

Abstract
Analysis of the human proteome has identified thousands of unique protein sequences that contain acetylated lysine residues in vivo. These modifications regulate a variety of biological processes and are reversed by the lysine deacetylase (KDAC) family of enzymes. Despite the known prevalence and importance of acetylation, the details of KDAC substrate recognition are not well understood. While several methods have been developed to monitor protein deacetylation, none are particularly suited for identifying enzyme‐substrate pairs of label‐free substrates across the entire family of lysine deacetylases. Here, we present a fluorescamine‐based assay which is more biologically relevant than existing methods and amenable to probing substrate specificity. Using this assay, we evaluated the activity of KDAC8 and other lysine deacetylases, including a sirtuin, for several peptides derived from known acetylated proteins. KDAC8 showed clear preferences for some peptides over others, indicating that the residues immediately surrounding the acetylated lysine play an important role in substrate specificity. Steady‐state kinetics suggest that the sequence surrounding the acetylated lysine affects binding affinity and catalytic rate independently. Our results provide direct evidence that potential KDAC8 substrates previously identified through cell based experiments can be directly deacetylated by KDAC8. Conversely, the data from this assay did not correlate well with predictions from previous screens for KDAC8 substrates using less biologically relevant substrates and assay conditions. Combining results from our assay with mass spectrometry‐based experiments and cell‐based experiments will allow the identification of specific KDAC‐substrate pairs and lead to a better understanding of the biological consequences of these interactions.
Keywords: lysine deacetylase, substrate specificity, fluorescamine, biological relevance, peptide substrates, deacetylation assay, sirtuins
Introduction
Analysis of mammalian proteomes has identified thousands of unique protein sequences that contain acetylated lysine residues in vivo.1, 2, 3, 4, 5, 6 This prevalent and reversible post‐translational modification is highly regulated and is important for a variety of biological processes. Lysine deacetylases (KDACs, also known as histone deacetylases, EC 3.5.1.98) are metal‐dependent enzymes that reverse this post‐translational modification, by catalyzing the hydrolysis of ε‐N‐acetyllysine residues in proteins via a conserved mechanism.7, 8, 9 Like acetylation itself, the activity of expressed KDACs has been directly linked to a wide variety of biological processes, including development and growth, memory formation, and regulation of metabolism.10, 11, 12, 13 KDAC activity has also been linked to numerous diseases, in particular chronic diseases such as asthma, cancers, muscular disorders, and diabetes.11, 14, 15, 16
Metal‐dependent lysine deacetylases, class I and class II KDACs, share a conserved reaction mechanism and catalytic domain, but differ widely in their intracellular distribution and cell‐type expression patterns.11, 17 However, with several thousand identified nonhistone acetylated proteins, it is highly unlikely that localization patterns alone are sufficient for the eleven metal‐dependent KDACs (several of which are primarily localized to the cell nucleus) and seven NAD‐dependent KDACs (class III KDACs or sirtuins) to react only with their intended substrates. Literature evidence indicates that substrates and inhibitors likely bind in different conformations, and product release is likely highly influenced by small conformational changes in KDACs.18, 19, 20 Therefore, it is essential to identify the particular contacts that are most important in determining binding, as dictated by substrate sequence and structure.
Despite the known prevalence and importance of acetylation, the details of KDAC substrate recognition are not well understood. Only a handful of the identified acetylated proteins have been matched to a particular KDAC, and the degree to which the substrate sets are discrete or overlapping among the KDAC isozymes is unknown, as are the factors that allow KDACs to discriminate between potential substrates.21, 22 The few studies which have definitively identified substrates of a particular KDAC were mostly directed studies using a cell‐based approach. For example, SMC3 was found to be a KDAC8 (also known as HDAC8)23 substrate by inhibiting KDAC8 expression in cells and monitoring the downstream effects specifically on SMC3 and related pathways.24 Studies such as this one are useful for linking KDACs to their substrates; however, they identify only a single enzyme‐substrate pair and are limited to situations where there is already a proposed enzyme‐substrate relationship. In addition, without supporting biochemical data, it is difficult to distinguish between a direct enzyme‐substrate relationship and an indirect effect. A recent development is the use of larger‐scale cell based experiments relying on pull‐down techniques and mass spectrometry in an attempt to link particular KDACs with potential substrates, which has led to the identification of several likely substrate proteins but has not yet helped clarify the differences in specificity between lysine deacetylases.25
In addition to these cell‐based methods, several in vitro biochemical assays have been developed to directly assess KDAC activity for particular substrates. KDAC8, and to a lesser extent a few other class I and some class II KDACs, have been screened using randomized substrate libraries to determine the effect of substrate sequence.26, 27, 28, 29, 30, 31, 32 However, all these systems present the enzyme with the substrate sequence in an unnatural context, either as a dye‐labeled conjugate presenting only sequence upstream of the lysine or as a peptide attached to a surface. Although experimentally convenient, these substrates likely behave significantly differently than natural substrates. An unbiased method for quantifying lysine deacetylase activity relies on radioactive labeling of substrates, but such labeling is expensive and not well suited for many applications.33, 34 HPLC quantification of deacetylated peptides has also been reported, but this method is cumbersome and not suited for high‐throughput applications.35 Another recent approach relies on the measurement of acetate production, and this method can be applied as either a stopped assay, similar to all other deacetylase assays, or as a continuous assay.36 However, this method still has limited sensitivity to small amounts of deacetylation, is considerably more complex than other methods, and cannot be used in any system where NADH is present (such as assays for the sirtuin family of lysine deacetylases). An analogous coupled assay for the sirtuins based on detecting the coupled production of nicotinamide has also been reported, but like the acetate coupled assay is relatively complex and involves the use of several enzymes working in parallel.37, 38 To date, no general purpose, relatively high‐throughput label‐free assay has been reported that works with all lysine deacetylases.
Based on these limitations, we developed an assay which would allow us to definitively identify substrates of a specific KDAC with adequate sensitivity to detect low levels of deacetylation and without requiring a fluorescent inhibitor or unnatural modification of substrate. The assay presented here utilizes fluorescamine, a compound originally used to detect and quantify protein, to detect free primary amines.39, 40, 41, 42 This molecule is ideal for detecting deacetylation, as it specifically reacts with the product of the deacetylation reaction (free lysine), but not the substrate (acetylated lysine), resulting in fluorescence. In fact, fluorescamine has been previously used to quantitatively monitor the deacetylation of N‐acetylglucosamine.43, 44 Based on these previous reports, we developed a fluorescamine‐based assay to measure lysine deacetylation by KDACs, which is comparable in sensitivity to existing assays that rely on fluorescently‐conjugated substrates and is applicable to all classes of KDACs. Using this assay, we measured KDAC8 activity against several potential peptide substrates, demonstrating that this enzyme does show substrate preference; however, this preference does not correlate with the results of previous screens using unnaturally modified substrates.
Results
A more biologically relevant fluorescamine assay
To address limitations of previously developed assays to detect deacetylation, we developed a novel assay using fluorescamine to detect deacetylation (Fig. 1). This assay, described in detail in the Materials and Methods, is based on previously described assays.44, 45 Briefly, KDACs were incubated with a substrate containing an acetylated lysine in a buffer that more closely approximated intracellular conditions than standard buffers used in deacetylase assays.46 Once the reaction was stopped, fluorescamine was added to the mixture, which reacts with primary amines (i.e., the free lysine formed as the product of the reaction) to generate a fluorescent product with a linear dependence on concentration.39, 44, 47 Under the conditions of our assay, we could reliably quantify amine concentrations of 0.2 to 100 µM (5–2500 pmol; Fig. 2). The assay is easily adapted for either endpoint characterization or generation of steady‐state parameters.
