

 This work is licensed under a
This work is licensed under a Summary
Lipoid congenital adrenal hyperplasia (lipoid CAH), the most severe form of CAH, is most commonly caused by mutations in steroidogenic acute regulatory protein (STAR), which is required for the movement of cholesterol from the outer to the inner mitochondrial membranes to synthesize pregnenolone. This study was performed to evaluate whether the salt-losing crisis and the adrenal inactivity experienced by a Scandinavian infant is due to a de novo STAR mutation. The study was conducted at the University of North Dakota, the Mercer University School of Medicine and the Memorial University Medical Center to identify the cause of this disease. The patient was admitted to a pediatric endocrinologist at the Sanford Health Center for salt-losing crisis and possible adrenal failure. Lipoid CAH is an autosomal recessive disease, we identified two de novo heterozygous mutations (STAR c.444C>A (STAR p.N148K) and STAR c.557C>T (STAR p.R193X)) in the STAR gene, causing lipoid CAH. New onset lipoid CAH can occur through de novo mutations and is not restricted to any specific region of the world. This Scandinavian family was of Norwegian descent and had lipoid CAH due to a mutation in S TAR exons 4 and 5. Overexpression of the STAR p.N148K mutant in nonsteroidogenic COS-1 cells supplemented with an electron transport system showed activity similar to the background level, which was ∼10% of that observed with wild-type (WT) STAR. Protein-folding analysis showed that the finger printing of the STAR p.N148K mutant is also different from the WT protein. Inherited STAR mutations may be more prevalent in some geographical areas but not necessarily restricted to those regions.
Learning points
STAR mutations cause lipoid CAH.
This is a pure population from a caucasian family.
Mutation ablated STAR activity.
The mutation resulted in loosely folded conformation of STAR.
Background
In addition to ambiguous genitalia development, the newest and the most recently characterized form of lipoid congenital adrenal hyperplasia (lipoid CAH) is the most severe form characterized by severe defects in the adrenal and the gonadal conversion of cholesterol to pregnenolone (1), the precursor of all steroids. Although a large number of clinical reports from Japan have described patients with large adrenal glands and babies born with abnormal female genitalia, more detailed information was missing (1). The hormonal disorder and levels of the hormonal reduction were first investigated by Prader et al. in the mid-1950s although the pathological descriptions of infants dying from lipoid CAH had appeared previously (1). Studies of affected adrenal or testicular tissues or their isolated mitochondria showed an inability to convert cholesterol to pregnenolone due to the severely reduced availability of mitochondrial cholesterol for the synthesis of pregnenolone, which occurs prior to the catalytic activity of the side chain cleavage enzyme P450scc (1). Patients with lipoid CAH have normal term delivery because placental biosynthesis of progesterone remains unaffected (1).
STAR is expressed in the adrenals and gonads, but not in the placenta, suggesting that mutations of this protein might be responsible for lipoid CAH (1). We proposed a two-hit model to explain the mechanism of action: first hit, in the absence of STAR as in early lipoid CAH, a mechanism independent of STAR can transfer some cholesterol molecules into the mitochondria, resulting in a low level of steroidogenesis. This increases corticotropin secretion and stimulates further production of cholesterol and its accumulation as cholesterol esters in lipid droplets. Second hit, as lipid droplets accumulate, they engorge the cell, damaging its cytoarchitecture through both physical displacement and by chemical action of cholesterol auto oxidation products, and all the steroidogenic capacity is destroyed.
Lipoid CAH is a rare disorder in most populations. An attenuated form of the disease, “non-classic lipoid CAH,” is caused by mutations in which 10–25% of normal STAR activity is retained (1, 2). These patients typically experience adrenal insufficiency several years after infancy, and the 46,XY individuals may masculinize normally, and mineralocorticoid secretion may be minimally affected (3). The most common mutation causing this phenotype is STAR p.R188C. Thus, the spectrum of clinical presentations of patients with STAR mutations is substantially broader than that of classic lipoid CAH, and many affected patients may not be diagnosed correctly (2).
