

Abstract
Nitrate reductases (NR) belong to the DMSO reductase family of Mo‐containing enzymes and perform key roles in the metabolism of the nitrogen cycle, reducing nitrate to nitrite. Due to variable cell location, structure and function, they have been divided into periplasmic (Nap), cytoplasmic, and membrane‐bound (Nar) nitrate reductases. The first crystal structure obtained for a NR was that of the monomeric NapA from Desulfovibrio desulfuricans in 1999. Since then several new crystal structures were solved providing novel insights that led to the revision of the commonly accepted reaction mechanism for periplasmic nitrate reductases. The two crystal structures available for the NarGHI protein are from the same organism (Escherichia coli) and the combination with electrochemical and spectroscopic studies also lead to the proposal of a reaction mechanism for this group of enzymes. Here we present an overview on the current advances in structural and functional aspects of bacterial nitrate reductases, focusing on the mechanistic implications drawn from the crystallographic data.
Keywords: molybdenum enzymes, nitrogen cycle, DMSO reductase family, periplasmic nitrate reductase, membrane‐bound nitrate reductase, crystal structure, enzymatic mechanism
Introduction
Molybdopterin enzymes
Molybdenum is the only second row transition element required by the majority of living organisms, being widespread in the environment because of the water solubility of its high‐valent oxides. Mo has been incorporated in a diverse range of biological systems complexed to a pyranopterin organic compound constituting the molybdenum cofactor (Moco).1, 2, 3
The majority of the Moco containing enzymes (molybdopterin or pyranopterin enzymes) catalyze redox reactions, taking advantage of the chemical versatility of the metal. An essential role of molybdenum is the catalysis of an oxo‐transfer reaction coupled to electron‐transfer between substrate and other cofactors such as Fe/S centers, hemes, or flavins. With the exception of the multinuclear MoFe7 cluster of bacterial nitogenases,4, 5 all other Mo (or W) dependent enzymes use the metal in a mononuclear form coordinated to the cis‐dithiolene group of one or two pyranopterins. In the catalytic center of these enzymes, Mo can be also coordinated to the side chain of a cysteine, selenocysteine, serine, or aspartate residue, and/or by oxygen or sulfur ligand in the oxo/hydroxyl or sulfide forms. Based on the large diversity of Mo coordinating ligands, their chemical structures and spectroscopic features, molybdopterin enzymes have been classified into three broad families: I—xanthine oxidase (XO) family; II—sulfite oxidase (SO) family; and III—dimethyl sulfoxide reductase (DMSOR) family.6 While eukaryotic Mo enzymes belong exclusively to the SO or XO families, the prokaryotic Mo/W enzymes may belong to all three families. Members of the DMSOR family present the highest variability in the Mo active site composition and the majority work under anaerobic conditions, whereby their respective cofactors serve as terminal electron acceptors in the respiratory metabolism.7, 8, 9
DMSO reductase family
Enzymes of the DMSOR family present considerable variations in the structure of the Mo active site, where the metal is coordinated by two pterin cofactors (referred to as molybdopterin guanine dinucleotide‐MGD, or pyranopterin guanine dinucleotide‐PGD).3, 10 Members of this family present great diversity and according to structural, spectroscopic, and biochemical evidences they have been classified into three subfamilies (I, II, and III) comprising respectively: subfamily I: periplasmic nitrate reductase (Nap) and formate dehydrogenase (Fdh); subfamily II: respiratory nitrate reductase (Nar) and ethylbenzene dehydrogenase (EBDH); subfamily III: DMSO reductase and trimethylamine N‐oxide (TMAO) reductase (Fig. 1).11 Other members of the DMSO reductase family cannot be included in any of the three subfamilies, as is the particular case of arsenite oxidase (Aio), since it does not have an amino acid side chain coordinating the Mo atom.12, 13
Figure 1.
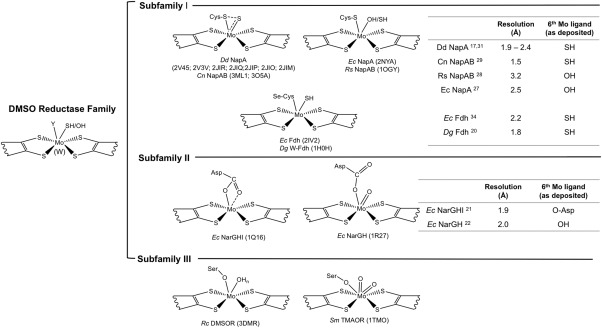
Active site structure of the DMSO reductase subfamilies members. The DMSO reductase family has been divided in three subfamilies according to the different elements coordinating the Mo atom at the active site, including: the dissimilatory nitrate reductase (Nap) and formate dehydrogenase (Fdh) (subfamily I) in which the active site is coordinated by a Cys (Nap) or a Se‐Cys (Fdh); the respiratory nitrate reductase (Nar) (subfamily II) where Mo is coordinated by one or two oxygen atoms from an aspartate residue, and the DMSO reductase and trimethylmanine N‐oxide (TMAO) reductase (subfamily III) with the side chain of a Ser residue in the coordinating position. Note that the scheme is not exhaustive and some members are not represented, as for example arsenite oxidase (Aio), which has also been assigned as belonging to the DMSO reductase family of Mo‐containing enzymes, but with some unusual properties when compared to the other members, namely the lack of an amino acid side chain as a metal ligand. The resolution of the crystal structures available for the Nap, Fdh, and Nar proteins (subfamilies I and II) as well as the 6th Mo ligand are also represented in the tables.
In contrast to the prokaryotic ones, eukaryotic NR found in plants, fungi, and algae (eukNR), belong to the SO family of Mo containing enzymes.14
The first crystal structures reported for members of the DMSOR family were the DMSO reductases from Rhodobacter (R.) sphaeroides 15 and from R. capsulatus 16 (subfamily III) but the knowledge on this complex family has increased largely with the subsequent crystallographic studies on new members (subfamily I): the periplamic nitrate reductase (Desulfovibrio (D.) desulfuricans ATCC 27774 Nap17) in 1999 followed by the first formate dehydrogenases (Escherichia (E.) coli FdhH18 and D. gigas FdhAB19, 20) in 1997 and 2001–2002, respectively. As to members of subfamily II, membrane‐bound nitrate reductases, the first and still only structures became available in 2003 and 2004 (E. coli NarGH21, 22).