Figure 1.
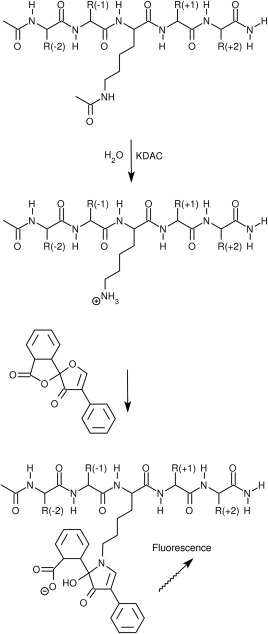
Fluorescamine reaction scheme. Peptide substrates, shown here as a 5‐mer, are initially deacetylated by a KDAC. After stopping the reaction, fluorescamine is added to generate a fluorescent product. Fluorescamine reacts more efficiently with primary amines than with secondary or tertiary amines, and only the product of a reaction with a primary amine is fluorescent. The N‐terminus and C‐terminus of the peptide are acetylated and amidated, respectively, to better mimic the presentation of a sequence within a longer protein sequence.
Figure 2.
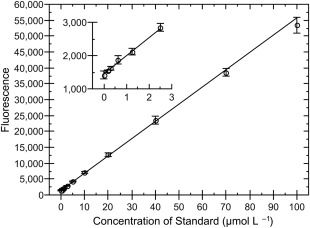
Sensitivity of fluorescamine assay. Several concentrations of unacetylated lysine (ac‐K‐NH2) in reaction buffer were treated in the same manner as reactions. Error bars represent the standard deviation of triplicate measurements. Line represents a weighted linear fit to the data. r 2 = 0.9998. Inset shows the same data zoomed to only the low concentrations, illustrating the fit to points at both extremes.
Standard buffer conditions for assessing KDAC activity against the Fluor‐de‐Lys substrate (50 mM tris pH 8.0, 2.7 mM KCl, 137 mM NaCl, 1 mg mL−1 BSA, 1 mM MgCl2) lack biological relevance and more closely resemble extracellular conditions than the cytosol.46, 48 In addition, the use of an amine‐based buffer is not suitable for fluorescamine due to the reaction of the fluorescamine with the buffer. Therefore, we chose a buffer for our assay (30 mM potassium phosphate pH 7.6, 100 mM KCl, 5% glycerol) that was closer to physiological intracellular conditions. While KDAC8 has been shown to be most active between pH 8 and 9,49 we decided that it was more appropriate to assess activity under a more physiologically relevant pH which still retains a reasonable degree of activity under in vivo conditions. Furthermore, phosphate buffer resulted in approximately 10× greater sensitivity than MOPS buffer or HEPES buffer (compare Fig. 2 to Supporting Information Fig. S1), even though MOPS and HEPES have tertiary amines that are not expected to react efficiently with fluorescamine. Borate buffers resulted in a slightly reduced sensitivity compared with phosphate buffer and are not effective buffers at physiologically relevant pH values (data not shown). For these reasons, we utilized the phosphate reaction buffer described above in our assay, which allowed us to use more physiological conditions than previous assays, with enough enzyme activity to maintain adequate sensitivity. Addition of up to 3 mM MOPS (from the protein storage buffer), 5 mM nicotinamide, 100 µM SAHA, 500 µM NAD+, and 100 µM NADH did not significantly affect the resulting signal (data not shown).
Several other parameters were also critical to the sensitivity and reproducibility of this assay. First, fluorescamine was dissolved in DMSO for detection of free amines. Although fluorescamine is soluble in several other solvents, they were not suitable for this assay. Using acetone and acetonitrile both resulted in precipitation when added to the reaction. Furthermore, DMSO has been previously shown to enhance the signal of fluorescamine when present to approximately 50% in the final solution.50 Adding NaCl to the reaction at a final concentration of 0.25 M before adding the fluorescamine prevented precipitation of phosphate when DMSO was used as the solvent. Removal of the enzyme by filtration before addition of fluorescamine, as reported by other investigators,36, 44 was not necessary under our reaction conditions and did not enhance signal intensity.
Note that as the fluorescamine solution aged, signal intensity was lost, equivalent to an approximately 20% decrease in signal intensity after 1 month; however, sensitivity was not greatly affected, with only a slight loss of low‐end sensitivity. For this reason, standards were included with each experiment to accurately quantitate activity. Signal intensity was stable from 20 min to at least 24 h after addition of fluorescamine solution to the reaction provided the fluorescamine solution was aged a day before use, as suggested by prior reports.50, 51 Detection of fluorescence several hours after addition of fluorescamine resulted in a slight improvement (∼5%) of low‐end sensitivity compared with measurement 20 min after fluorescamine addition (data not shown).
Additionally, we performed all enzymatic reactions at 25°C, as the enzyme does not appear to be stable at 37°C under our reaction conditions. A direct comparison of enzymatic activity at these two temperatures, using two peptide substrates, indicated that KDAC8 lost most of its activity after 1.5 to 2 h incubation at 37°C, but remained active at 25°C well past that point (Supporting Information Fig. S2). To determine whether the decrease in activity at higher temperatures could be attributed to protein instability, we used circular dichroism to monitor the enzyme at both temperatures over time. Consistent with the activity data, structural changes were observed during the timeframe of the reaction when the enzyme was incubated at 37°C, but not at 25°C (Supporting Information Fig. S3), with significant changes in structure evident after 1 h at 37°C but not at 25°C. To ensure that this effect was not due simply to a poor choice of buffer conditions, we also performed similar assays using a Fluor‐de‐Lys substrate in the recommended buffer and observed a similar loss of activity over time at 37°C (data not shown).
Peptides corresponding to known acetylated proteins are deacetylated in vitro by KDAC8
Because our assay is not dependent on a fluorescently labeled substrate, we were able to easily assess several potential substrates. In this way, we were able to investigate KDAC8 activity in a more biologically relevant manner, allowing us to address substrate specificity. Based on previous work, mostly utilizing mass spectrometry approaches, thousands of acetylation sites have been identified in vivo.1, 2, 3, 4, 5, 6 To begin to investigate whether any of these acetylation sites could be deacetylated by KDAC8, we designed a limited panel of 5‐mer peptides, each from a known acetylated protein, with the sequence ac‐X‐X‐{K‐ac}‐X‐X‐am (Fig. 1). N‐terminal acetylation and C‐terminal amidation were utilized to better mimic an internal protein sequence, so as to avoid possible effects from N‐terminal and C‐terminal charges on the peptide that would not be found in the source protein. We began with 5‐mer peptides specifically to compare the activity with our label‐free substrates to the predictions from prior work examining the impact of the sequence adjacent to the acetylated lysine in a labeled substrate. Of the peptides tested, only three had previously been linked specifically to KDAC8: ac‐IS{K‐ac}FD‐am, ac‐AR{K‐ac}ST‐am, and ac‐PV{K‐ac}FI‐am (in the context of a 9‐mer).25, 52 Two others (ac‐YS{K‐ac}GF‐am and ac‐YQ{K‐ac}WD‐am) are from proteins known to be deacetylated by KDAC8; however, it is either not known which lysine within these proteins is the true KDAC8 substrate or another acetylated lysine has recently been identified as the likely KDAC8 substrate.21, 24, 53 We performed the fluorescamine assay described above with each of these peptides to determine whether KDAC8 could deacetylate any of the potential substrates. After estimating initial reaction rates from endpoint experiments, we found that several of the potential substrates were deacetylated by KDAC8 in vitro (Table 1). Not surprisingly, there was a fairly large range of activity associated with the various peptide substrates, indicating that the sequence immediately surrounding the acetylated lysine is an important determinant for KDAC8 activity. As expected, circular dichroism spectra of the peptides did not indicate any regular secondary structure of the substrates (data not shown).
