factor receptor‐bound protein 2 (Grb2) is an adaptor protein consisting of one central SH2 domain flanked by two Src homology 3 (SH3) domains. Schiering et al. described the dimerization of Grb2 SH2 via chain segment exchange as illustrated in Fig. 8(b).5 In the process of dimerization, the conformational changes in the EF loop trigger the opening of the monomer, and the αB‐helix extends onto the symmetric intertwined unit. The βF secondary structure element is not present anymore and this region is involved in stabilizing the dimer. The authors presented the dimeric form in complex with a tetra peptide. Remarkably, the two binding pockets are still available for peptide binding and the dimerization is not substantially altering the interaction surface. While the Fyn SH2 dimer discussed here is not compatible with the specific‐pY peptide bound state, the segment exchange of Grb2 SH2 domain reduces the ligand affinity compared with the monomer and has potential influence on its specificity.7 In case of Grb2 SH2 dimer, the peptide addition does not induce the disruption of the dimer and the peptide binding event is modified with respect to the monomer. The differences between Fyn SH2 and Grb2 SH2 binding events reside mainly in the fact that they have different specificity binding positions. In Fyn SH2, the specificity pocket from position pTyr+3 is not accessible in Grb2 and consequently, the specificity interaction of the latter protein occurs at position pTyr+2. This phenomenon is explained by the presence of a tryptophan residue which blocks the pTyr+3 position in SH2 from Grb2 and therefore the peptide is forced to reverse in a β‐turn. Consequently, the peptide binding has different impact on the dimers of the two SH2 domains.
A similar mechanism as for Grb2 SH2, from the structural point of view, was observed in the case of the SH2 from Interleukin‐2 tyrosine kinase (Itk).8 The EF loop and βF‐strand are the hinge regions involved in the dimerization [Fig. 8(c)]. In contrast with Grb2, in the Itk the dimer is more compact having a distance of about 15 Å between the central β‐sheet of the two monomer units, while in Grb2 this distance is ∼35 Å. The specificity pocket of Itk SH2 is formed, but due to the dimerization interface, this site cannot accommodate the peptide at position pTyr+3 anymore. However, in both cases the canonical SH2 fold is recovered.
A different SH2 interface was observed in the case of the adaptor protein Nck1.6 The DE loop is extended and makes the interactions with the second unit, while the βE‐strand, EF loop, and βF‐strand are all keeping their initial structures [Fig. 8(d)]. Additionally, the canonical SH2 fold is reconstituted by βE, βF‐strands and αB‐helix from the symmetric monomer unit. On the other hand, in the cases of Grb2, Itk and Fyn this classical SH2 fold is reconstituted only by the αB‐helix. Due to its dimerization interface, the Nck1 SH2 dimer has both binding pockets accessible.
Structurally, the dimer of the Fyn SH2 domain reported here [Fig. 8(a)] is different from all the three cases discussed before. The main distinction resides in the binding surface, which is substantially altered by the dimerization process. The two symmetrical units are twisted with respect to each other and this angle inhibits the formation of the specificity pocket and also blocks the access to this region. A slight twist between the units is observed in all the SH2 dimers, but it is less pronounced and, in case of Grb2 and Nck1, it does not impede the traditional binding interface.
The residues composing the hinge regions and their disposition can be visualized in a multiple sequence alignment [Fig. 8(e)]. Ignoring the exact location of the hinge area (EF loop for Grb2, Itk and Fyn or DE loop for Nck1), this region is extended in all the SH2 dimers. Moreover, the hinge corresponds to highly conserved loop regions within the classical monomer SH2 fold. Interestingly, only in the case of Fyn SH2 dimer, the hinge region becomes structured by connecting the pre‐existing two secondary structure elements βE and βF. On contrary, the dimerization in Itk and Grb2 SH2 domains promoted the unfolding of βF‐strand. To our knowledge, the Fyn SH2 is the only SH2 dimer where the hinge region becomes structured via the dimerization process.