Nitrate reductases (NR)
Nitrate reductases belong to the DMSOR family of Mo containing enzymes, and are responsible for the initial reductive steps of the nitrogen cycle, according to the reaction:
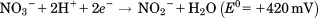 |
Nitrate reductases possess diverse cellular location, active site structure and function and have been classified into three distinct types: dissimilatory periplasmic nitrate reductases (Nap), respiratory membrane‐bound nitrate reductases (Nar), and assimilatory cytoplasmic nitrate reductases (Nas).23, 24, 25, 26
The periplasmic nitrate reductase NapA from D. desulfuricans was the first to be structurally analyzed by crystallography to 1.9 Å resolution in 1999,17 and other studies followed with different crystal structures determined in the past few years: NapA from E. coli (part of the NapAB complex),27 NapAB from R. sphaeroides,28 NapAB from C. necator (formerly known as R. eutropha),29 and NapB from H. influenzae.40 The 3D structure of the oxidized and partially reduced forms of the C. necator NapAB enzyme29 as well as of several D. desulfuricans NapA complexes with bound substrate and inhibitors were also reported,30 and have revealed the true Mo atom coordination at the active site thus providing important clues towards the deduction of novel enzymatic mechanisms. The implications of these findings will be further presented in this review.
The crystal structures of the membrane‐bound nitrate reductases from E. coli (NarGHI and NarGH21, 22) have been solved, revealing novel features for the DMSOR family that define subfamily II, in particular the presence of an aspartate side chain as the Mo ligand. Additional structural and spectroscopic data of a complex of E. coli NarGHI with a quinol‐analog as well as of several variants26 provided important details on the ubiquinol oxidation at the bacterial membrane.
The crystallographic studies revealed that the catalytic subunits of Nap and Nar are very similar in terms of global fold, domain organization, and metal cofactor content, but that considerable differences are observed in the region at and surrounding the active site, as well as in the funnel leading to it.3 To date no crystal structure has been reported for the Nas protein, so that structural comparisons cannot be made.
Membrane‐Bound Nitrate Reductases
Crystallographic studies
The membrane‐bound nitrate reductases (Nar) have been isolated from a variety of organisms and are responsible for generating the proton motive force across the cytoplasmatic membrane of the cells.25, 31 The crystal structures of E. coli NarGHI and NarGH were independently solved by two research groups at 1.921 and 2.0 Å22 resolution, respectively (PDB ID: 1Q16 and 1R27). The Nar protein is constituted by three different subunits (NarG‐140 kDa, NarH‐58 kDa, and NarI‐26 kDa) and can be purified as a heterotrimer (NarGHI) or as a heterodimer (NarGH) depending if it is solubilized in detergent32 or in water, respectively.33 The NarGH complex is located in the cytoplasm, anchored to the inner surface of the cytoplasmatic membrane by the NarI subunit (Fig. 2).
Figure 2.
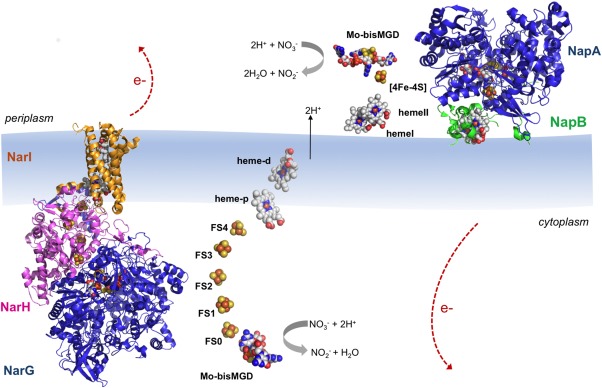
Overall structures of E. coli NarGHI (PDB ID: 1Q16) and C. necator NapAB (PDB ID: 3ML1) according to their localization in the cell. The catalytic subunits NarG and NapA are represented in blue; the NarH is represented in pink; and the heme containing subunits NarI and NapB are represented in orange and green, respectively. The protein cofactors (Mo bis‐MGD, FeS clusters and heme groups) are represented color‐coded. Note that the electron transfer pathway in both enzymes follows cofactor localization: electrons generated at the active site flow through the FeS centers until they reach the heme groups, from where they are dissipated. The five FeS clusters in NarGHI are referred to as FS0–FS4 with increasing distance from the Mo bis‐MGD.
The largest NarG catalytic subunit contains the Moco active site and a [4Fe–4S] center (FS0). It is organized in four conserved domains grouped around the cofactor, similarly to the other Mo‐bisMGD enzymes, namely NapA (Fig. 2). The presence of the FS0 was previously undetected in EPR studies and it was found that this FeS center is coordinated by one histidine and three cysteines—HxxxCxxC(x)nC, which is an unusual coordination pattern when comparing to the FeS centers in other nitrate reductases—CxxCxxC(x)nC.22, 34 Crystallographic studies performed by Rothery35 (PDB IDs: 3IR5, 3IR6, and 3IR7) demonstrated that FS0 assembly is an important prerequisite for the Mo‐bisMGD cofactor insertion during the holoenzyme maturation. In the active site the Mo atom is coordinated by six ligands in a distorted trigonal prism geometry [Fig. 3(A)]. Four of such ligands are sulfurs from the dithiolenes of the two MGD cofactors (Mo‐bisMGD, referred as P‐ and Q‐pterins according to their localization as proximal or distal to FS0). The other ligands coordinating the Mo atom are the carboxylate side chain of an aspartate residue in a monodentate fashion [NarG‐Asp222, Fig. 3(A)] and an oxo group, which is absent in the NarGHI structure.21 In this structure (PDB ID: 1Q16), both oxygen atoms from Asp222 side chain coordinate the Mo atom. Also, the Q‐pterin is present in an open bicyclic dihydropterin structure, rather than in the usual tricyclic pyranopterin form present in other Mo enzymes. In NarGH (PDB ID: 1R27), although the deposited coordinates show an open bicyclic form, the authors found unclear the distinction between the bicyclic and tricyclic forms of the pterin.22
Figure 3.