Table 1.
Endpoint Activity for Selected Peptides with KDAC8
| Peptide sequence | Activity (pmol min−1 µg−1) | Source protein(s) | Ref a |
|---|---|---|---|
| ac‐FR{K‐ac}RW‐am | 19.5 ± 3.6 | Arf‐GAP with dual PH domain‐containing protein 1 (ADAP1) | 3 |
| ac‐SL{K‐ac}FG‐am | 12.1 ± 2.4 | Succinate dehydrogenase [ubiquinone] flavoprotein subunit, mitochondrial (SDHD) | 3, 6 |
| ac‐IS{K‐ac}FD‐am | 6.9 ± 2.0 | AT‐rich interactive domain‐containing protein 1A (ARID1A) | 25 |
| ac‐LT{K‐ac}SP‐am | 5.9 ± 2.8 | Non‐muscle caldesmon (CALD1) | 5 |
| ac‐FA{K‐ac}WR‐am | 4.5 ± 2.1 | Fructose‐bisphosphate aldolase A (ALDOA); Fructose‐bisphosphate aldolase C (ALDOC) | 3, 6 |
| ac‐PV{K‐ac}FI‐am | 3.5 ± 0.7 | Cysteine‐rich protein 2‐binding protein (CSRP2BP) | 3, 6 |
| ac‐YS{K‐ac}GF‐am | 2.8 ± 1.4 | Src substrate cortactin (SRC) | 3, 6 |
| ac‐YQ{K‐ac}WD‐am | 2.5 ± 0.8 | Structural maintenance of chromosomes protein 3 (SMC3) | 3, 6 |
| ac‐AR{K‐ac}ST‐am | 2.2 ± 0.3 | Histone H3.1t (HIST3H3); Histone H3.3 (H3F3A); Histone H3.1 (HIST1H3A) | 3, 52 |
| ac‐FS{K‐ac}AF‐am | 2.2 ± 0.8 | Nucleolar RNA helicase II (DDX21) | 3, 5, 6 |
| ac‐TG{K‐ac}TF‐am | 1.6 ± 0.3 | Actin‐related protein 2/3 complex subunit 2 (ARPC2) | 3, 5 |
| ac‐VI{K‐ac}GF‐am | 1.3 ± 0.1 | Transferrin receptor protein 1 (TFRC) | 4 |
| ac‐LA{K‐ac}HA‐am | 0.9 ± 0.8 | Histone H2B type 1‐K (HIST1H2BK) | 3, 5, 6 |
| ac‐LH{K‐ac}LL‐am | 0.6 ± 0.5 | Nuclear receptor co‐activator 3 (NCOA3); Nucleoprotein TPR (TPR) | 3, 5, 6, 25 |
| ac‐SD{K‐ac}TI‐am | None detected | Tubulin alpha‐3 chain (TUBA3A) | 5 |
Reference source that the sequence is acetylated, which usually does not coincide with a report that the sequence is a substrate for KDAC8.
To further validate the measured activity using our assay, we compared the endpoint activity of KDAC8 with a 9‐mer peptide (ac‐STPV{K‐ac}FISR‐am) as measured by our assay and a previously reported coupled assay for acetate.36 Under our assay conditions, KDAC8 showed lower activity than what was previously reported for this substrate (5.4 ± 1.7 pmol min−1 µg−1 compared with a predicted value of 21 pmol min−1 µg−1 based on the reported k cat and K M).25 To determine whether this difference was due to differences in buffer and reaction temperature, we repeated our assay using a HEPES‐based buffer and increased the temperature to 30°C to mimic the conditions under which this reaction was measured previously. Under these conditions, the activity increased to 24 ± 5 pmol min−1 µg−1, which is in agreement with the activity measured using the coupled acetate assay, thus validating our quantification of product formation. To confirm the apparent buffer‐specific differences in activity, we performed a Fluor‐de‐Lys assay using KDAC8 and the Fluor‐de‐Lys HDAC8 substrate in either our phosphate‐containing reaction buffer at 25°C or the HEPES‐containing buffer at 30°C. Not surprisingly, KDAC8 was more active in the HEPES‐containing buffer than the phosphate‐containing buffer in this assay, 46 ± 5 pmol min−1 µg−1 and 23.6 ± 3.0 pmol min−1 µg−1, respectively. Unfortunately, it is not possible to assay the Fluor‐de‐Lys reaction directly using fluorescamine due to interference from the intense fluorescence of the 7‐amino‐4‐methylcoumarin attached to the substrate in the 50% DMSO mixture.
Local sequence affects both binding affinity and catalytic rate
Differences in KDAC8 activity against different substrates may be due either to changes in the binding affinity of the substrate to the active site or changes in the rate of catalysis. A major limitation of the endpoint experiments (Table 1) is that they cannot distinguish between these two possibilities. We used the fluorescamine assay to perform analysis of the steady‐state kinetics on selected peptide substrates (Fig. 3 and Table 2). Those peptides which showed clear activity in the endpoint experiment and which were sufficiently soluble in reaction buffer were characterized in this manner. Of the peptides subjected to this analysis, three had K M > 2 mM. Based on experimental limitations, accurate KM values could not be worked out for these substrates, as the substrate could not be added to the reaction at a high enough concentration to adequately saturate the reaction. Furthermore, exact catalytic rates were also not able to be determined for these substrates. The other four substrates had weak, but measurable K M values (ranging from 730 µM to 1.5 mM). Interestingly, we also noticed a range of catalytic rates, ranging from 0.03 to 0.18 molecules per second. These data suggest that the sequence surrounding the acetylated lysine affects binding affinity and catalytic rate independently. The catalytic efficiencies ranged from a maximum of 171 ± 13 M−1 s−1 to at least 10‐fold lower, even for those peptides with sufficient activity to obtain a reliable efficiency value, indicating that KDAC8 has significant substrate preferences for catalyzing some peptides over others. We did not observe substrate or product inhibition effects even at very high substrate concentrations (5 mM).
Figure 3.
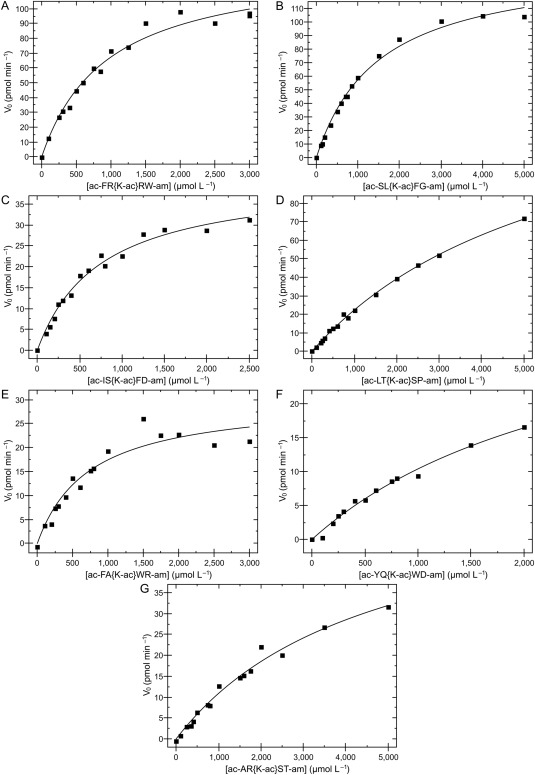
KDAC8 activity with selected peptides. KDAC8 was incubated with several concentrations of each peptide. Aliquots were removed and stopped at various timepoints to determine the initial deacetylation rate for each reaction. For each substrate, concentration was plotted against initial rate and non‐linearly fit to the Michaelis‐Menten equation. Steady‐state kinetics parameters (Table II) were calculated from these data. Peptide ac‐YQ{K‐ac}WD‐am (F) was not soluble above 2 mM.