One can postulate that the Src‐related homologs of Fyn SH2 may all have the same intertwined dimer forms. This postulate is supported by two observations. First, as the multiple sequence alignment of the eight Src family members reveals in Figure 8(f), the amino‐acid composition of the hinge region identified for Fyn SH2 appears to be very conserved. Second, the amyloid prediction tool Waltz,33 which specifically predicts the propensity of a sequence to adapt a cross‐β arrangement typically observed in amyloid fibrils, identified a strong amyloid nucleating segment in the hinge region of Fyn SH2 (Fig. 2), suggesting this region may nucleate amyloid formation under appropriate conditions. Applying Waltz on the other seven SH2 sequences reveals that exactly the same region can be identified in all of them [see Fig. 8(f)]. It would be interesting to test this hypothesis in order to understand whether the slight amino acid differences have a strong effect on the dimer structure as well as the balance between dimer and monomer populations. Moreover, given the high sequence similarity one can wonder whether dimers could be formed between the SH2 domains of different Src‐family members and what biological implications this could have.
Structural implications for the entire protein
The biological relevance of the SH2 dimerization is less investigated. Most SH2 dimers reported were detected through crystallization, like those of Itk and Nck1.5, 6, 8 These dimers were not further studied in other (solution) conditions, which may explain why none of them were shown to have biological or functional relevance. The Fyn SH2 dimer described in this study, however, was identified in both crystal and solution conditions. Size exclusion chromatography, CD and NMR experiments clearly indicate that this dimer is not simply the result of crystallographic packing.
The physiological significance of the Fyn SH2 dimer is less studied. In light of the observations described here, it is worth investigating the impact of such dimerization on the entire kinase structure. This attempt is motivated by the observation that the kinase structure is sterically compatible with the dimer. As the structure of the complete Fyn protein is not available, the closely related human Src kinase was used to generate a model. We used the reported structure of the Src kinase (PDB code: 2SRC34) in its closed conformation. In this state, the C‐terminal regulatory tail is phosphorylated at the tyrosine residue, which is bound to the SH2 high affinity pocket.
Fyn activity, like all other Src‐like kinases, is regulated through the SH3‐SH2 domain combination.35 Activation will occur when (i) the tail p‐Tyr is dephosphorylated, removing the binding between with the SH2 domain, (ii) competitively binding the SH2 domain with a peptide that has a higher affinity than the p‐Tyr tail, and (iii) binding the SH3 domain without dephosphorylating the tail. The model proposed here does not seem to exclude any of these mechanisms. Note also that for Fyn SH2 the interaction between the inhibitory C‐terminal region and SH2 domain can be shown to occur only at the affinity pocket (discussed in detail in another manuscript currently under review). This binding manner does not engage the specificity pocket. Therefore, the inhibitory C‐terminal region bound to the SH2 dimer could be compatible. This hypothesis has still to be validated, but our preliminary analysis suggests such compatibility.
Nonetheless, such a phenomenon was already reported in case of Grb2 SH2 dimer.5, 7 This dimer was shown to be metastable in solution by analytical gel filtration5 and was identified in both free and peptide‐bound states. A reduction in ligand affinity of the dimer, when compared with the monomer state was highlighted by isothermal titration calorimetry studies.7 The authors found that this dimer is compatible with the peptide binding, when the classical pTyr+3 specificity pocket is not engaged in the interaction with the ligand. The Grb2 is an adaptor protein composed by a SH2 domain flanked by two SH3 domains. In an attempt to understand the possible biological significance of the dimerization, a model of the entire Grb2 dimer was proposed.5 In this simulation, the process assures the dimerization and all the SH2 and SH3 domains are fully accessible. Moreover, the exchange modifies the ligand affinity, which can then modulate the interaction in the signaling pathways of this adaptor protein.
Another important piece of information shown in our study is the Fyn SH2 dimer shift towards the monomer upon the interaction with the high affinity peptide involving both SH2 binding pockets. Based on these results and the kinase dimer model, it is tempting to speculate the existence of a sensitive dimerization mechanism. This mechanism can be modulated by the SH2 ligand‐binding event. In other words, in the kinase active form the SH2 is bound to the natural high affinity target and the kinase dimer is disrupted. On the other hand, the inactive kinase is compatible with the dimer formation, if we assume that the SH2 dimerization is not impeded by binding to phosphorylated C‐terminal negative regulatory tail of the kinase.
