

Abstract
Lpg0406, a hypothetical protein from Legionella pneumophila, belongs to carboxymuconolactone decarboxylase (CMD) family. We determined the crystal structure of lpg0406 both in its apo and reduced form. The structures reveal that lpg0406 forms a hexamer and have disulfide exchange properties. The protein has an all‐helical fold with a conserved thioredoxin‐like active site CXXC motif and a proton relay system similar to that of alkylhydroperoxidase from Mycobacterium tuberculosis (MtAhpD), suggesting that lpg0406 might function as an enzyme with peroxidase activity and involved in antioxidant defense. A comparison of the size and the surface topology of the putative substrate‐binding region between lpg0406 and MtAhpD indicates that the two enzymes accommodate the different substrate preferences. The structural findings will enhance understanding of the CMD family protein structure and its various functions.
Keywords: Legionella pneumophila, carboxymuconolactone decarboxylase family, lpg0406, hexameric ring structure, alkylhydroperoxidase
Short abstract
Interactive Figure 1 | PDB Code(s): 5dik; 5dip
Introduction
The carboxymuconolactone decarboxylase (CMD) family includes alkylhydroperoxidase (AhpD), γ‐CMD, and a distinct member protein TTHA0727 from Thermus thermophiles HB8. AhpD participates in bacterial antioxidant defense by regenerating its partner AhpC for further reduction of peroxides into alcohols. It has a conserved thioredoxin‐like active site CXXC motif critical for the peroxidase activity. γ‐CMD catalyzes the conversion of γ‐Carboxymucolactone to β‐ketoadipate enol‐lactione in the β‐ketoadipate pathway,1 which is a key part of the degradation process of aromatic compounds in bacteria and in some eukaryotes. However, the active site residues of γ‐CMD have not been identified. TTHA0727 lacks peroxidase activity due to the absence of the CXXC motif and its function is unknown.2
Known structures for the CMD family include Mycobacterium tuberculosis AhpD (MtAhpD, PDB codes 1knc, 1gu9, 1me5),3, 4, 5 the protein TTHA0727 (PDB code 2cwq),2 protein PA0269 from Pseudomonas aeruginosa (PDB code 2o4d)6 and a number of unpublished structures (PDB codes 1p8c, 1vke, 3d7i, 2qeu, 2af7, and 3bey). According to the previous studies,2, 3, 4, 5, 6, 7 the CMD family proteins mediate a various biochemical reactions and contain a conserved structural motif, CMD core, which is involved in the formation of the structural core region and stable multimerization.
Lpg0406 from Legionella pneumophila is 113 amino acids long, with a molecular mass of 12.2 kDa. Size‐exclusion chromatography of the protein expressed in Escherichia coli suggests that lpg0406 is present as an oligomeric, possibly hexameric, species. Similar to AhpD, the lpg0406 sequence has a CXXC motif. The two cysteines are separated by a glycine and a proline. In order to better understand the details of the biological functions, we determined the crystal structure of lpg0406 in its apo‐state and in reduced form. Our structural analyses suggest that lpg0406 might function as an enzyme with peroxidase activity and involved in antioxidant defense.
Results and Discussion
The refined model contains three lpg0406 monomers in the asymmetric unit. The lpg0406 protomer has an AhpD‐like fold consisting of six α helices (α1‐α6), a V‐shaped N‐terminal helical hairpin (α1, α2) followed by the three‐helical CMD core (α3‐α5, residues 44‐94) and C‐terminal helix (α6).[Fig. 1(A)]
Figure 1.