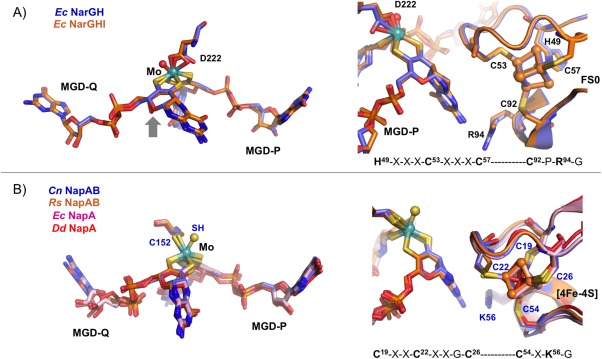
Comparison of the active site (left) and the electron transfer pathways (right) between: (A) E. coli NarGH (blue, PDB ID: 1R27) and NarGHI (orange, PDB ID 1Q16). (B) C. necator NapAB (blue, PDB ID: 3ML1), R. sphaeroides NapAB (orange, PDB ID: 1OGY), E. coli NapA (pink, PDB ID: 2NYA), and D. desulfuricans NapA (red, PDB ID: 2NAP), showing the respectively Mo ligands and the nearest [4Fe–4S] center. The electron transfer pathway also includes conserved residues (Arg94 in Nar and Lys56 in Nap, respectively) which possible role is to mediate the electron transfer between the pterin (via its exocyclic NH2) and the FeS center.
The NarH is the electron transfer subunit and contains three [4Fe–4S] centers (FS1, FS2, FS3) plus a [3Fe–4S] center (FS4), all ferredoxin type. According to their redox properties the FS centers belong to two different classes: the high potential class with positive midpoint potentials (+130 mV for FS1 and +180 mV for FS4), and the low‐potential class with negative potentials (−420 mV for FS2 and −55 mV for FS3).21, 31 The NarH subunit consists of a core structure containing the FS centers, one linker region connecting the NarG and NarH subunits and one C‐terminal extension (Fig. 2).
The small NarI subunit anchors NarGH at the cytoplasmatic side of the membrane, providing the binding site (Q‐site) for the oxidation of the physiological electron donor quinol, a process through which protons are translocated to the periplasm. The existence of the Q‐site has been confirmed by the crystal structure of E. coli NarGHI in complex with the quinol binding inhibitor pentachlorophenol (PCP) (PDB ID: 1Y4Z) where the electron density map showed a PCP molecule located in a binding pocket near the heme groups.26 NarI is composed by five transmembrane helices and harbors two b‐type hemes, termed proximal (b p) and distal (b d) to indicate their positions relative to the catalytic site (Fig. 2). The disposition of the redox cofactors in NarGHI promotes efficient electron transfer from the quinol‐binding site in NarI to the molybdenum guanine dinucleotide cofactor in NarG, where nitrate is reduced to nitrite (Fig. 2). The C‐terminus of NarI faces the cytosol being composed by highly conserved residues involved in hydrogen and electrostatic interactions, important for the NarGHI heterotrimer formation.21
The Marinobacter hydrocarbonoclasticus (Mh) (formerly Pseudomonas nautica)36, 37 and the Paracoccus pantotrophus (Pp)38 membrane bound nitrate reductases are closely related to the Ec NarGHI protein and have been extensively biochemically and spectroscopically characterized but no crystal structure has been reported so far.
Reaction mechanism
The reaction mechanism of membrane‐bound NR has been mainly proposed based on enzyme kinetics, electron paramagnetic resonance (EPR) and protein film voltammetry (PFV) studies. It has been found that the oxidation state of the Mo ion changes between Mo(VI) and Mo(V) during the oxo‐transfer reaction, and an oxygen ligand (OH/H2O) is released upon cofactor reduction to Mo(IV). The Mo(V) form of the enzyme exhibits a pH‐dependent equilibrium between high‐pH and low‐pH forms. The PFV results on Mh NarGH,36, 37 Pp NarGH,38 and Ec NarGHI39, 40 revealed different catalytic voltammetric responses for MhNarGH in comparison to the other two enzymes, which led to different interpretations for the corresponding catalytic cycles.
In Pp NarGH and Ec NarGHI the results obtained suggested a reaction mechanism based on the availability of the substrate (Scheme 1). In the Pp NarGH protein the authors propose the existence of two alternative reaction pathways determined by the rate of electron and substrate addition to the active site: in one pathway Mo(VI) is reduced to Mo(V) and the electrons needed to complete the redox reaction are provided upon nitrate binding; in the other pathway instead, Mo(VI) can be directly reduced to Mo(IV) followed by the binding of the substrate, which is immediately converted to the product while Mo is reoxidized to Mo(VI) (Scheme 1).38 The results obtained for the Ec NarGHI suggest that with low levels of nitrate, the substrate binds to Mo(V) and the addition of an electron/proton couple releases the product; if, in contrast, nitrate is highly abundant it binds to the Mo(IV) and a higher rate of turnover becomes dominant.40
Scheme 1.
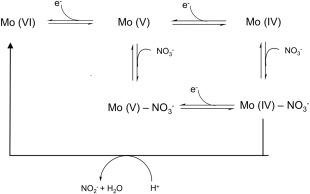
Proposed mechanism for nitrate reduction by respiratory nitrate reductases, based on enzyme kinetics and protein film voltammetry as proposed by Anderson et al. for Pp NarGH38 and by Elliot et al. for Ecoli NarGHI.40
In the case of Mh NarGH37 the results show that the two catalytically distinct forms were affected differently by the type of oxidizing substrate used, indicating that the enzyme–substrate complex formation must occur before any redox event and therefore suggesting an alternative proposal for substrate reduction by Nars.