Dimerization has been shown to play a regulatory role in other kinases like those belonging to the P21 activated kinases (PAK) type I family.36 Upon crystallization of PAK1 it was observed that the kinase forms a dimer and the results were confirmed by different in vitro experiments.37, 38 This observation led Parrini et al.39 to investigate whether the dimer also appears in vivo. They were able to confirm the presence of the dimer in vivo, revealing also that PAK1 is auto‐inhibited in trans via an auto‐inhibitory domain. Cdc42 and Rac1 induce dissociation of the PAK1 dimer and the activation of the PAK1 kinase. The results indicate that PAK type I activity is negatively regulated through dimerization. It is therefore interesting to question whether such a scenario is also meaningful for other kinds of kinases. One could hypothesize that, just as PAK1, Fyn dimerization may play a role in improving the efficiency of inhibition and that dimer dissociation leads to activation.39 Yet, at this point there is no experimental evidence that would point to such a scenario for Fyn or other Src‐family members.
In conclusion, our data do not exclude the possibility of Fyn kinase dimerization based on the SH2 intertwining mechanism, but the relevance of this association phenomenon remains unclear. It is therefore worth investigating if (i) such a SH2‐mediated dimerization process of Fyn or close related kinases can be found in the cell and (ii) if the case, what would be the functional relevance of such a process.
Materials and Methods
Sample preparation
The SH2 domain, corresponding to residues 149–248, of human Fyn kinase was obtained in high concentration and purity as previously described.24 Briefly, the construct was over‐expressed in two different expression vectors: pET15b (Novagen) and pColdII (TaKaRa). Both vectors place an N‐terminal six His‐tag, but only pET15b contains a thrombin cleavage site between the tag and the construct of interest. The Fyn SH2 domain was obtained in Escherichia coli BL21 (DE3) cells and purified from cell lysate by affinity chromatography followed by the size exclusion separation according to the details mentioned elsewhere.24 The molecular‐weight standards used for gel‐filtration consist of bovine thyroglobulin (670 kDa), bovine γ‐globulin (158 kDa), chicken ovalbumin (44 kDa), horse myoglobin (17 kDa), and vitamin B12 (1.35 kDa).
Peptide
The specific phosphotyrosine peptide (denoted specific‐pY), corresponds to residues 321–331 of hamster middle‐T antigen. The peptide has the following sequence: EPQ‐pY‐EEIPIYL and it has been shown to bind with high affinity to the Fyn SH2 domain.26, 40 The peptide residues are numbered relative to the phosphotyrosine (pTyr), which is considered position 0. The residues located N‐terminal in respect to the pTyr carry negative numbers, while the C‐terminal ones have positive positions. The peptide was purchased from JPT Peptide Technologies GmbH with more than 95% purity.
Circular dichroism (CD)
The far‐UV circular dichroism (CD) spectra were recorded on a J‐715 spectro‐polarimeter (Jasco) in the 200–250 nm spectral region. The measurements were performed at 25°C in 50 mM sodium phosphate buffer, pH 6.50, 5 mM DTT. The protein concentration was adjusted at 0.2 mg/mL. The DTT has high absorbance in the far‐UV region, which prevented the recording of reliable spectra at wavelengths below 210 nm. As a consequence, the measurements can be used for comparisons, but the secondary structure calculations are not accurate and are not taken into account. The data were acquired using a cell with a path length of 0.1 cm, a scanning speed of 50 nm/min, a 2 s integration time and 1 nm bandwidth. The mean residue ellipticity ([θ], deg cm2 mol−1) was obtained as follows: [θ] = (θ • M rw)/(c • l), where θ is the observed ellipticity, M rw is the mean residue molecular weight, c is the concentration, and l is the path length.41, 42
Isothermal titration calorimetry (ITC)
The ITC measurements were performed on a VP‐ITC calorimeter (MicroCal, GE Healthcare). The samples were degassed for 20 min under vacuum prior to the experiments. The measurements were done at 25°C in 50 mM sodium phosphate buffer, pH 6.50, 5 mM DTT. The peptide was initially dissolved in the reaction buffer to obtain stock solutions and then diluted to obtain the concentrations used in the titration. The data were fitted to a single site‐binding model using MicroCal Origin. The ITC experiments were performed with the SH2 domain obtained from pCold II expression vector.
Crystallography
The crystals of the free Fyn SH2 dimer were obtained from the non‐His‐tagged protein (making use of the pET15b expression vector), while the Fyn SH2:specific‐pY complex was prepared by adding an excess of specific‐pY peptide into Fyn SH2 His‐tagged domain (from pColdII expression vector). The crystallization conditions, the data collection and analysis were previously detailed.24 The diffraction data of the dimer were integrated with iMOSFLM43 and reduced with SCALA,44 and data from the Fyn SH2:specific‐pY complex with the HKL2000 suite.45 In both cases he structure was solved by molecular replacement using the coordinates from 1G83 as search model as implemented in PHASER.46 Successive rounds of manual building in Coot47 and refinement cycles in phenix.refine 48 were performed to complete both structural models. Statistics for the data collection and refinement are shown in Table 1.