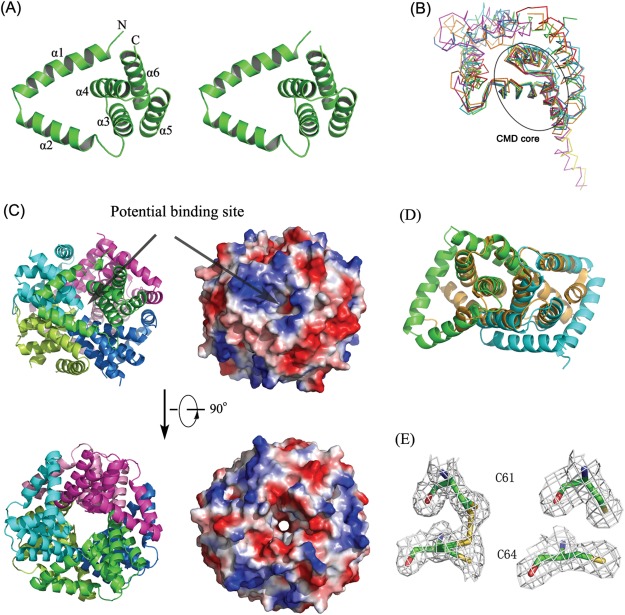
A: Cartoon representation of lpg0406 structure (Stereo view). The N and C termini and each of the six alpha helices are labeled. An interactive view is available in the electronic version of the article. B: Superposition of lpg0406 structure (green), TM1620 from T. maritima (PDB code 1p8c) (red), MJ0742 from M. jannaschii (PDB code 3d7i) (cyan), putative CMD from B. xenovorans LB400 (PDB code 2qeu; orange), γ‐CMD from M. thermoautotrophicum (PDB code 2af7; blue), O27018 from M. thermoautotrophicum (PDB code 3bey; yellow), and TTHA0727 from T. thermophilus HB8 (PDB code 2cwq; magenta). C: Ribbon representation and the electrostatic surface plots of the hexamer organization of lpg0406 with each of the subunits colored separately in green (chain A), cyan (chain B), magenta (chain C), limon (chain A′), blue (chain B′), and pink (chain C′). The positive surface was drawn in blue and negative surface in red. Arrows point to the putative substrate binding site. An interactive view is available in the electronic version of the article. (D) Superposition of the lpg0406 dimer (colored in cyan and green) and MtAhpD monomer (orange). E: Electron density map contoured at 1.0δ for lpg0406 active‐site cysteines in apo form (left 1.9 A) and reduced (right, 2.1 A). 83 × 81mm (300 × 300 DPI).
Structure homology search by the DALI server8 revealed that 0406 has the highest similarity to TM1620 from Thermotoga maritima (PDB codes 1p8c and 1vke), with a Z score of 15.5 and a root‐mean‐square deviation (RMSD) of 1.8 Å for 110 common Cα atoms. The superimposition of lpg0406 monomer structure and its homologs showed that the architecture of the CMD core motif is remarkably conserved, and supported that the three α‐helix structure feature is the common structural motif for the CMD family proteins. Despite of the high similarity in CMD core, there are some significant differences in the size and orientation of the other parts [Fig. 1(B)].
The structures of the three lpg0406 chains are essentially similar, with RMSD (root mean square deviations) of only 0.06 Å for main chain atoms. The three monomers form a homohexamer with their crystallographic symmetry (CS) mates [Fig. 1(C)]. The interactions between helices α2‐α5, including the typical CMD‐CMD contacts, results in three tight dimers formation (Chains A‐B′, B‐A′, and C‐C′). The interactions between chains A‐A′, B‐C′, C‐B′ by using α2 further stabilize the hexamer ring structure. Hydrophobic interactions make the major contribution to the stability of the whole ring architecture. Structural superposition of MtAhpD and lpg0406 structures indicated that lpg0406 dimer and hexamer are structurally related to MtAhpD monomer and trimer, respectively [Fig. 1(D)]. Similar to MtAhpD and TTHA0727 structures, there is a small tunnel in the center of lpg0406 hexameric ring [Fig. 1(C)].
In apo‐form structure, Cys61 and Cys64 are modeled in two alternative conformations for oxidized and reduced states, which indicate lpg0406 probably has disulfide exchange properties [Fig. 1(E)]. To test if the disulfides could be fully converted into sulfhydryls, we resolved the lpg0406 structure in reduced form. When soaked for 5 min in a solution consisting of 2mM DTT, these crystals were found to be reproducibly changed to the space group I213 with an intact dimer in the asymmetric unit. However, this dimer appears to trimerize by the crystallographic 3‐fold symmetry axis to form a homohexameric ring structure. The hexamer remained intact, and overlap very well after aligning with its apo‐form structure. As expected, the cysteine residues in the active site CXXC motif were present in the reduced form, in which the sulfhydryls of the two cysteines lie 3.6 Å apart [Fig. 1(E)]. Except this, the reduction induced no other notable conformational change.