The available Ec NarGHI and NarGH crystal structures are likely to represent the oxidized Mo (VI) form of the enzymes since crystals of the two proteins were grown aerobically, although partial reduction due to the X‐ray exposure cannot be excluded. The proposed reaction mechanism based on the combination of PVF results and on the crystallographic evidence suggests that in NarGH the active low‐pH form (Mo VI) leads to the Mo(V) form with an Asp‐O and HO groups as Mo ligands, upon one electron reduction. In this structure Asp222 is hydrogen bonded to the Nϵ of a conserved His546, which might become deprotonated in the transition to the inactive high‐pH Mo form. This would cause the rotation of the Asp222 carboxylate side chain into a bidentate coordination, thus blocking nitrate binding to the active site. The conserved His residue could be the ionizable residue responsible for the transition between the high‐pH inactive and the low‐pH active states of the enzyme.22
Nars are complex enzymes and further investigation is necessary to establish a general reaction mechanism common for membrane‐bound nitrate reductases and in particular the elucidation of more crystal structures would constitute a major advance in this direction.
Periplasmic Nitrate Reductases
Crystallographic studies
The physiological role of dissimilatory periplasmic nitrate reductases (Nap) is diverse. Nap has been implicated in denitrification and nitrate reduction processes and in the maintenance of the cellular redox potential and nitrate scavenging. Besides, it is also linked to the quinol oxidation, but does not transduce the free energy from the quinol/nitrate couple into proton motive force, as in the case of Nar.23 With one exception (the monomeric NapA from D. desulfuricans) all other Nap proteins that have been studied are found in an heterodimeric form NapAB, being located in the periplasm compartment of the cell (Fig. 2).3
Several crystal structures of periplasmic nitrate reductases have been reported. The first was from the monomeric NapA from D. desulfuricans at 1.9 Å resolution,17 while the structure from NapAB from R. sphaeroides was the first of a heterodimeric Nap protein to be solved.28 Crystal structures of the individual NapA from E. coli (Ec),27 as well as of a proteolyzed fragment of Haemophilus (H.) influenza NapB40 were also reported, the later corresponding to a globular domain of H. influenza NapB obtained by proteolysis of the recombinant protein.41, 42 The most recent crystal structures reported correspond to the heterodimeric NapAB from Cupriavidus (C.) necator solved at 1.5 Å resolution in different redox states.29 Six D. desulfuricans NapA complexes with bound substrate and/or inhibitors have also been published.30
The 3D structure of Nap is very much conserved. The large catalytic subunit (NapA, 80 kDa) contains the Mo atom bound to two MGD cofactors and a [4Fe–4S] center and can be organized into four subdomains composed by discontinuous stretches of the polypeptide chain all involved in the Mo‐bisMGD binding. The catalytic core is deeply buried in the protein with a substrate channel oriented opposite to the heterodimer contact interface and extending ∼15 Å away from the protein surface. The Mo atom in the active site is coordinated by six sulfur ligands in a distorted trigonal prismatic geometry (Fig. 1). Four of the sulfurs are provided by the two dithiolenes (Mo‐bisMGD), while the other two correspond to the Sγ atom of a cysteine residue and a sulfur terminal ligand. In the original Dd NapA structure, the sixth ligand was assigned as an hydroxo/water ligand but additional studies on the same enzyme reacted with reducing agents, substrates and inhibitors, provided new evidence on the Mo active site details, allowing to unequivocally assess the sixth Mo ligand as a sulfur atom30 thus correcting the originally reported structure.
Also, the crystal structure of the C. necator NapAB, the only Nap structure obtained at nearly atomic resolution, 1.5 Å, corroborated this new evidence.29 In addition, C. necator NapAB could also be crystallized in the (partially) reduced form, which provided valuable information. These crystallographic data had major mechanistic implications as discussed below. The NapB subunit is characterized by low structural complexity and contains two c‐type hemes, responsible for electron transfer to the catalytic subunit NapA. It is composed by three short α‐helices linked by two large loops. As in the case of the Nar protein, cofactors disposition in a single chain in Nap is related with the flow of electrons during the catalytic cycle. The electrons received at NapB through the heme groups, are then transferred to the [4Fe–4S] centers and later to the Mo atom at the NapA subunit.
In NapA, the side chain of a conserved Tyr (Tyr58 in C. necator NapAB) lies between the c‐type heme in NapB and the [4Fe–4S] center in NapA, at the interface of the two subunits. This tyrosine is conserved (Fig. 4) in all heterodimeric NR (with similar orientation), but not in the monomeric NapA from D. desulfuricans, where a serine residue can be found instead, strongly suggesting its role in electron transfer mediation between NapA and NapB.29, 30 In the heterodimeric NapAB from C. necator and R. spheroides both the N‐ and C‐terminal parts of NapB are fully structured forming two extended arms embrace the NapA subunit thus stabilizing the complex. In the structure of NapB from H. influenza the two regions were cleaved during purification.
Figure 4.

Comparison of the amino acid sequence of NapA from C. necator (Cn), R. sphaeroides (Rs), E. coli (Ec), and D. desulfuricans (Dd) (A), and NapB from C. necator, R. sphaeroides, E. coli, and H. influenza (Hi) (B). The [4Fe–4S] binding Cys residues are marked as (*) and the cysteine that is a Mo ligand is marked in green. In the NapB subunit the His residues coordinating the two Fe atoms from the heme groups are marked in orange. Note that the two signal peptide sequences from NapA (29 residues) and NapB (35 residues) were omitted from the amino acid sequence comparison. Underlined in orange are the two surface loops (267–297 and 543–596 in C. necator NapAB numbering) conserved in the heterodimeric proteins but not in the monomeric D. desulfuricans NapA. Alignment results were obtained with the CLUSTAL OMEGA multiple sequence alignment program at the EMBL EBI web server and the figure prepared using the program Jalview.