We identified a common mutation among Japanese, Korean and Palestinian patients that caused lipoid CAH (1). In addition, three groups have reported five Japanese alleles carrying STAR p.A218V (1). However, other STAR mutations appear in individual families rather than being widespread in an ethnic group. Here, we report for the first time STAR mutations identified in an infant of Norwegian (Scandinavian) descent that causes lipoid CAH. Besides the novel point mutation, the patient also has a premature stop codon, resulting in a truncated protein. The vast majority of the current population of Scandinavia originated from a small region in the southern Scandanvia thought to have been occupied by northern Germanic tribes in the Early Middle Ages. The original population is thought to have started about 12 000 years ago and the cross-cultural settlement is currently unknown (4). Thus it is considered a pure ethnic origin. However, neither the patient's father nor mother carried the same mutation, suggesting that although lipoid CAH is considered autosomal, it may also appear independent of genetic origin. We examined whether the novel mutation is solely responsible for the lipoid CAH by recreating the mutations in STAR cDNA, determining activity, folding and expression by comparison with the WT unaffected protein sequence.
Case presentation
A 2.5 month old infant girl was admitted to the pediatric intensive care unit (PICU) at the Sanford Health Center due to adrenal salt wasting crisis. The infant was the full-term first child of parents of Norwegian descent. The baby presented with the normal female external genitalia, including the absence of gonads in the labial folds, and also showed no abnormality in a routine normal newborn screen. She had hyperpigmented areolas, labia majora and skin relative to her parents. Due to the urgency to obtain steroid precursor before starting glucocorticoid replacement and limited blood volume, we concentrated to immediate steroid analysis. We did not measure DHEA and androstenedione because of the low blood volume. Newborn screen found normal 17-hydroprogesterone.
Laboratory analysis revealed that the patient had sodium levels of 117 mEqu/l (∼140 mEqu/l normal) and potassium levels of 8.9 mEqu/l (3.7–5.2 mEqu/l) and clinically dehydrated. In addition, her adrenocorticotropic hormone (ACTH) level was 5995 pg/ml (normal <60 pg/ml), renin was 120 ng/ml per h (normal <7.8 ng/ml per h), pregnenolone was <16 ng/dl and 17-hydroxypregnenolone was <16 ng/dl. Random cortisol was 3.6 μg/dl before the first dose of glucocorticoid was given. An ultrasound of the patient's adrenal glands revealed they were enlarged; a second ultrasound showed her right adrenal gland measured 2.8×1.4×1.3 cm, which was just over half the size of her corresponding kidney. The left adrenal gland measured 1.0×1.1×1.3 cm, which was approximately one-third the size of the corresponding kidney. Ovaries or uterus could not be identified on the ultrasound, although resolution was poor due to infant's inability to keep her bladder full. Array comparative genomic hybridization (aCGH) analysis indicated a single X and a single Y with normal autosomes and no imbalances observed.
Investigation
Methods
Informed consent was obtained from each family member, and blood samples collected in ethylenediaminetetraacetate were used to prepare genomic DNA using a genomic DNA isolation kit (DNeasy Blood and Tissue Kit, Qiagen). The DNA of the patient and the parents was first amplified with the oligonucleotides, S3 and AS1, and was cut with Tsp45 I and BstN1 to screen for the common STAR p.Q258X and STAR p.R182L mutations, as previously described (1). Then, using a variety of primer combinations, we completed the amplification of exon 5 with JK5S (5′TCG TGA TTC TGG TTC CCC ATG GCC TGG TAG3′) and JK5AS (5′GGG TTT GGA GCC TGC TGC CCG TAT TAC3′) primers and exon 4 with EX4S (TGC TGG GAT TAT AGG CGT GAA C) and Ex4AS (GCT AGG GGT CCT CTC TTT GAT ACA G) and PCR conditions as previously described (1). The PCR products were sequenced directly using commercial resources (MC Lab, South San Francisco, CA, USA). The steroid conversion was analyzed following our laboratory procedure and characterized by liquid chromatography equipped with mass spectrometry (LC–MS) (5).
Cell culture, transfection, western blotting and activity
The cell culture, transfection, western blotting and activity were determined by following the procedure, which was developed in the lab (6). Metabolic conversion of cholesterol to pregnenolone was performed following the transfection of the whole COS-1 cells. Reactions were initiated with NADPH and 14C-cholesterol.