In MtAhpD structure, the proton relay system consists of five residues, Glu118, Cys130, His132, Cys133, and His137, and one water molecule.4, 5 Lpg0406 shares the CXXC signature with AhpD. The motif is located at the fourth helix (α4) and near the central cavity of the hexamer. Structure‐based alignment revealed that the active site of lpg0406 is structurally similar to that of MtAhpD [Fig. 2(A)]. His68 forms hydrogen bond with Cys64 via a structurally conserved water molecule. The carboxylate anion of Glu49 is hydrogen‐bonded to the nitrogen of His68. These hydrogen‐bonding interactions provide a reasonable mechanism for deprotonation of Cys64, the residue thought to react with the O—O bond of peroxides [Fig. 2(B)]. In MtAhpD, His132 could serve as a base for deprotonation of either cysteine. The analogues position in lpg0406 is occupied by Gly63, which does not have the same effect. Based on the analysis of PA0269 structure, the catalytic role of His132 in MtAhpD could be played by tyrosine.6 In lpg0406 structure, the location of Tyr100* (from a neighboring subunit) 4.6 Å from Cys61, and Tyr91′ (from another neighboring subunit) 6.5 Å from Cys64 and 3.8 Å from His68 suggested that they might be involved in the proton relay system [Fig. 2(B)]. Therefore, the hexameric assembly may be of importance in maintaining the structure of the active site, suggesting the hexameric form is the functioning biological unit.
Figure 2.
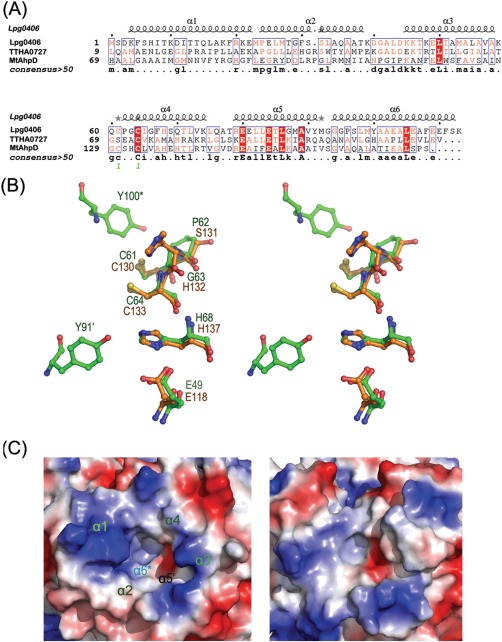
A: Structure‐based sequence alignment of lpg0406, MtAhpD (PDB code 1knc) and TTHA0727 (PDB code 2cwq). B: Superposition of the putative proton relay system of lpg0406 (green) and MtAhpD (orange). The “′” and “*” indicate the residues from the different neighboring subunits. C: Comparison of the electrostatic surface plots of putative substrate‐binding pocket between lpg0406 (left) and MtAhpD (right). The positive surface was drawn in blue and negative surface in red. α1, α2, α3, and α4 are from a single subunit, α5′ and α6* are from different neighboring subunits.
On the basis of the structures of MtAhpD and protein TTHA0727, the putative substrate binding site is a cleft region which extends from the outer, solvent‐exposed edge to the active site CXXC motif [Figs. 1(C) and 2(C)]. All helixes appear to be involved in the binding site construction. The lpg0406 substrate‐binding site is a pocket, with positively charged outer edges and internal hydrophobic region, formed by residues from α1‐α4 of an individual subunit, and α5', α6* of two different neighboring subunits. Despite of the high similarity in active site, there are some notable differences in the size and the surface topology of the putative substrate‐binding region between lpg0406 and MtAhpD [Fig. 2(C)]. Substrate‐binding pocket of lpg0406 is much shallower and smaller than that of MtAhpD, suggesting that the two enzymes accommodate the different substrate preferences.