Structural analysis and comparison
Comparison of the two heterodimeric NapAB crystal structures (C. necator and R. sphaeroides) shows that the two proteins share a similar overall globular shape, folding and cofactor localization [Fig. 5(A)]. The buried surface area as well as the number and type of inter‐subunit contacts are also very similar. The Cn NapA and Rs NapA catalytic subunits share 71% sequence identity and the same number of residues (802). Both structures superimpose with an rmsd of 0.67 Å for 780 Cα aligned. The monomeric NapA from D. desulfuricans (723 residues) shares only 36% sequence identity with the Cn NapA, but the two structures are very similar, with an rmsd of 0.9 Å for 590 Cα superimposed. The main difference between the Dd NapA and the corresponding subunit of the NapAB complex from Cn, Rs, and Ec is the existence of two exposed loops at the surface (residues 267–297 and 543–596, in Cn NapA numbering) conserved in the heterodimeric proteins but not in the monomeric one where those segments are absent [Fig. 5(B)].
Figure 5.
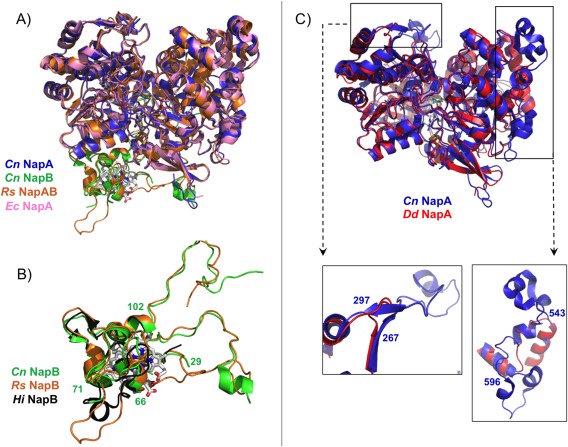
Superpositioning of the crystal structures of the periplasmic nitrate reductases deposited in the PDB: (A) Comparison between heterodimeric proteins: NapAB from C. necator (NapA in blue and NapB in green, PDB ID: 3ML1), NapAB from R. sphaeroides (orange, PDB ID: 1OGY), and NapA from E. coli (pink, PDB ID: 2NYA). (B) Highlight of the comparison between Cn (green), Rs (orange), and Hi (black, PDB ID: 1JNI) small subunit NapB, with evidence of the similarities in the core structure of the three proteins (Cn NapB numbering). (C) Comparison between the catalytic subunits from Cn NapAB and Dd NapA, with a closeup of the two loops that differ considerably between heterodimeric and monomeric proteins (Cn NapAB numbering).
In contrast to the catalytic subunit NapA, the electron transfer subunit NapB from C. necator has lower sequence identity (52%) with R. sphaeroides NapB, and the two structures superimpose with an rsmd of 0.96 Å (86 superimposed out of 134 Cα atoms). The structure of the H. influenza NapAB proteolyzed fragment superimposes onto Cn NapB with an rsmd of 0.78 Å for 49 Cα atoms. These two proteins share only 37% sequence identity. The globular, core domain of the three NapB structures is very similar, with the exception of a short α‐helix present in the Hi NapB [residues 65–72, Fig. 5(C)]. This corresponds to an exposed polypeptide chain that appears disordered in the Cn NapB structure (residues Arg66–Gln71) showing no continuous electron density. In Rs NapB this same region is unstructured and protrudes towards the solvent as shown in Figure 5(C).
Structure‐derived reaction mechanism for periplasmic nitrate reductases
The revised coordination sphere of Mo, with a total of six sulfur atoms and no oxygen ligand directly bound to the metal, was first revealed by the detailed re‐analysis of the Dd NapA crystal structure and later confirmed with the Cn NapAB structures obtained at high resolution and for two redox states.29, 30 This new information had important implications for the deduction and clarification of the reaction mechanism that had to be completely reformulated.
The correct interpretation of the Mo coordination sphere on Dd NapA and on Cn NapAB was possible due to high resolution of the data as well as to a careful inspection of electron density maps and B‐factors distribution. When the sixth Mo ligand was refined as oxygen, positive residual electron density was observed in the difference electron density map, strongly suggesting that a heavier atom should occupy the position [Fig. 6(A) for Cn NapAB]. If a sulfur atom was refined at the same position, the positive electron density completely disappeared [Fig. 6(B)]. The unambiguous assignment of the sixth Mo ligand was also made on the basis of the B‐factor analysis, if an oxygen atom was refined at the same position, the resulting B‐factor was lower than the average B‐factors of the surrounding atoms. If, in contrast, the sixth position was refined as a S atom, the resulting B‐factor was in accordance with the B‐factor values of the nearest atoms, revealing the correctness of the solution. Moreover, the distance between the sulfur ligand and the Sγ atom of the Cys is significantly shorter than the van der Waals contact distances (∼3.3 Å), which also demonstrates that an O atom would not fit appropriately (Fig. 6).
Figure 6.
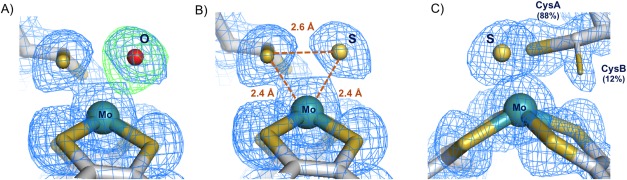
Closeup of the Mo coordination sphere for the C. necator NapAB (PDB ID: 3O5A) with the 2mFo–DFc electron density maps contoured at 1σ (blue) and the mFo–DFc map contoured at 3σ (green). (A) Refinement of the 6th Mo ligand as oxygen yields positive electron density on the mFo–DFc map showing that a heavier element occupies that position. (B) The positive electron density completely disappeared when the 6th Mo ligand was refined as a sulfur atom, revealing the correctness of the solution. (C) The Mo site of the partially reduced form of the enzyme, showing the two alternate conformations of Cys152: CysA bound (88% occupancy), and CysB unbound (12% occupancy).