Table 2.
Steady‐State Kinetics Parameters for Peptides with KDAC8
| Peptide sequence | k cat (s−1) | K M (µM) | k cat/K M (M−1 s−1) | Source protein(s) |
|---|---|---|---|---|
| ac‐FR{K‐ac}RW‐am | 0.162 ± 0.007 | 950 ± 110 | 171 ± 13 | ADAP1 |
| ac‐SL{K‐ac}FG‐am | 0.178 ± 0.006 | 1500 ± 120 | 118 ± 6 | SDHD |
| ac‐IS{K‐ac}FD‐am | 0.051 ± 0.002 | 750 ± 80 | 69 ± 4 | ARID1A |
| ac‐LT{K‐ac}SP‐am | >0.10 | >2000 | 32.1 ± 0.8 | CALD1 |
| ac‐FA{K‐ac}WR‐am | 0.037 ± 0.004 | 730 ± 170 | 51 ± 8 | ALDOA; ALDOC |
| ac‐YQ{K‐ac}WD‐am | >0.03 | > 2000 | 17.4 ± 1.0 | SMC3 |
| ac‐AR{K‐ac}ST‐am | >0.03 | > 2000 | 16.3 ± 1.1 | HIST3H3; H3F3A; HIST1H3A |
Applicability to other KDAC classes
To demonstrate the utility of our assay for a range of KDACs other than the class I metal‐dependent KDAC8, we tested peptides previously reported as substrates for class II (metal‐dependent) and class III (NAD‐dependent sirtuins) enzymes (Table 3) with a representative enzyme from each of these classes. Our measured specific activity for Sirt1 and its substrate is slightly less than half the previously reported value, as expected based on the low purity of the commercially available Sirt1 we utilized (Supporting Information Fig. S4) and the presence of the GST tag compared with highly purified full‐length and tag‐free Sirt1.54 While each of the three enzymes reacted in a statistically identical manner with respect to the ARID1A peptide, their selectivity for the other two peptides tested in this study were variable (Table 3). Notably, KDAC6 was more active with the three peptides identified as KDAC8 substrates than the previously reported KDAC6 substrate.
Table 3.
Endpoint Activity for Selected Peptides with Sirt1 and KDAC6
| Activity (pmol min−1 µg−1) | ||||
|---|---|---|---|---|
| Peptide sequence | Sirt1 | KDAC6 | KDAC8d | Source protein |
| ac‐QLS{K‐ac}WP‐am | 21.2 ± 2.9a | —b | —b | FOXO3 |
| ac‐DGQMPSD{K‐ac}TIGGGD‐am | —b | 1.2 ± 0.2c | —b | TUBA3A |
| ac‐FR{K‐ac}RW‐am | 38 ± 6 | 16 ± 6 | 19.5 ± 3.6 | ADAP1 |
| ac‐SL{K‐ac}FG‐am | 18.6 ± 1.4 | 4.3 ± 0.8 | 12.1 ± 2.4 | SDHD |
| ac‐IS{K‐ac}FD‐am | 7.3 ± 0.4 | 7.2 ± 2.2 | 6.9 ± 2.0 | ARID1A |
Discussion
The assay presented here can be used to monitor deacetylation of a variety of substrates, including peptides or even proteins, without the need for modification of substrates, such as attachment of a fluorophore. It is quantitative, and, because standards are used, activity is expressed as an actual rate, allowing direct comparisons between enzymes or substrates, even when other assays were used to determine activity. A limitation of this approach is that the substrate cannot contain high levels of other free amines, as they will also react with fluorescamine and lead to high background signals. Under our reaction conditions, with substrate:enzyme ratios of at least 300:1, background from the enzyme was not a significant factor even though KDAC8 contains 22 free amines (i.e., as long as substrate was at least 10–15 more concentrated than other amines on a molar basis).
This assay remains linear over a range of approximately three orders of magnitude and is highly sensitive. A sensitivity of 0.2 µM (5 pmol) was easily achieved when performing the assay. In contrast, other assays using fluorescamine to detect deacetylated lysine report much lower sensitivities (approximately 10–12 µM).44, 45 A recently reported continuous, coupled assay for metal‐dependent KDACs measuring acetate production was able to achieve 1 to 2 µM sensitivity; however, that assay has the disadvantage that it is much more complex than the assay presented here and it is not suitable when NADH or other interfering fluorophores are present.36 In fact, the only previously reported assays which show comparable sensitivities and linear range to the assay presented here involve substrates modified with fluorescent dye, such as the commercially available Fluor‐de‐Lys substrates.27 Our assay conditions also did not show any evidence of substrate or product inhibition for any of the tested peptides, unlike prior reports using alternate assay conditions,36 and we were able to quantify activity using substantially lower enzyme concentrations than typically reported in prior work.
Despite being at least equivalent to our assay in terms of sensitivity and linear range, there are several reasons why it is desirable to avoid assays utilizing fluorescently conjugated substrates. Based on previous reports that conjugating coumarin dye molecules to the substrate positively affects KDAC activity,32, 36 it is obviously advantageous to be able to evaluate enzymatic activity in the absence of these molecules. Additionally, while convenient for adapting to high‐throughput applications such as screening for small molecules which affect KDAC activity, fluorescently labeled substrates are not well‐suited for other studies. They require more involved synthesis than unlabeled substrates, making them less suitable for applications where several substrates are being queried. Also, the presence of the conjugated dye limits the ability to study the sequence surrounding the lysine, making these substrates less amenable for studies of substrate specificity. In contrast, our approach has increased versatility and better represents biological substrates compared with labeled substrates while retaining comparable sensitivity.
The fluorescamine method is inexpensive compared with coupled assays involving multiple additional enzymes, and can be used with all lysine deacetylases. As our method relies solely on detection of lysine, it should also be usable for other lysine‐modifying enzymes and the removal of functional groups other than acetate. However, these advantages do come with one significant limitation: the need to minimize the presence of other primary amines in the reaction. This limitation has two practical effects. First, the N‐terminus of the substrate should be “capped,” such as by an acetyl group. This limitation will typically only be of minor concern, as acetylating the N‐terminus also removes the positive charge of the N‐terminus and therefore better mimics most internal protein sequences. Therefore, this limitation is only significant for peptides that include the true N‐terminal sequence from the source protein. The second limitation is that the substrate cannot contain any other unmodified lysine residues without dramatically increasing the background signal. Note that the presence of arginine or modified lysine residues is not a concern. For a sufficiently active substrate, the presence of one additional lysine would not necessarily preclude measurement of the activity, but the sensitivity would drop at least an order of magnitude (comparable to changing from a phosphate buffer to MOPS or HEPES). Additional lysine residues would further reduce the sensitivity. As is true for all deacetylation assays, if multiple acetylated lysine residues are present and simultaneously deacetylated by a KDAC, the interpretation of activity may be more complex. As a significant number (but by no means a majority) of reported acetylated lysine residues include a second lysine residue nearby,1, 2, 3, 4, 5, 6 our assay is not the best choice to screen every known acetylated sequence. However, fluorescamine would be appropriate as a first approach to screen a majority of potential substrates with as many enzymes as possible and to take advantage of its high sensitivity. As the activity measurements are consistent with other assays, coupled assays could then be used for the sequences not easily measured by fluorescamine, thereby substantially reducing overall costs and increasing throughput compared with utilizing coupled assays for all sequences.