Taken together, the structural findings suggested that lpg0406 is probably an enzyme with peroxidase activity and involved in antioxidant defense. Therefore, lpg0406 is more functionally related to AhpD than γ‐CMD and protein TTHA0727.
Materials and Methods
Cloning, expression, purification, and crystallization were described as shown in the electronic Supporting Information.
Data collection and processing
The apo‐lpg0406 X‐ray data were collected at 100 K from a single crystal diffracting X‐ray to 1.9 Å resolution at a wavelength of 0.97915 Å. A total of 250 images were recorded with 1 s exposure using an oscillation range of 1°. While the SeMet‐lpg0406 X‐ray data were collected at 100 K from a single crystal diffracting X‐ray to 2.3 Å resolution at a wavelength of 0.97911 Å. A total of 265 images were recorded with 0.5s exposure at a crystal‐to‐detector distance of 250 mm using an oscillation range of 1°. Reduced_lpg0406 crystals were obtained by soaking apo‐lpg0406 crystals with 2 mM DTT and the diffraction were collected in a similar way as described above but at a wavelength of 0.97861 Å.
The SeMet‐lpg0406 data were processed with Mosflm9 and scaled using the CCP4 suite.10 Native data sets for apo and reduced crystals were processed and scaled with HKL‐200011 and programs from the CCP4 package.10 All crystal parameters and data collection statistics are summarized in Table 1.
Table 1.
Data Collection and Refinement Statistics
| Apo | Reduced | Se‐peak | |
|---|---|---|---|
| Wavelength (Å) | 0.97915 | 0.97861 | 0.97911 |
| Space group | C2221 | I213 | C2221 |
| Cell parameters | |||
| a/b/c (Å) | 62.80/104.81/106.75 | 145.71/145.71/145.71 | 62.65/104.75/106.41 |
| Resolution range (Å) | 50–1.9(1.97–1.90) | 50–2.1 (2.14–2.10) | 48–2.3 (2.42–2.30) |
| No. of unique reflections | 27,567 (2759) | 30,286 (1523) | 15,238 (2304) |
| R merge a (%) | 11.1 (41.4) | 11.2 (42.5) | 10.6 (17.6) |
| I/σ(I) | 9.0 (3.6) | 29.1 (10.6) | 15.5 (10.8) |
| Redundancy | 2.9 (2.9) | 20.3 (20.8) | 9.8 (10.3) |
| Completeness (%) | 98.6 (99.9) | 100 (100) | 95.6 (100) |
| Anomalous completeness | 95.5 (100) | ||
| Anomalous multiplicity | 5.2 (5.3) | ||
| Refinement summary | |||
| R factorb (%) | 18.3 | 13.4 | |
| Free R factorc (%) | 20.4 | 15.8 | |
| rmsdd in bond lengths (Å) | 0.011 | 0.007 | |
| rmsd in bond angles (°) | 1.38 | 1.12 | |
| No. of protein atoms/ASU | 2575 | 1762 | |
| No. of water molecules/ASU | 79 | 210 | |
| Ramachandran plot (%) | |||
| Ramachandran favored | 99 | 99 | |
| Ramachandran outliers | 0 | 0 | |
| Average B‐factor(Å2) | 24.60 | 24.8 | |
| PDB ID code | 5DIK | 5DIP |
Values in parentheses refer to the highest resolution shell.
Rmerge=∑|Ihkl ‐ < Ihkl >|/< Ihkl >, where Ihkl is a single value of the measured intensity of the hkl reflection and < Ihkl > is the mean of all measured values of the intensity of the hkl reflections.
R‐factor =∑h | |Fobs|−|Fcalc| |/∑|Fobs|, where |Fobs| and |Fcalc| are the observed and calculated structure factor amplitudes, respectively. Summation includes all reflections used in the refinement.
Free R factor = ∑| |Fobs|−|Fcalc| |/∑|Fobs|, evaluated for a randomly chosen subset of 5% of the diffraction data not included in the refinement.