This revealed that, in both structures, the sulfur atom from the coordinating Cys152 is making a partial disulfide bond with the terminal sulfido ligand [Fig. 6(B,C)]. In the earlier study of the Dd NapA, the lower resolution of the data did not permit a similar conclusion. This new interpretation, based on the improved crystallographic data, required a revision of the Nap reaction mechanism. In this context, three alternative proposals were considered, each with attention to substrate binding and reaction pathway. These proposals combined molybdenum and sulfur redox chemistry rather than the previously and usually accepted redox chemistry, based solely on the Mo atom.30 Two of the possible mechanisms involve direct binding of nitrate to the Mo atom, in the first coordination sphere, while a third hypothesis considered the binding of nitrate to the terminal sulfur ligand, in the second coordination sphere of the metal (Fig. 7). Subsequent theoretical and computational studies were performed by independent authors to further investigate which of the three alternative pathways was energetically more favored.43, 44, 45, 46 In these studies, it was found that the interaction of the substrate with the active site promotes the displacement of the Cys152 residue, which remains indirectly bound to the Mo atom through a persulfide bond with the sixth sulfur ligand.43, 44 This evidence was experimentally validated by the crystal structure of the partially reduced form of the NapAB from C. necator, in which the displacement of the sulfur ligand at the active site was observed.29
Figure 7.
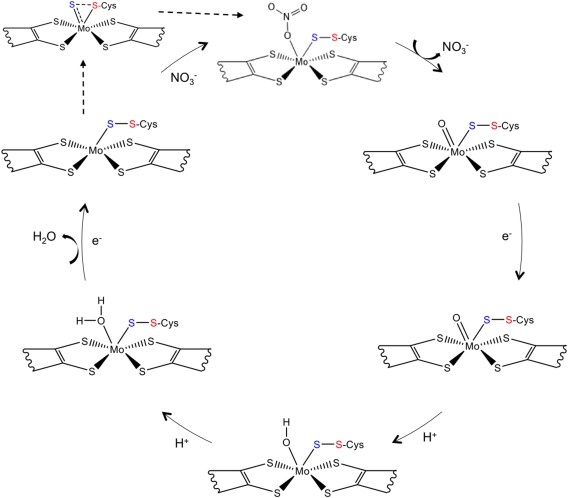
Representation of the currently proposed reaction mechanism for the periplasmic nitrate reductase, adapted from Cerqueira et al.48 The two electrons and the two protons required by the reaction are provided by external reductants and from the solvent, respectively.
The combination of structural and theoretical evidence indicates that the reaction mechanism must occur through a first‐shell or first coordination‐sphere type of mechanism, in which the substrates binds directly to the Mo atom. This is only possible due to the conformational re‐arrangement of the Mo coordination sphere, which occurs when the substrate is available at the active site. This mechanism was called the S‐shift (with analogy to the carboxylate shift observed in Zn enzymes),47 and results on a free coordination position that is used to bind the substrate. Once the catalytic reaction is complete, and the product is released, the free position is again available and two different pathways are possible, depending on the availability of the substrate: if nitrate is still available a new cycle of reaction proceeds, but if, in contrast, no more substrate is available, the inverse of the S‐shift mechanism occurs, allowing the metal to return to the hexa‐coordinated initial form. The theoretical studies show that this is a very energetically favorable mechanism, and that it can be seen as an activation mechanism independent of the reaction mechanism.44, 48
Concluding Remarks
In this review, we have presented a summary of major structural and mechanistic advances in the field of bacterial nitrate reductases, particularly for the membrane‐bound and periplasmic nitrate reductases. The detailed analysis of the respective crystal structures and the elucidation of the reaction mechanism for this group of Mo containing enzymes is essential for a better understanding of the function of the cellular nitrogen cycle.
The evidence for new reaction mechanisms, partially contradicting what had been assumed before was possible due to a more careful analysis of crystallographic data, in some cases, due to higher resolution data and in others due to improved refinement protocols.
Is worth mentioning the impact of the technological advances in the X‐ray crystallography field, in particular the use of microfocus beams and advanced detectors at latest generation synchrotron sources as well as the availability of complex protein expression systems which allowed to produce fully active recombinant proteins. Nevertheless, and despite the progress made since the elucidation of the first Nap crystal structure in 1999, the knowledge regarding this group of enzymes is only partly understood, still providing challenging opportunities for future research on the topic of nitrate reductases.
Acknowledgments
The authors acknowledge the continuous financial support from the Portuguese Fundação para a Ciência e Tecnologia (FCT‐MEC) through projects UID/Multi/04378/2013, POCI/QUI/57641/2004, PTDC/QUI/64733/2006.
Grant number: SFRH/BPD/84581/2012.