Previous attempts to address substrate specificity of KDAC8 have been conducted using panels of peptide substrates that are either conjugated to a fluorophore or are attached at one end to a solid surface.30, 31 Using these methods, the effect of residues in the positions either directly upstream (−2, −1) or directly downstream (+1, +2) on substrate specificity was analyzed independently. Using the techniques presented in those reports, it was not possible to simultaneously address the residues on either side of the lysine. A major advantage of the assay presented here is that it was possible to simultaneously investigate the effects of residues on either side of the acetylated lysine on substrate preference. We compared the normalized activity of the residues in these assays to the normalized activity of the peptides studied in our assay (Fig. 4). Surprisingly, the activity of KDAC8 against the peptides used in this study did not correlate to the predictions from either of the previous studies we analyzed in this way. In particular, we did not observe the strong bias toward aromatic residues in the +1 position that has been previously reported,31 although our substrate sequence diversity is too great to draw specific conclusions regarding substrate specificity using our limited sample size. Thus, the previously performed screens to address substrate specificity cannot accurately predict activity in a more physiological context. These discrepancies highlight the importance of using more biologically relevant substrates and assay conditions, as the context in which the acetylated lysine is presented to the enzyme affects the substrate preference. To compare activity in our assay to activity obtained in these previous studies without adding additional complexity, we focused on 5‐mer peptides lacking regular secondary structure in this study. Future investigations will be needed to further explore how amino acids more distant from the acetylated lysine and/or secondary structure affects KDAC activity. As described in a recent review of KDAC8 substrates, the low catalytic efficiency with the tested substrates, compared with typical values of 105 to 106 M−1 s−1, suggests that either better substrates exist but have yet to be identified or that the enzyme requires an unidentified cofactor or participation in a multiprotein complex for maximal activity.22
Figure 4.
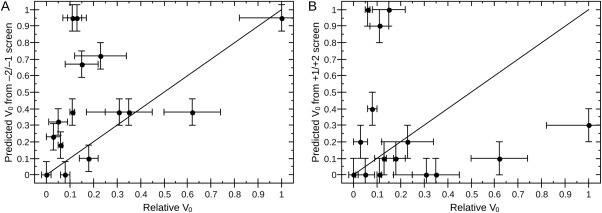
Screens with labeled substrates do not reflect activity in the fluorescamine assay. Normalized activity of endpoint assays (Table I) does not correlate with (A) a screen of all possible −2 and −1 sequences using fluorophore‐labeled peptides (r 2 = 0.18)31 or (B) a screen of all possible +1 and +2 sequences using surface‐attached peptides (r 2 = 0.02).30 Lines represent a hypothetical perfect correlation; deviation from the line indicates disagreement between observed and predicted results. Error bars come from experimental results (x‐axis) or are extrapolated based on the reported precision of values used for predictions (y‐axis).
Interestingly, one of the peptides with which KDAC8 showed the highest activity in our assay (ac‐IS{K‐ac}FD‐am) corresponds to ARID1A, a tumor suppressor which was previously identified as a KDAC8 substrate in cells and confirmed in vitro.25 We obtained a k cat/K M value that was approximately 10‐fold less than the value previously reported; however, our value was determined with the 5‐mer peptide and the previously reported value was determined with the 9‐mer. Our K M value for the 5‐mer peptide is approximately two‐fold lower than the value obtained using the 9‐mer peptide in the previous study.25 This difference in K M values for the 5‐mer and 9‐mer peptide suggests that the additional amino acids significantly influence the activity; however, the majority of the reduction is presumably due to a reduced catalytic activity with the 5‐mer peptide compared with the 9‐mer, which can largely or entirely be attributed to the greater activity of KDAC8 in the previously reported HEPES‐containing buffer. The CSRP2BP 9‐mer also exhibited higher endpoint activity than the corresponding 5‐mer, suggesting that the effect of peptide length and residues more remote from the acetylated lysine should be a focus for future studies. However, our measured catalytic efficiencies are near those reported for several potential substrates identified by a pull‐down assay.25 The K M values determined for the KDAC8 substrates are also within the range of previous reports, which are usually reported as in excess of 1 mM and often only reported as a lower limit.9, 25, 36, 49 As described previously, our assays were all conducted at 25°C in phosphate buffer at pH 7.6, which is similar to conditions used for the previously reported coupled sirtuin assay.37, 38 In contrast, the prior characterization of KDAC8 substrates was conducted in HEPES pH 8.0 and 30°C, which is closer to the pH and temperature optimum for the enzyme but less physiologically relevant. The change in reaction conditions to our phosphate buffer does result in a decreased rate. However, our phosphate buffer is a better mimic of intracellular conditions (pH <8, high potassium, low sodium) than previously reported buffers.48, 55 In addition, the use of a phosphate buffer, with a pK a substantially lower than amine‐type buffers, means that the reaction buffer pH could be lowered to reflect the conditions in different intracellular organelles down to a pH of approximately 6 and therefore cover almost the entire range of relevant intracellular pH values.56
Peptides from the other previously identified KDAC8 substrates tested in our experiments [Histone 3 lysine9 (H3K9), cortactin, and structural maintenance of chromosomes protein 3 (SMC3)] were also deacetylated by KDAC8 in our assay, albeit with a lower activity than the ARID1A peptide. These substrates were identified from cell lysate experiments in which the acetylation status of these proteins were affected by changes in KDAC8 expression in cells.21, 24, 52, 53 As the previous studies did not directly test activity of KDAC8 against these substrates in vitro, this study represents the first evidence that these sequences are substrates for KDAC8 and that the observed in vivo effects are due to direct deacetylation by KDAC8 rather than indirect effects. These results demonstrate that the assay developed here will be a powerful complementary approach to confirm deacetylation targets identified using cell‐based approaches for any lysine deacetylase. By combining results from our assay with mass spectrometry‐based experiments and cell‐based experiments as done for the examples above, we can begin to truly identify specific KDAC‐substrate pairs and understand the biological consequences of these interactions.
The assay presented here is an excellent tool for understanding KDAC substrate specificity. It is relatively simple compared with other reported deacetylase assays. Additionally, it is very sensitive and is linear over a wide range. This assay is particularly amenable for probing substrate specificity, as substrates do not require any special modification, such as a fluorophore, and the assay is designed to be easily conducted in 96‐well format, allowing multiple potential substrates to be assayed simultaneously. These advantages are critical for identifying substrates for individual KDACs, which will facilitate a major long‐term goal of the field to translate the substrate identification into greater understanding of the biological pathways regulated by KDACs. The best substrates identified here are near the activity for the best labeled substrate available for KDAC8 (compare Table 1 with 23.6 ± 3.0 pmol min−1 µg−1 for the Fluor‐de‐Lys HDAC8 substrate), and so our method could serve as replacement tool for screening for inhibitors using a more biologically relevant substrate and at substantially reduced cost compared with screens against labeled substrates. Furthermore, the techniques presented here could potentially be used to investigate nonacetyl modifications that may be removed by KDACs, as has recently been suggested.13
Materials and Methods
KDAC8 expression and purification
pJExpress401 vector (DNA 2.0) containing codon‐optimized human KDAC8, fused to a tobacco‐etch virus (TEV) protease cleavage site and His6 tag (pJExpress‐KDAC8), was used to express KDAC8 in BL21 Escherichia coli. Cells were grown in 2× YT broth at 37°C with shaking at 250 rpm. When cells reached an OD600 = 0.8–1.0, 50 µM ZnCl2 and 1 mM IPTG were added, followed by an additional 3.5 h of growth at 37°C. After induction, cells were harvested by centrifugation at 3500 rpm for 20 min at 4°C. Cells pellets were stored at −20°C until lysis.