Nuclear magnetic resonance
The 15N transverse relaxation rates (R 2) were measured at 25°C on a Varian NMR Direct‐Drive System 600 MHz spectrometer. The R 2 rates were obtained from series of 2D experiments16 recorded with delays of 10, 30, 50, 70, 90, 110, 130, 150, and 170 ms. To permit error estimation, a duplicate spectrum was recorded for 10‐ms delay. All NMR data were processed with NMRPipe49 and analyzed with CCPNMR software.50 R 2 values were obtained by fitting the peak intensity to a single exponential decay function, making use of the CCPNMR built in application. The errors were estimated directly from the variation between the corresponding experiments with different relaxation delays.
The full 1H, 15N, and 13C assignment of the backbone and side‐chain of Fyn SH2 domain have been reported,31 for both the free and specific‐pY peptide bound states. These assignments have been used to identify the resonances of the free state (dimer) and the additional signals attributed to the dimerization behavior. To obtain the Fyn SH2 bound state, for NMR purposes, the specific‐pY peptide was added in excess to the Fyn SH2 free state to a molar ratio of 3 : 1.
NMR chemical shift perturbation
Chemical shift differences were calculated as absolute values between the peptide bound form and the free form, in case of Cα, Cβ, and Hα resonances. For the glycine residues, Hα was represented as the average of the two Hα chemical shifts. The combined HN, N chemical shift deviations were calculated using the following equation:
The threshold for a significant chemical shift deviation is defined as the average of all chemical shift differences for the corresponding atom type plus one standard deviation.
Accession Numbers
Structural data are available in the wwPDB database under the accession numbers: 4U17 for free Fyn SH2 domain (dimer state) and 4U1P for Fyn SH2 in complex with a high affinity phosphotyrosine‐containing peptide (monomer state).
References
- 1. Liu BA, Jablonowski K, Raina M, Arce M, Pawson T, Nash PD (2006) The human and mouse complement of SH2 domain proteins‐establishing the boundaries of phosphotyrosine signaling. Mol Cell 22:851–868. [DOI] [PubMed] [Google Scholar]
- 2. Liu BA, Shah E, Jablonowski K, Stergachis A, Engelmann B, Nash PD (2011) The SH2 domain‐containing proteins in 21 species establish the provenance and scope of phosphotyrosine signaling in eukaryotes. Sci Signal 4:ra83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaneko T, Joshi R, Feller SM, Li SS (2012) Phosphotyrosine recognition domains: the typical, the atypical and the versatile. Cell Commun Signal 10:32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu BA, Engelmann BW, Nash PD (2012) The language of SH2 domain interactions defines phosphotyrosine‐mediated signal transduction. FEBS Lett 586:2597–2605. [DOI] [PubMed] [Google Scholar]
- 5. Schiering N, Casale E, Caccia P, Giordano P, Battistini C (2000) Dimer formation through domain swapping in the crystal structure of the Grb2‐SH2‐Ac‐pYVNV complex. Biochemistry 39:13376–13382. [DOI] [PubMed] [Google Scholar]
- 6. Frese S, Schubert WD, Findeis AC, Marquardt T, Roske YS, Stradal TE, Heinz DW (2006) The phosphotyrosine peptide binding specificity of Nck1 and Nck2 Src homology 2 domains. J Biol Chem 281:18236–18245. [DOI] [PubMed] [Google Scholar]
- 7. Benfield AP, Whiddon BB, Clements JH, Martin SF (2007) Structural and energetic aspects of Grb2‐SH2 domain‐swapping. Arch Biochem Biophys 462:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Joseph RE, Ginder ND, Hoy JA, Nix JC, Fulton DB, Honzatko RB, Andreotti AH (2012) Structure of the interleukin‐2 tyrosine kinase Src homology 2 domain; comparison between X‐ray and NMR‐derived structures. Acta Cryst F68:145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mackinnon SS, Malevanets A, Wodak SJ (2013) Intertwined associations in structures of homooligomeric proteins. Structure 21:638–649. [DOI] [PubMed] [Google Scholar]