Protein finger printing
[35S]-Methionine (Met)-labeled WT and STAR p.N148K were synthesized in a cell-free system (CFS) using a TNT-rabbit reticulocyte (Promega). Ribosomes and their associated polypeptide chains were removed by centrifugation at 150 000g for 15 min at 4 °C. We performed limited proteolysis of the WT and mutant STAR using sequencing grade trypsin (Thermo fisher).
Outcome and follow-up
STAR is a 7.5 kb gene with seven exons, but exons 5–7 comprise 40% of the protein. Most STAR mutations causing lipoid CAH were found in exons 5–7 of the STAR gene (1). In addition, we have identified several mutants in exons 3 and 4 (1). Therefore, we amplified a 2.1-kb fragment of genomic DNA isolated from the patient spanning exons 5–7 with S3 and AS1 primers as well as exon 4 with Ex4S and Ex4AS primers using previously described procedures (1) followed by sequencing each exon (1). Genetic data showed that the patient had a mutation in exon 5 at nucleotide 557, which was changed from cytosine (C) to thymine (T), resulting from arginine 193 to a stop codon. The single nucleotide polymorphism database (dbSNP) number for Arg193Ter is rs387907235. Also, another mutation in exon 4 at nucleotide 444 from cytosine (C) to adenine (A) (dbSNP – rs775130992), resulted in the STAR p.N148K mutation (Fig. 1), which was observed before in an Italian patient (7). The premature stop codon truncated the protein with 21 kDa or 193 amino acids. Given that deletion of ten amino acids from the C-terminus of STAR destroyed 50% of its activity (1), we expect that this mutation reduces pregnenolone synthesis.
Figure 1.
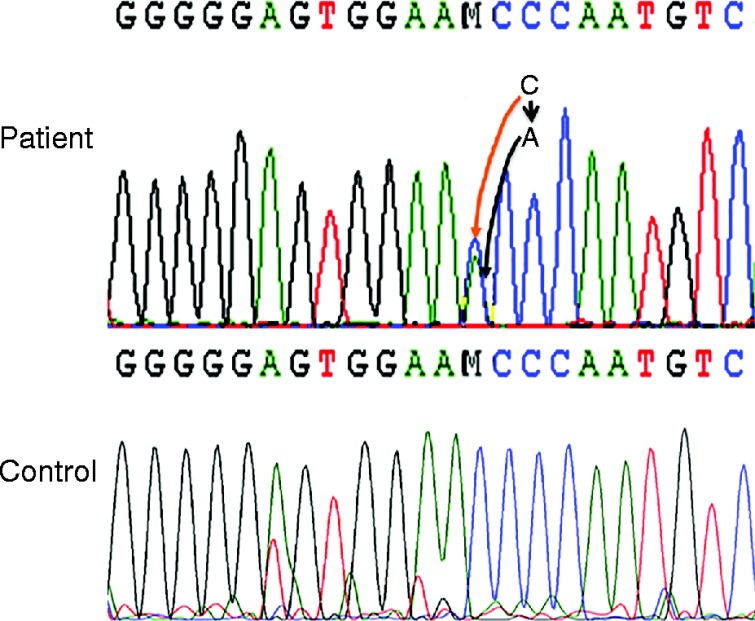
Sequencing of the genomic DNA isolated from the patient and control (Unaffected individual) with Ex4S and Ex4AS primer. The change in nucleotide (C–A) is indicated by arrowhead.
A similar analysis with the genomic DNA of each parent revealed the absence of either mutation, suggesting that both are de novo mutations. Some STAR mutations, including STAR p.M225T in exon 4, induce only a 45% reduction in activity as compared with others in which 10–25% of activity is retained. To examine the impact of these mutations on STAR activity, we overexpressed the mutant cDNA in nonsteroidgenic COS-1 cells that were also transfected with ferrodoxin, ferrodoxin reductase and P450scc as a single fusion protein, F2 (1). As shown in Fig. 2, analysis of pregnenolone synthesis revealed that the WT vector synthesized threefold more pregnenolone as compared with the mutant STAR protein (Fig. 2A and B). The mutant synthesized pregnenolone similar to the vector control, confirming that indeed the identified mutation completely destroyed the activity. The quantitative analysis is presented in Fig. 2B. The authenticity of the converted steroid was analyzed first by liquid chromatography (Fig. 2C) followed by confirmation through mass spectrometry analysis (Fig. 2D). As expected the mutant and the WT processed into the mitochondria as evidenced by the western blotting of the transfected COS-1 cells (Fig. 3). Because the mutants and the WT have the same mitochondrial targeting sequence, thus both the proteins were imported in to the mitochondria in a similar fashion (1).