Cells were resuspended in lysis buffer (30 mM MOPS pH 8.0, 150 mM KCl, 5% glycerol, 5 mM imidazole, 2 mM MgCl2, 1× HALT protease inhibitor [Thermo Scientific], 0.5 mg mL−1 egg white lysozyme) and incubated with rocking for 30 min on ice (typically 10 mL of lysis buffer was used per 1 L cells harvested). Cell suspensions were sonicated three times at 30% amplitude for 10 sec (Fisher Scientific Sonic Dismembrator Model 120, 1/8″ probe), followed by 30 sec on ice. Lysates were clarified by centrifugation at 27,000g for 20 min at 4°C.
Clarified lysate was added to TALON resin (Clontech) equilibrated with column buffer (30 mM MOPS pH 8.0, 150 mM KCl, 5% glycerol, 5 mM imidazole) and incubated on ice for 15 min with rocking (resin bed volume of 1 mL per 10 mL lysis buffer). Resin was pelleted by centrifugation at 700g for 5 min and washed twice with 10 bed volumes of column buffer each time. After final centrifugation, resin was transferred to column housing and washed with an additional 10 bed volumes of column buffer. KDAC8 was eluted (30 mM MOPS pH 8.0, 150 mM KCl, 5% glycerol, 150 mM imidazole) and collected in fractions. TEV protease was added (1:25) to fractions containing protein, and the mixture was dialyzed in TEV cleavage buffer (30 mM MOPS pH 8.0, 150 mM KCl, 5% glycerol, 1 mM 2‐mercaptoethanol, 0.3 mM EDTA pH 7.0) overnight at 4°C with one buffer change. This was followed by dialysis into buffer containing 30 mM MOPS pH 8.0, 150 mM KCl, and 5% glycerol overnight at 4°C with one buffer change.
Following cleavage with TEV protease, protein was flowed over TALON resin equilibrated with the final dialysis buffer for secondary purification. Purified KDAC8 (flow‐through) was collected. Glycerol and tris(2‐carboxyethyl)phosphine (TCEP) were added to final concentrations of 25% and 1 mM, respectively. ZnCl2 was added to an equimolar ratio to KDAC8 and protein was stored at −20°C. Using this storage method, KDAC8 activity was stable for at least several months. This protocol typically yielded approximately 1 mg KDAC8 per liter of culture, at a purity of >95% (assessed by SDS‐PAGE and stained with GelCode Blue (Thermo Scientific); Supporting Information Fig. S5).
TEV protease expression and purification
His6‐tagged TEV protease was expressed and purified from E. coli transformed with pRK793 (AddGene 8827) using metal affinity chromatography.57 Briefly, cells were grown in 2× YT broth at 37°C with shaking at 250 rpm until OD600 reached approximately 1.0 and then induced with 1 mM IPTG. After 4 h, cells were harvested and TEV protease was purified using TALON resin, similarly to the protocol above for KDAC8. The protein of interest was eluted (30 mM MOPS pH 8.0, 150 mM KCl, 5% glycerol, 150 mM imidazole) and fractions containing protein were dialyzed into storage buffer (30 mM MOPS pH 8.0, 150 mM KCl, 25% glycerol, 1 mM TCEP) and stored at −20°C.
Fluorescamine assay
Peptides were custom synthesized with acetylated lysine in the middle position, as well as an acetylated N‐terminus and an amidated C‐terminus (Genscript). All peptide masses were confirmed by mass spectrometry, purified to >95% by HPLC, and stored as concentrated stocks in buffer or DMSO as determined by solubility. For endpoint assays, 100 µM peptide substrates were incubated with 200 nM KDAC8, 200 nM human Sirtiun1(193‐741)‐GST (Sirt1, BPS Bioscience), or 20 nM human HDAC6‐GST (KDAC6, BPS Bioscience) at 25°C for 60 min in reaction buffer (30 mM potassium phosphate pH 7.6, 100 mM KCl, 5% glycerol) or HEPES reaction buffer (50 mM HEPES pH 8.0, 137 mM NaCl, 2.7 mM KCl) in a reaction volume of 90 µL. Reactions with Sirt1 also included 500 µM NAD+. Reactions were stopped by addition of suberoylanilide hydroxamic acid (SAHA) to a concentration of 100 µM for metal‐dependent KDACs or 5 mM nicotinamide for Sirt1. For each reaction, an identical inhibited reaction was prepared which contained 100 µM SAHA or 5 mM nicotinamide preincubated with enzyme before addition of substrate. Additional enzyme‐only and substrate‐only controls were also subjected to the same reaction conditions to verify the effectiveness of the inhibitor. The reaction mixture was diluted 1:1 with 0.5 M NaCl and 50 µL aliquots were added to black polypropylene 96‐well plates (Corning) in triplicate. 50 µL of 0.1 mg mL−1 fluorescamine in spectroscopic grade DMSO was then added to each well. Plates were incubated at room temperature for at least 20 min. Fluorescence was detected in a microplate reader (BioTek) at 390 nm excitation and 485 nm emission.39
To create a standard curve, several concentrations (ranging from 100 µM to 0.16 µM; 2500–4 pmol per well) of unacetylated lysine (N‐α‐acetyl‐l‐lysine amide hydrochloride, ac‐Lys‐NH2; Chem‐Impex International) were prepared and incubated in the same manner as reactions, the fluorescence was measured, and pmol versus fluorescence was linearly fit. For each reaction, the initial rate (V 0) was calculated by subtracting the average raw fluorescence of the inhibited reaction from the average raw fluorescence of the reaction. This number was divided by the slope of the standard curve and corrected for NaCl dilution and to scale from the volume of a single well to the total reaction volume. The resulting pmol were then divided by the reaction time and enzyme concentration to report specific activity, which allowed for direct comparison between experiments. Specific activity values were averaged for at least three reactions with each peptide. Statistical outliers, calculated using the Grubbs method,58 were tested for and excluded when detected in any data sets of at least four independent measurements.
Fluor‐de‐Lys assay
Assays were performed identically to the fluorescamine assay except for the following changes. The substrate used was the Fluor‐de‐Lys HDAC8 substrate (Enzo Life Sciences), which has the sequence ac‐RH{K‐ac}{K‐ac}‐7‐amino‐4‐methylcoumarin. Assays were performed in either reaction buffer or HEPES reaction buffer. After stopping the reactions, 2 mg mL−1 trypsin (MP Biomedical) in reaction buffer was added instead of 0.5 M NaCl, and the mixtures were incubated at 37°C for 15 min. The mixtures were then added in triplicate to the 96‐well plate and fluorescence measured with excitation at 360 nm and emission at 460 nm. Fluorescence was converted to rates using a standard curve of 7‐amino‐4‐methylcoumarin (Alfa Aesar).