References
- 1. Mendel RR, Bittner F (2006) Cell biology of molybdenum. Biochim Biophys Acta 1763:621–635. [DOI] [PubMed] [Google Scholar]
- 2. Hille R (1996) The mononuclear molybdenum enzymes. Chem Rev 96:2757–2816. [DOI] [PubMed] [Google Scholar]
- 3. Romão MJ (2009) Molybdenum and tungsten enzymes: a crystallographic and mechanistic overview. Dalton Trans 21:4053–4068. [DOI] [PubMed] [Google Scholar]
- 4. Lawson DM, Smith BE (2002) Molybdenum nitrogenases: a crystallographic and mechanistic view. Met Ions Biol Syst 39:75–119. [PubMed] [Google Scholar]
- 5. Allen RM, Chatterjee R, Madden MS, Ludden PW, Shah VK (1994) Biosynthesis of the iron‐molybdenum cofactor of nitrogenase. Crit Rev Biotechnol 14:225–249. [DOI] [PubMed] [Google Scholar]
- 6. Hille R (2002) Molybdenum enzymes containing the pyranopterin cofactor: an overview. Met Ions Biol Syst 39:187–226. [PubMed] [Google Scholar]
- 7. Schwarz G, Mendel RR, Ribbe MW (2009) Molybdenum cofactors, enzymes and pathways. Nature 460:839–847. [DOI] [PubMed] [Google Scholar]
- 8. Hille R, Nishino T, Bittner F (2011) Molybdenum enzymes in higher organisms. Coord Chem Rev 255:1179–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gonzalez PJ, Rivas MG, Mota C, Brondino CD, Moura I, Moura JJG (2013) Periplasmic nitrate reductases and formate dehydrogenases: Biological control of the chemical properties of Mo and W for fine tuning of reactivity, substrate specificity and metabolic role. Coord Chem Rev 257:315–331. [Google Scholar]
- 10. Grimaldi S, Schoepp‐Cothenet B, Ceccaldi P, Guigliarelli B, Magalon A (2013) The prokaryotic Mo/W‐bisPGD enzymes family: a catalytic workhorse in bioenergetic. Biochim Biophys Acta 1827:1048–1085. [DOI] [PubMed] [Google Scholar]
- 11. McDevitt CA, Hugenholtz P, Hanson GR, McEwan AG (2002) Molecular analysis of dimethyl sulphide dehydrogenase from Rhodovulum sulfidophilum: its place in the dimethyl sulphoxide reductase family of microbial molybdopterin‐containing enzymes. Mol Microbiol 44:1575–1587. [DOI] [PubMed] [Google Scholar]
- 12. Ellis PJ, Conrads T, Hille R, Kuhn P (2001) Crystal structure of the 100 kDa arsenite oxidase from Alcaligenes faecalis in two crystal forms at 1.64 A and 2.03 A. Structure 9:125–132. [DOI] [PubMed] [Google Scholar]
- 13. Warelow TP, Oke M, Schoepp‐Cothenet B, Dahl JU, Bruselat N, Sivalingam GN, Leimkühler S, Thalassinos K, Kappler U, Naismith JH, Santini JM (2013) The respiratory arsenite oxidase: structure and the role of residues surrounding the rieske cluster. PLoS One 8:e72535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fischer K, Barbier GG, Hecht HJ, Mendel RR, Campbell WH, Schwarz G (2005) Structural basis of eukaryotic nitrate reduction: crystal structures of the nitrate reductase active site. Plant Cell 17:1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schindelin H, Kisker C, Hilton J, Rajagopalan KV, Rees DC (1996) Crystal structure of DMSO reductase: redox‐linked changes in molybdopterin coordination. Science 272:1615–1621. [DOI] [PubMed] [Google Scholar]
- 16. Schneider F, Löwe J, Huber R, Schindelin H, Kisker C, Knäblein J (1996) Crystal structure of dimethyl sulfoxide reductase from Rhodobacter capsulatus at 1.88 A resolution. J Mol Biol 263:53–69. [DOI] [PubMed] [Google Scholar]
- 17. Dias JM, Than ME, Humm A, Huber R, Bourenkov GP, Bartunik HD, Bursakov S, Calvete J, Caldeira J, Carneiro C, Moura JJ, Moura I, Romão MJ (1999) Crystal structure of the first dissimilatory nitrate reductase at 1.9 A solved by MAD methods. Structure 7:65–79. [DOI] [PubMed] [Google Scholar]
- 18. Boyington JC, Gladyshev VN, Khangulov SV, Stadtman TC, Sun PD (1997) Crystal structure of formate dehydrogenase H: catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science 275:1305–1308. [DOI] [PubMed] [Google Scholar]
- 19. Raaijmakers H, Teixeira S, Dias JM, Almendra MJ, Brondino CD, Moura I, Moura JJ, Romão MJ (2001) Tungsten‐containing formate dehydrogenase from Desulfovibrio gigas: metal identification and preliminary structural data by multi‐wavelength crystallography. J Biol Inorg Chem 6:398–404. [DOI] [PubMed] [Google Scholar]
- 20. Raaijmakers H, Macieira S, Dias JM, Teixeira S, Bursakov S, Huber R, Moura JJ, Moura I, Romão MJ (2002) Gene sequence and the 1.8 A crystal structure of the tungsten‐containing formate dehydrogenase from Desulfovibrio gigas . Structure 10:1261–1272. [DOI] [PubMed] [Google Scholar]
- 21. Bertero MG, Rothery RA, Palak M, Hou C, Lim D, Blasco F, Weiner JH, Strynadka NC (2003) Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nat Struct Biol 10:681–687. [DOI] [PubMed] [Google Scholar]
- 22. Jormakka M, Richardson D, Byrne B, Iwata S (2004) Architecture of NarGH reveals a structural classification of Mo‐bisMGD enzymes. Structure 12:95–104. [DOI] [PubMed] [Google Scholar]
- 23. Sparacino‐Watkins C, Stolz JF, Basu P (2014) Nitrate and periplasmic nitrate reductases. Chem Soc Rev 43:676–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stolz JF, Basu P (2002) Evolution of nitrate reductase: molecular and structural variations on a common function. Chembiochem 3:198–206. [DOI] [PubMed] [Google Scholar]
- 25. Richardson DJ, Berks BC, Russell DA, Spiro S, Taylor CJ (2001) Functional, biochemical and genetic diversity of prokaryotic nitrate reductases. Cell Mol Life Sci 58:165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bertero MG, Rothery RA, Boroumand N, Palak M, Blasco F, Ginet N, Weiner JH, Strynadka NC (2005) Structural and biochemical characterization of a quinol binding site of Escherichia coli nitrate reductase A. J Biol Chem 280:14836–14843. [DOI] [PubMed] [Google Scholar]