Figure 2.
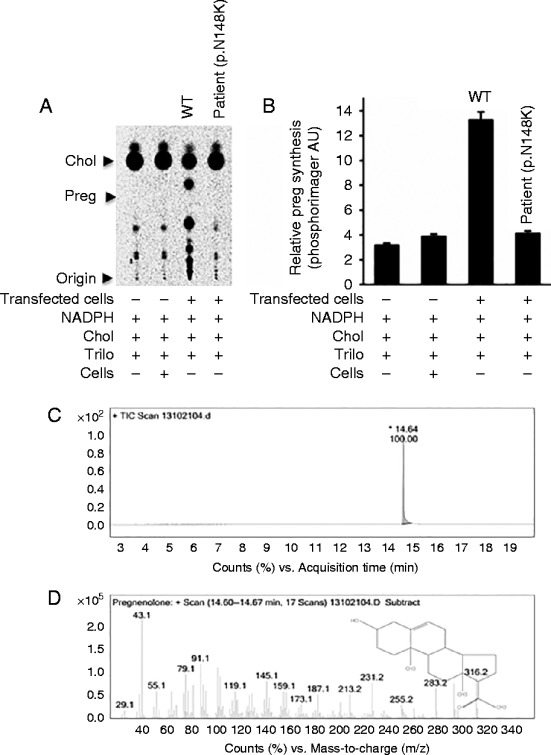
Measurement of pregnenolone synthesis by the STAR p.N148K mutant. (A) The pregnenolone synthesis by the STAR p.N148K mutant was compared with WT STAR after their expression in nonsteroidogenic COS-1 cells. (B) Quantitative analysis of the amount of steroid synthesized through phosphorimager analysis. Y-axis is presented in arbitrary units (AU) developed by phosphoimager program. (C and D), GC/MS analysis of the pregnenolone identified from the TLC plate, where panel (C) is the liquid chromatogram and (D) is the mass spectrometric identification of the steroid, pregnenolone. Data represented in panel B is the mean±s.e.m. of three independent experiments performed at three different times.
Figure 3.
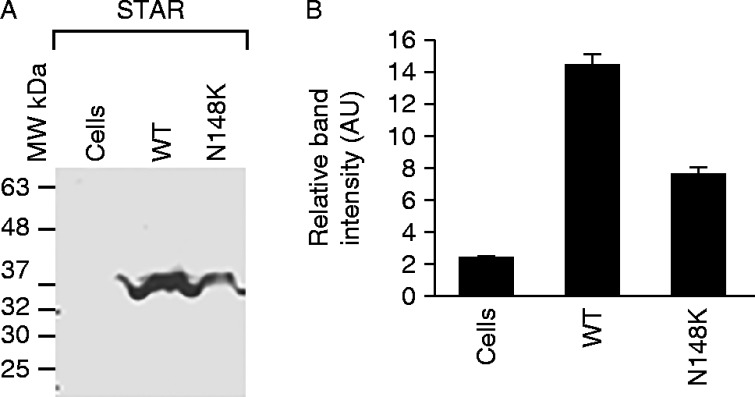
Determination of WT and mutant STAR expression. Protein levels were determined by western blot analysis with STAR antiserum. The right hand panel shows the quantitative estimation of the intensity of bands from the western blot experiment and data represented in the left hand panel is the mean±s.e.m. of three independent experiments performed at three different times.