Fluorescamine steady‐state kinetics assay conditions
Fifteen concentrations of peptide substrates were incubated with 150 nM KDAC8 in reaction buffer at 25°C. Aliquots were taken from the reactions at several timepoints (0, 10, 20, 40, 60 min), and the reactions were stopped by addition of SAHA to 100 µM. Aliquots continued to incubate at 25°C until the final timepoint. Each aliquot was processed as described above for stopped reactions. A standard curve was also generated for each experiment as described above.
For each reaction, the slope resulting from fluorescence versus time was obtained. These slopes were converted to the rate of substrate conversion (pmol min−1) by dividing by the slope of the standard curve. Rates for each substrate concentration were plotted, and Michaelis‐Menten steady‐state parameters were calculated using QTIplot software by nonlinearly fitting the Michaelis‐Menten equation. Catalytic efficiency was calculated directly from velocity data using a derivative of the Briggs‐Haldane equation as described elsewhere,36 even for substrates for which K M and k cat could not be reliably determined individually.
Temperature stability assays
A steady‐state kinetics fluorescamine assay was performed as described above with a few modifications to address enzyme stability at higher temperature. 200 µM of each peptide substrate or Fluor‐de‐Lys reagent (Enzo Life Sciences) was incubated with 125 nM KDAC8 in reaction buffer at either 25°C or 37°C. Aliquots of each reaction were stopped at 0, 0.5, 1, 1.5, 2, 2.5, and 3 h and worked up as described above for the corresponding assay. Stability was also monitored by CD spectrophotometry using a J‐1500 spectrapolarimeter (Jasco). 500 nM KDAC8 in reaction buffer was incubated at either 25°C or 37°C. Spectra were obtained from 200 to 245 nm at a scan rate of 5 nm min−1 with a 16 s integration every 1.0 nm in a 2 mm quartz cuvette with a 1.0 nm bandwidth. One buffer‐corrected spectrum was taken every 10 min for 3 h at each temperature.
Supporting information
Supporting Information
References
- 1. Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang X‐J, Zhao Y (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23:607–618. [DOI] [PubMed] [Google Scholar]
- 2. Basu A, Rose KL, Zhang J, Beavis RC, Ueberheide B, Garcia BA, Chait B, Zhao Y, Hunt DF, Segal E, Allis CD, Hake SB (2009) Proteome‐wide prediction of acetylation substrates. Proc Natl Acad Sci USA 106:13785–13790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M (2009) Lysine acetylation targets protein complexes and co‐regulates major cellular functions. Science 325:834–840. [DOI] [PubMed] [Google Scholar]
- 4. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan K‐L (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327:1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lundby A, Lage K, Weinert BT, Bekker‐Jensen DB, Secher A, Skovgaard T, Kelstrup CD, Dmytriyev A, Choudhary C, Lundby C, Olsen JV (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep 2:419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schölz C, Weinert BT, Wagner SA, Beli P, Miyake Y, Qi J, Jensen LJ, Streicher W, McCarthy AR, Westwood NJ, Lain S, Cox J, Matthias P, Mann M, Bradner JE, Choudhary C (2015) Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat Biotechnol 33:415–423. [DOI] [PubMed] [Google Scholar]
- 7. Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP (1999) Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401:188–193. [DOI] [PubMed] [Google Scholar]
- 8. Chen K, Zhang X, Wu Y‐D, Wiest O (2014) Inhibition and mechanism of HDAC8 revisited. J Am Chem Soc 136:11636–11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gantt SL, Gattis SG, Fierke CA (2006) Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry 45:6170–6178. [DOI] [PubMed] [Google Scholar]
- 10. Aka JA, Kim G‐W, Yang X‐J (2011) K‐acetylation and its enzymes: overview and new developments. Handb Exp Pharmacol 206:1–12. [DOI] [PubMed] [Google Scholar]
- 11. Yao Y‐L, Yang W‐M (2011) Beyond histone and deacetylase: an overview of cytoplasmic histone deacetylases and their nonhistone substrates. J Biomed Biotechnol 2011:146493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sando R, 3rd Gounko N, Pieraut S, Liao L, Yates J, III, Maximov A (2012) HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 151:821–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Olsen CA (2014) An update on lysine deacylases targeting the expanding “acylome.”. ChemMedChem 9:434–437. [DOI] [PubMed] [Google Scholar]
- 14. Peng L, Seto E (2011) Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handb Exp Pharmacol 206:39–56. [DOI] [PubMed] [Google Scholar]
- 15. Tang J, Yan H, Zhuang S (2013) Histone deacetylases as targets for treatment of multiple diseases. Clin Sci 124:651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X, Liu J, Zhen J, Zhang C, Wan Q, Liu G, Wei X, Zhang Y, Wang Z, Han H, Xu H, Bao C, Song Z, Zhang X, Li N, Yi F (2014) Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int 86:712–725. [DOI] [PubMed] [Google Scholar]
- 17. Tiwari S, Dharmarajan S, Shivanna M, Otteson DC, Belecky‐Adams TL (2014) Histone deacetylase expression patterns in developing murine optic nerve. BMC Dev Biol 14:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang D‐F, Wiest O, Helquist P, Lan‐Hargest H‐Y, Wiech NL (2004) On the function of the 14 A long internal cavity of histone deacetylase‐like protein: implications for the design of histone deacetylase inhibitors. J Med Chem 47:3409–3417. [DOI] [PubMed] [Google Scholar]
- 19. Wang D‐F, Helquist P, Wiech NL, Wiest O (2005) Toward selective histone deacetylase inhibitor design: homology modeling, docking studies, and molecular dynamics simulations of human class I histone deacetylases. J Med Chem 48:6936–6947. [DOI] [PubMed] [Google Scholar]
- 20. Haider S, Joseph CG, Neidle S, Fierke CA, Fuchter MJ (2011) On the function of the internal cavity of histone deacetylase protein 8: R37 is a crucial residue for catalysis. Bioorg Med Chem Lett 21:2129–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI, Cristea IM (2013) The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol 9:672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wolfson NA, Ann Pitcairn C, Fierke CA (2013) HDAC8 substrates: histones and beyond. Biopolymers 99:112–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Europe‐Finner GN, Karolczak‐Bayatti M, Taggart MJ (2015) The multifaceted KDAC8: a smooth muscle contractile regulator. Trends Pharmacol Sci 36:493 [DOI] [PubMed] [Google Scholar]
- 24. Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E, Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T, Lombardi PM, Magnaghi‐Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems PJ, Prigent C, Gillessen‐Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, Krantz ID, Shirahige K (2012) HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 489:313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Olson DE, Udeshi ND, Wolfson NA, Pitcairn CA, Sullivan ED, Jaffe JD, Svinkina T, Natoli T, Lu X, Paulk J, McCarren P, Wagner FF, Barker D, Howe E, Lazzaro F, Gale JP, Zhange Y‐L, Subramanian A, Fierke CA, Carr SA, Holson EB (2014) An unbiased approach to identify endogenous substrates of “histone” deacetylase 8. ACS Chem Biol 2014:2210–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heltweg B, Jung M (2002) A microplate reader‐based nonisotopic histone deacetylase activity assay. Anal Biochem 302:175–183. [DOI] [PubMed] [Google Scholar]