Root‐mean square‐deviation from ideal values.
Structure determination and refinement
The initial phase was calculated using AutoSol, and an initial model was built using AutoBuild from PHENIX.12 Using the native data set and the initial model as a search coordinate, the structure of apo‐lpg0406 was determined by molecular replacement with the Phaser program.13 The model was completed by iterative manual building in Coot14 and refined with REFMAC15 and PHENIX.12 The reduced‐lpg0406 structure was solved by molecular replacement using the apo‐lpg0406 monomer structure as a search model. Both structures contain all the residues except the recombinant 6‐his tag and the initiation methionine. All final crystallographic models were evaluated using MolProbity.16 The refinement statistics are summarized in Table 1. The coordinates and structure factors have been deposited in the Protein Data Bank under the accession code 5dik and 5dip.
Sequence analysis and structural presentation
Amino acid sequences were aligned by Multalin,17 and the figure of structure‐based sequence alignment was generated using ESPript.18 All illustrations were prepared with PyMOL.19
Supporting information
Supporting Information
Acknowledgments
The authors thank Professor Maikun Teng and Dr. Zhongliang Zhu at USTC for generous assistance, and Dr. Zhaoqing Luo (Purdue University) for L. pneumophila genomic DNA. We also thank the staff at SSRF beamline BL17U1 for assistance with synchrotron data collection.
References
- 1. Parke D, Meagher RB, Ornston LN (1973) Relationships among enzymes of the beta‐ketoadipate pathway. 3. Properties of crystalline gamma‐carboxymuconolactone decarboxylase from Pseudomonas putida. Biochemistry 12:3537–3542. [DOI] [PubMed] [Google Scholar]
- 2. Ito K, Arai R, Fusatomi E, Kamo‐Uchikubo T, Kawaguchi S, Akasaka R, Terada T, Kuramitsu S, Shirouzu M, Yokoyama S (2006) Crystal structure of the conserved protein TTHA0727 from Thermus thermophilus HB8 at 1.9 A resolution: A CMD family member distinct from carboxymuconolactone decarboxylase (CMD) and AhpD. Protein Sci 15:1187–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bryk R, Lima CD, Erdjument‐Bromage H, Tempst P, Nathan C (2002) Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin‐like protein. Science 295:1073–1077. [DOI] [PubMed] [Google Scholar]
- 4. Nunn CM, Djordjevic S, Hillas PJ, Nishida CR, Ortiz de Montellano PR (2002) The crystal structure of Mycobacterium tuberculosis alkylhydroperoxidase AhpD, a potential target for antitubercular drug design. J Biol Chem 277:20033–20040. [DOI] [PubMed] [Google Scholar]
- 5. Koshkin A, Nunn CM, Djordjevic S, Ortiz de Montellano PR (2003) The mechanism of Mycobacterium tuberculosis alkylhydroperoxidase AhpD as defined by mutagenesis, crystallography, and kinetics. J Biol Chem 278:29502–29508. [DOI] [PubMed] [Google Scholar]
- 6. Clarke TE, Romanov V, Chirgadze YN, Klomsiri C, Kisselman G, Wu‐Brown J, Poole LB, Pai EF, Chirgadze NY (2011) Crystal structure of alkyl hydroperoxidase D like protein PA0269 from Pseudomonas aeruginosa: homology of the AhpD‐like structural family. BMC Struct Biol 11:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee HY, Yang JK (2009) Crystallization and preliminary X‐ray crystallographic analysis of gamma‐carboxymucolactone decarboxylase from Sulfolobus solfataricus. Acta Crystallogr Sect F Struct Biol Cryst Commun 65:1197–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holm L, Rosenstrom P (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG (2011) iMOSFLM: a new graphical interface for diffraction‐image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Method Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- 12. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 15. Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen VB, Arendall WB, Headd JJ, III, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Corpet F (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16:10881–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gouet P, Courcelle E, Stuart DI, Metoz F (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308. [DOI] [PubMed] [Google Scholar]
- 19. DeLano WL (2002) The PyMOL Molecular Graphics System. Palo Alto, CA: DeLano Scientific.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information