Analysis of the impact of STAR mutations on STAR protein folding previously showed that the STAR mutants were folded in a manner similar to that of the WT protein. For example, STAR p.M225T is folded similarly to WT protein. Thus, we determined whether the N148K mutant was folded differently from the WT protein by expressing it in a cell-free rabbit reticulocyte system, which expresses proteins in a fashion similar to that of cells and then analyzed protein conformation by protein finger printing. The resulting proteolytic protein digestion pattern can be used to examine the conformation of a protein, since it depends on the accessibility of the cleavage sites to the protease. If the accessibility of the cleavage site varies due to changes in conformation, then different proteolytic patterns will emerge. As shown in Fig. 4, the WT and the STAR p.N148K mutant have different proteolysis patterns, suggesting that the STAR p.N148K mutant is folded differently from the WT STAR and possibly less likely to interact with the mitochondrial receptor, voltage-dependent anion channels 1and 2 (VDAC1 and VDAC2) (6).
Figure 4.
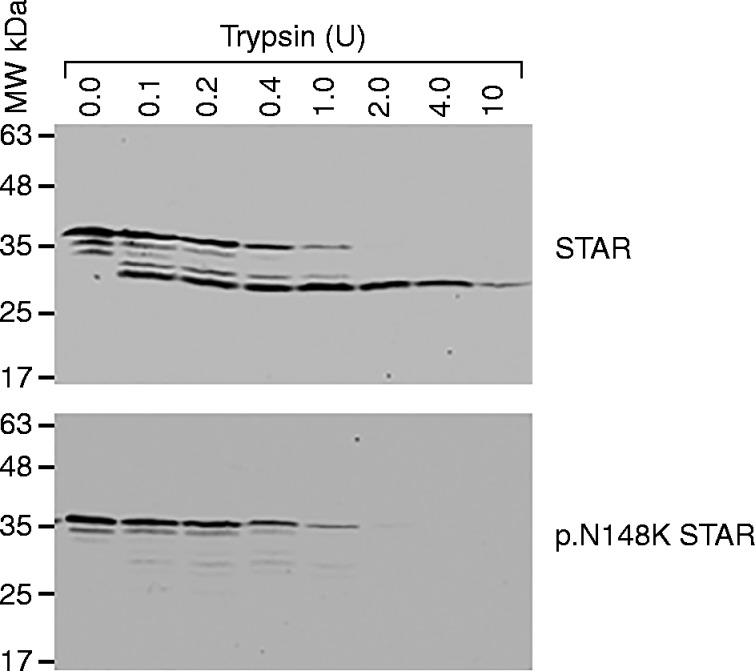
Analysis of STAR p.N148K mutant conformation by protein finger printing. Cell-free synthesized, radiolabeled WT and mutant STAR p.N148K were proteolyzed, and the proteolytic pattern was visualized by autoradiography.
The patient was treated with i.v. hydrocortisone and discharged on oral hydrocortisone 15 mg/m2 per day, fludrocortisone 0.1 mg twice daily with salt supplementation. At 6 month of age, the patient underwent an exploratory laparotomy with bilateral gonadodectomy. Pathological examination of the gonads confirmed the presence of testes. At the time of surgery, renin levels had normalized into the normal range and ACTH was just above the normal level.
Discussion
STAR gene mutations in at least 190 patients with lipoid CAH have now been described, which includes 100 patients available in Japanese literature. These studies identified some patients, who were siblings or were the products of known consanguinity; however, these patients represent a small number of alleles. The only consistent genetic clusters identified to date include the STAR p.Q258X mutation in Japanese and Korean populations (1) and the STAR p.R182L mutation among Palestinian Arabs (1). The present study is the first to identify the STAR p.N148K mutation in a patient of Scandinavian origin and was carefully confirmed in independent samples to eliminate the possibility of a mix-up of samples in the laboratory. This missense mutation resulting in the loss-of-function as observed by analyzing pregnenolone synthesis, is similar to the level obtained using the empty vector or buffer control. In addition to the STAR p.N148K mutation, a second mutation STAR p.R193X, a nonsense mutation, was identified.