- 10. Bennett MJ, Eisenberg D (1994) Domain swapping: Entangling alliances between proteins. Proc Natl Acad Sci USA 91:3127–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bennett MJ, Schlunegger MP, Eisenberg D (1995) 3D Domain swapping: a mechanism of oligomer assembly. Protein Sci 4:2455–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu Y, Eisenberg D (2002) 3D domain swapping: as domains continue to swap. Protein Sci 11:1285–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rousseau F, Schymkowitz JW, Itzhaki LS (2003) The unfolding story of three‐dimensional domain swapping. Structure 11:243–251. [DOI] [PubMed] [Google Scholar]
- 14. Bennett MJ, Eisenberg D (2004) The evolving role of 3D domain swapping in proteins. Structure 12:1339–1341. [DOI] [PubMed] [Google Scholar]
- 15. Bennett MJ, Sawaya MR, Eisenberg D (2006) Deposition diseases and 3D domain swapping. Structure 14:811–824. [DOI] [PubMed] [Google Scholar]
- 16. Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman‐Kay JD, Kay LE (1994) Backbone dynamics of a free and phosphopeptide‐complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33:5984–6003. [DOI] [PubMed] [Google Scholar]
- 17. Kay LE, Muhandiram DR, Farrow NA, Aubin Y, Forman‐Kay JD (1996) Correlation between dynamics and high affinity binding in an SH2 domain interaction. Biochemistry 35:361–368. [DOI] [PubMed] [Google Scholar]
- 18. Pintar A, Hensmann M, Jumel K, Pitkeathly M, Harding SE, Campbell ID (1996) Solution studies of the SH2 domain from the fyn tyrosine kinase: secondary structure, backbone dynamics and protein association. Eur Biophys J 24:371–380. [DOI] [PubMed] [Google Scholar]
- 19. Zhang W, Smithgall TE, Gmeiner WH (1998) Self‐association and backbone dynamics of the hck SH2 domain in the free and phosphopeptide‐complexed forms. Biochemistry 37:7119–7126. [DOI] [PubMed] [Google Scholar]
- 20. Finerty PJ, Jr. , Muhandiram R, Forman‐Kay JD (2002) Side‐chain dynamics of the SAP SH2 domain correlate with a binding hot spot and a region with conformational plasticity. J Mol Biol 322:605–620. [DOI] [PubMed] [Google Scholar]
- 21. Siegal G, Davis B, Kristensen SM, Sankar A, Linacre J, Stein RC, Panayotou G, Waterfield MD, Driscoll PC (1998) Solution structure of the C‐terminal SH2 domain of the p85 alpha regulatory subunit of phosphoinositide 3‐kinase. J Mol Biol 276:461–478. [DOI] [PubMed] [Google Scholar]
- 22. Mulhern TD, Shaw GL, Morton CJ, Day AJ, Campbell ID (1997) The SH2 domain from the tyrosine kinase Fyn in complex with a phosphotyrosyl peptide reveals insights into domain stability and binding specificity. Structure 5:1313–1323. [DOI] [PubMed] [Google Scholar]
- 23. Machida K, Thompson CM, Dierck K, Jablonowski K, Karkkainen S, Liu B, Zhang H, Nash PD, Newman DK, Nollau P, Pawson T, Renkema GH, Saksela K, Schiller MR, Shin DG, Mayer BJ (2007) High‐throughput phosphotyrosine profiling using SH2 domains. Mol Cell 26:899–915. [DOI] [PubMed] [Google Scholar]
- 24. Huculeci R, Buts L, Lenaerts T, van Nuland NA, Garcia‐Pino A (2012) Purification, crystallization and preliminary X‐ray diffraction analysis of the Fyn SH2 domain and its complex with a phosphotyrosine peptide. Acta Cryst F68:359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gorrec F (2009) The MORPHEUS protein crystallization screen. J Appl Cryst 42:1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bradshaw JM, Grucza RA, Ladbury JE, Waksman G (1998) Probing the “two‐pronged plug two‐holed socket” model for the mechanism of binding of the Src SH2 domain to phosphotyrosyl peptides: a thermodynamic study. Biochemistry 37:9083–9090. [DOI] [PubMed] [Google Scholar]
- 27. Taylor JD, Ababou A, Fawaz RR, Hobbs CJ, Williams MA, Ladbury JE (2008) Structure, dynamics, and binding thermodynamics of the v‐Src SH2 domain: implications for drug design. Proteins 73:929–940. [DOI] [PubMed] [Google Scholar]