- 27. Wegener D, Wirsching F, Riester D, Schwienhorst A (2003) A fluorogenic histone deacetylase assay well suited for high‐throughput activity screening. Chem Biol 10:61–68. [DOI] [PubMed] [Google Scholar]
- 28. Riester D, Hildmann C, Schwienhorst A, Meyer‐Almes F‐J (2007) Histone deacetylase inhibitor assay based on fluorescence resonance energy transfer. Anal Biochem 362:136–141. [DOI] [PubMed] [Google Scholar]
- 29. Gurard‐Levin ZA, Mrksich M (2008) The activity of HDAC8 depends on local and distal sequences of its peptide substrates. Biochemistry 47:6242–6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gurard‐Levin ZA, Kilian KA, Kim J, Bahr K, Mrksich M (2010) Peptide arrays identify isoform‐selective substrates for profiling endogenous lysine deacetylase activity. ACS Chem Biol 5:863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Riester D, Hildmann C, Grunewald S, Beckers T, Schwienhorst A (2007) Factors affecting the substrate specificity of histone deacetylases. Biochem Biophys Res Commun 357:439–445. [DOI] [PubMed] [Google Scholar]
- 32. Gurard‐Levin ZA, Kim J, Mrksich M (2009) Combining mass spectrometry and peptide arrays to profile the specificities of histone deacetylases. Chembiochem 10:2159–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buggy JJ, Sideris ML, Mak P, Lorimer DD, McIntosh B, Clark JM (2000) Cloning and characterization of a novel human histone deacetylase, HDAC8. Biochem J 350 Pt 1:199–205. [PMC free article] [PubMed] [Google Scholar]
- 34. Van den Wyngaert I, de Vries W, Kremer A, Neefs J, Verhasselt P, Luyten WH, Kass SU (2000) Cloning and characterization of human histone deacetylase 8. FEBS Lett 478:77–83. [DOI] [PubMed] [Google Scholar]
- 35. Smith BC, Denu JM (2007) Acetyl‐lysine analog peptides as mechanistic probes of protein deacetylases. J Biol Chem 282:37256–37265. [DOI] [PubMed] [Google Scholar]
- 36. Wolfson NA, Pitcairn CA, Sullivan ED, Joseph CG, Fierke CA (2014) An enzyme‐coupled assay measuring acetate production for profiling histone deacetylase specificity. Anal Biochem 456:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith BC, Hallows WC, Denu JM (2009) A continuous microplate assay for sirtuins and nicotinamide‐producing enzymes. Anal Biochem 394:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hubbard BP, Sinclair DA (2013) Measurement of sirtuin enzyme activity using a substrate‐agnostic fluorometric nicotinamide assay. Methods Mol Biol 1077:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Udenfriend S, Stein S, Bohlen P, Dairman W, Leimgruber W, Weigele M (1972) Fluorescamine: a reagent for assay of amino acids, peptides, proteins, and primary amines in the picomole range. Science 178:871–872. [DOI] [PubMed] [Google Scholar]
- 40. Chen RF, Smith PD, Maly M (1978) The fluorescence of fluorescamine‐amino acids. Arch Biochem Biophys 189:241–250. [DOI] [PubMed] [Google Scholar]
- 41. De Bernardo S, Weigele M, Toome V, Manhart K, Leimgruber W, Bohlen P, Stein S, Udenfriend S (1974) Studies on the reaction of fluorescamine with primary amines. Arch Biochem Biophys 163:390–399. [DOI] [PubMed] [Google Scholar]
- 42. Böhlen P, Stein S, Dairman W, Udenfriend S (1973) Fluorometric assay of proteins in the nanogram range. Arch Biochem Biophys 155:213–220. [DOI] [PubMed] [Google Scholar]
- 43. Blair DE, Schuttelkopf AW, MacRae JI, van Aalten DMF (2005) Structure and metal‐dependent mechanism of peptidoglycan deacetylase, a streptococcal virulence factor. Proc Natl Acad Sci USA 102:15429–15434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang X, Hernick M (2011) A fluorescence‐based assay for measuring N‐acetyl‐1‐d‐myo‐inosityl‐2‐amino‐2‐deoxy‐alpha‐d‐glucopyranoside deacetylase activity. Anal Biochem 414:278–281. [DOI] [PubMed] [Google Scholar]
- 45. Wang W, Maniar M, Jain R, Jacobs J, Trias J, Yuan Z (2001) A fluorescence‐based homogeneous assay for measuring activity of UDP‐3‐O‐(R‐3‐hydroxymyristoyl)‐N‐acetylglucosamine deacetylase. Anal Biochem 290:338–346. [DOI] [PubMed] [Google Scholar]
- 46. Godt RE, Maughan DW (1988) On the composition of the cytosol of relaxed skeletal muscle of the frog. Am J Physiol 254:C591–C604. [DOI] [PubMed] [Google Scholar]
- 47. Stein S, Bohlen P, Udenfriend S (1974) Studies on the kinetics of reaction and hydrolysis of fluorescamine. Arch Biochem Biophys 163:400–403. [DOI] [PubMed] [Google Scholar]
- 48. Freund J, Kalbitzer HR (1995) Physiological buffers for NMR spectroscopy. J Biomol NMR 5:321–322. [DOI] [PubMed] [Google Scholar]
- 49. Schultz BE, Misialek S, Wu J, Tang J, Conn MT, Tahilramani R, Wong L (2004) Kinetics and comparative reactivity of human class I and class IIb histone deacetylases. Biochemistry 43:11083–11091. [DOI] [PubMed] [Google Scholar]
- 50. Froechlich PM, Cunningham TD (1976) The enhancement of the fluorescence of fluorescamine derivatives in mixed dimethylsulfoxide‐water solvents. Anal Chim Acta 84:427–430. [Google Scholar]
- 51. De Silva JA, Strojny N (1975) Spectrofluorometric determination of pharmaceuticals containing aromatic or aliphatic primary amino groups as their fluorescamine (fluram) derivatives. Anal Chem 47:714–718. [DOI] [PubMed] [Google Scholar]
- 52. Fu Y, Zhang P, Ge J, Cheng J, Dong W, Yuan H, Du Y, Yang M, Sun R, Jiang H (2014) Histone deacetylase 8 suppresses osteogenic differentiation of bone marrow stromal cells by inhibiting histone H3K9 acetylation and RUNX2 activity. Int J Biochem Cell Biol 54:68–77. [DOI] [PubMed] [Google Scholar]
- 53. Li J, Chen S, Cleary RA, Wang R, Gannon OJ, Seto E, Tang DD (2014) Histone deacetylase 8 regulates cortactin deacetylation and contraction in smooth muscle tissues. Am J Physiol Cell Physiol 307:C288–C295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cao D, Wang M, Qiu X, Liu D, Jiang H, Yang N, Xu R‐M (2015) Structural basis for allosteric, substrate‐dependent stimulation of SIRT1 activity by resveratrol. Genes Dev 29:1316–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Seidel D, Schmidt‐Gayk H (1980) Innere Medizin, 5th ed Thieme: Stuttgart, pp 548–562. [Google Scholar]
- 56. Demaurex N (2002) pH Homeostasis of cellular organelles. News Physiol Sci Int J Physiol Prod Jointly Int Union Physiol Sci Am Physiol Soc 17:1–5. [DOI] [PubMed] [Google Scholar]
- 57. Kapust RB, Tözsér J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS (2001) Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild‐type catalytic proficiency. Protein Eng 14:993–1000. [DOI] [PubMed] [Google Scholar]
- 58. Grubbs F (1969) Procedures for detecting outlying observations in samples. Technometrics 11:1–21. [Google Scholar]
- 59. Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Matthias P (2003) HDAC‐6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J 22:1168–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information