- 27. Jepson BJ, Mohan S, Clarke TA, Gates AJ, Cole JA, Butler CS, Butt JN, Hemmings AM, Richardson DJ (2007) Spectropotentiometric and structural analysis of the periplasmic nitrate reductase from Escherichia coli . J Biol Chem 282:6425–6437. [DOI] [PubMed] [Google Scholar]
- 28. Arnoux P, Sabaty M, Alric J, Frangioni B, Guigliarelli B, Adriano JM, Pignol D (2003) Structural and redox plasticity in the heterodimeric periplasmic nitrate reductase. Nat Struct Biol 10:928–934. [DOI] [PubMed] [Google Scholar]
- 29. Coelho C, González PJ, Moura JG, Moura I, Trincão J, Romão MJ (2011) The crystal structure of Cupriavidus necator nitrate reductase in oxidized and partially reduced states. J Mol Biol 408:932–948. [DOI] [PubMed] [Google Scholar]
- 30. Najmudin S, González PJ, Trincão J, Coelho C, Mukhopadhyay A, Cerqueira NM, Romão CC, Moura I, Moura JJ, Brondino CD, Romão MJ (2008) Periplasmic nitrate reductase revisited: a sulfur atom completes the sixth coordination of the catalytic molybdenum. J Biol Inorg Chem 13:737–753. [DOI] [PubMed] [Google Scholar]
- 31. Guigliarelli B, Magalon A, Asso M, Bertrand P, Frixon C, Giordano G, Blasco F (1996) Complete coordination of the four Fe–S centers of the beta subunit from Escherichia coli nitrate reductase. Physiological, biochemical, and EPR characterization of site‐directed mutants lacking the highest or lowest potential [4Fe–4S] clusters. Biochemistry 35:4828–4836. [DOI] [PubMed] [Google Scholar]
- 32. Jormakka M, Törnroth S, Abramson J, Byrne B, Iwata S (2002) Purification and crystallization of the respiratory complex formate dehydrogenase‐N from Escherichia coli . Acta Crystallogr D58:160–162. [DOI] [PubMed] [Google Scholar]
- 33. Rothery RA, Bertero MG, Spreter T, Bouromand N, Strynadka NC, Weiner JH (2010) Protein crystallography reveals a role for the FS0 cluster of Escherichia coli nitrate reductase A (NarGHI) in enzyme maturation. J Biol Chem 285:8801–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blasco F, Guigliarelli B, Magalon A, Asso M, Giordano G, Rothery RA (2001) The coordination and function of the redox centres of the membrane‐bound nitrate reductases. Cell Mol Life Sci 58:179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Correia C, Besson S, Brondino CD, González PJ, Fauque G, Lampreia J, Moura I, Moura JJ (2008) Biochemical and spectroscopic characterization of the membrane‐bound nitrate reductase from Marinobacter hydrocarbonoclasticus 617. J Biol Inorg Chem 13:1321–1333. [DOI] [PubMed] [Google Scholar]
- 36. Marangon J, Paes de Sousa PM, Moura I, Brondino CD, Moura JJ, González PJ (2012) Substrate‐dependent modulation of the enzymatic catalytic activity: reduction of nitrate, chlorate and perchlorate by respiratory nitrate reductase from Marinobacter hydrocarbonoclasticus 617. Biochim Biophys Acta 1817:1072–1082. [DOI] [PubMed] [Google Scholar]
- 37. Magalon A, Asso M, Guigliarelli B, Rothery RA, Bertrand P, Giordano G, Blasco F (1998) Molybdenum cofactor properties and [Fe–S] cluster coordination in Escherichia coli nitrate reductase A: investigation by site‐directed mutagenesis of the conserved his‐50 residue in the NarG subunit. Biochemistry 37:7363–7370. [DOI] [PubMed] [Google Scholar]
- 38. Anderson LJ, Richardson DJ, Butt JN (2001) Catalytic protein film voltammetry from a respiratory nitrate reductase provides evidence for complex electrochemical modulation of enzyme activity. Biochemistry 40:11294–11307. [DOI] [PubMed] [Google Scholar]
- 39. Elliott SJ, Hoke KR, Heffron K, Palak M, Rothery RA, Weiner JH, Armstrong FA (2004) Voltammetric studies of the catalytic mechanism of the respiratory nitrate reductase from Escherichia coli: how nitrate reduction and inhibition depend on the oxidation state of the active site. Biochemistry 43:799–807. [DOI] [PubMed] [Google Scholar]
- 40. Brigé A, Leys D, Meyer TE, Cusanovich MA, Van Beeumen JJ (2002) The 1.25 A resolution structure of the diheme NapB subunit of soluble nitrate reductase reveals a novel cytochrome c fold with a stacked heme arrangement. Biochemistry 41:4827–4836. [DOI] [PubMed] [Google Scholar]
- 41. Brigé A, Leys D, Van Beeumen JJ (2001) Crystallization and preliminary X‐ray analysis of the recombinant dihaem cytochrome c (NapB) from Haemophilus influenzae. Acta Crystallogr D57:418–420. [DOI] [PubMed] [Google Scholar]
- 42. Brigé A, Cole JA, Hagen WR, Guisez Y, Van Beeumen JJ (2001) Overproduction, purification and novel redox properties of the dihaem cytochrome c, NapB, from Haemophilus influenzae . Biochem J 356:851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cerqueira NM, Gonzalez PJ, Brondino CD, Romão MJ, Romão CC, Moura I, Moura JJ (2009) The effect of the sixth sulfur ligand in the catalytic mechanism of periplasmic nitrate reductase. J Comput Chem 30:2466–2484. [DOI] [PubMed] [Google Scholar]
- 44. Cerqueira NM, Fernandes PA, Gonzalez PJ, Moura JJ, Ramos MJ (2013) The sulfur shift: an activation mechanism for periplasmic nitrate reductase and formate dehydrogenase. Inorg Chem 52:10766–10772. [DOI] [PubMed] [Google Scholar]
- 45. Hofmann M (2009) Density functional theory study of model complexes for the revised nitrate reductase active site in Desulfovibrio desulfuricans NapA. J Biol Inorg Chem 14:1023–1035. [DOI] [PubMed] [Google Scholar]
- 46. Xie HJ, Cao ZX (2010) Enzymatic reduction of nitrate to nitrite: Insight from density functional calculations. Organometallics 29:436–441. [Google Scholar]
- 47. Sousa SF, Cerqueira NMFSA Brás NF, Fernandes PA Ramos MJ (2014) Enzymatic “tricks”: Carboxylate shift and sulfur shift. Intl J Quant Chem 114:1253–1256. [Google Scholar]
- 48. Cerqueira NM, Pakhira B, Sarkar S (2015) Theoretical studies on mechanisms of some Mo enzymes. J Biol Inorg Chem 20:323–335. [DOI] [PubMed] [Google Scholar]