- 28. Kaneko T, Huang H, Zhao B, Li L, Liu H, Voss CK, Wu C, Schiller MR, Li SS (2010) Loops govern SH2 domain specificity by controlling access to binding pockets. Sci Signal 3:ra34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Waksman G, Shoelson SE, Pant N, Cowburn D, Kuriyan J (1993) Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide‐free forms. Cell 72:779–790. [DOI] [PubMed] [Google Scholar]
- 30. Kaneko T, Huang H, Cao X, Li X, Li C, Voss C, Sidhu SS, Li SS (2012) Superbinder SH2 domains act as antagonists of cell signaling. Sci Signal 5:ra68 [DOI] [PubMed] [Google Scholar]
- 31. Huculeci R, Buts L, Lenaerts T, van Nuland NA (2011) 1H, 13C and 15N backbone and side‐chain chemical shift assignment of the Fyn SH2 domain and its complex with a phosphotyrosine peptide. Biomol NMR Assign 5:181–184. [DOI] [PubMed] [Google Scholar]
- 32. Jarymowycz VA, Stone MJ (2006) Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem Rev 106:1624–1671. [DOI] [PubMed] [Google Scholar]
- 33. Maurer‐Stroh S, Debulpaep M, Kuemmerer N, Lopez de la Paz M, Martins IC, Reumers J, Morris KL, Copland A, Serpell L, Serrano L, Schymkowitz JW, Rousseau F (2010) Exploring the sequence determinants of amyloid structure using position‐specific scoring matrices. Nat Methods 7:237–242. [DOI] [PubMed] [Google Scholar]
- 34. Xu W, Doshi A, Lei M, Eck MJ, Harrison SC (1999) Crystal structures of c‐Src reveal features of its autoinhibitory mechanism. Mol Cell 3:629–638. [DOI] [PubMed] [Google Scholar]
- 35. Engen JR, Wales TE, Hochrein JM, Meyn MA, 3rd Banu Ozkan S, Bahar I Smithgall TE (2008) Structure and dynamic regulation of Src‐family kinases. Cell Mol Life Sci 65:3058–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rane CK, Minden A (2014) P21 activated kinases: structure, regulation, and functions. Small GTPases 5: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lei M, Lu W, Parrini M‐C, Eck MJ, Mayer BJ, Harrison SC (2000) Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell 102:387–397. [DOI] [PubMed] [Google Scholar]
- 38. Buchwald G, Hostinova E, Rudolph MG, Kraemer A, Sickmann A, Meyer HE, Scheffzek K, Wittinghofer A (2001) Conformational switch and role of phosphorylation in PAK activation. Mol Cell Biol 21:5179–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Parrini MC, Lei M, Harrison SC, Mayer BJ (2002) Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol Cell 9:73–83. [DOI] [PubMed] [Google Scholar]
- 40. Cantley LC, Auger KR, Carpenter C, Duckworth B, Graziani A, Kapeller R, Soltoff S (1991) Oncogenes and signal transduction. Cell 64:281–302. [DOI] [PubMed] [Google Scholar]
- 41. Andrade MA, Chacon P, Merelo JJ, Moran F (1993) Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng 6:383–390. [DOI] [PubMed] [Google Scholar]
- 42. Sreerama N, Venyaminov SY, Woody RW (1999) Estimation of the number of alpha‐helical and beta‐strand segments in proteins using circular dichroism spectroscopy. Protein Sci 8:370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Leslie AGW, Powell HR. Processing diffraction data with mosflm. In: Read RJ, Sussman JL, Eds. (2007) Evolving Methods for Macromolecular Crystallography. Springer, Netherlands, pp 41–51. [Google Scholar]
- 44. Evans P (2006) Scaling and assessment of data quality. Acta Cryst D 62:72–82. [DOI] [PubMed] [Google Scholar]
- 45. Otwinowski Z, Minor M (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- 46. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Cryst 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 48. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive python‐based system for macromolecular structure solution. Acta Cryst D 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293. [DOI] [PubMed] [Google Scholar]
- 50. Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59:687–696. [DOI] [PubMed] [Google Scholar]