STAR missense mutations causing lipoid CAH are mostly concentrated from exons 5–7, and all other mutations are present throughout the gene sequence. As expected all the frameshift mutations generates truncated STAR protein resulting complete inactivity. Deletion of only ten carboxyl-terminal amino acids reduces STAR activity by half, and deletion of only 28 carboxyl-terminal amino acids deletes all activity (8). However, deletion of the N-terminal 62 amino acids does not impact activity. The small number of missense mutations that cause lipoid CAH all lie between amino acids 169 and 275 (1), and these tend to be the residues most highly conserved in the related protein, START (1) for STAR-related lipid binding protein. In the absence of STAR expression, steroidogenic cells still produce 10–14% pregnenolone; however, the mechanism of STAR independent steroidogenesis is not clearly understood, may be minimum level of steroid is available with START domain proteins other than STAR. The STAR c.C444A nucleotide is conserved across species, and the mutation possibly would destroy any steroidogenic capacity in all mammalian system originated from proteins with START domains.
STAR does not bind cholesterol, but fosters it into the mitochondria. In addition, the mitochondrial import process of STAR is different from the other mitochondrial-targeted proteins, so it is likely that mutant STAR interacts with VDAC2 (6) in a different fashion as compared with the WT. Analysis of the three-dimensional (3D) crystal structure or solution structure of STAR has showed that missense mutations alter STAR folding (1). We have found that residues 63–193 of STAR slow the protein's transit into the mitochondria, permitting the biologically active carboxyl-terminus to have more interactions with the OMM-associated VDAC2 (6). STAR requires interaction with VDAC1 prior to reaching VDAC2 for entry into the mitochondria, which is dependent on its appropriate folding facilitated immediately after its release from the ribosome by the MAM-associated GRP78 (6). Given that we have previously shown the requirement of a specific orientation between mitochondria, cholesterol and STAR to foster 220 molecules of cholesterol, it is possible that the altered conformation that results from the STAR p.N148K mutation prevents its interaction with VDAC1 and, therefore, VDAC2, inhibiting cholesterol fostering. The exact nature of the STAR-GRP78 interaction remains to be understood, and might occur via multiple variations as START domain proteins can transfer cholesterol to different intracellular membranes (6).
The difference between 21-hydroxylase deficiency and lipoid CAH is easy to confirm; lipoid CAH patients have normal looking external female genitalia of both the XX and XY karyotypes and have extremely low levels of all steroid hormones. However, the 21-hydroxylase deficiency patients have higher level of 21-deoxysteroids, especially 17-hydroxyprogesterone, and affected 46,XX not the 46,XY patients are virilized. The lipoid CAH may be confused with 3β-hydroxysteroid dehydrogenase (3βHSD) deficiency due to higher level of 17-hydroxypregnenolone (1). The most difficult is to differentiate between lipoid CAH and congenital adrenal hypoplasia, because of the normal looking XY female genitalia is likely to be a lipoid CAH patient. However, the diagnosis of lipoid CAH in 46,XX patients generally depends on the radiographic demonstration of massively enlarged adrenals (1) which are not found in adrenal hypoplasia. The diagnosis of lipoid CAH in the infant is now more common than it had been previously appreciated.
De novo mutations have been widely characterized in neurological disorders, especially in Schnophonia and epilepsy (9). Three different kinds of models have been discussed to explain de novo mutations (10). For a specific mutation rate, which is due to random variations, the number of actual mutations is expected to have a Poisson distribution (10). In the mathematical program and accounting for Poisson variation, a linear fit shows (effect=2.01 mutations per year) that father's age can explain up to 94% of the remaining variation in the observed mutation counts. In the second model that also considers maternal mutation, the mutation count increases by 3.23% per year. This second model explains 96.6% of the remaining variation. The third model assumes that the maternal mutation rate is constant at 14.2, and the parental mutations increase exponentially. This explains 97.1% of the remaining variation, and the rate of paternal mutations is estimated to increase by 4.28% per year, which corresponds to doubling every 16.5 years and increasing by eightfold in 50 years. Hence, although it seems that the number of paternal de novo mutations increases at a rate that accelerates with the father's age, more data at the upper age are needed to better evaluate the nature of the acceleration (10). We could not confirm consanguinity following a long discussion and parents confirmed it was their first marriage. Also during discussion both the mother and the father confirmed that this was their first and only biological newborn baby. However, we do not have a spectrum of patients; thus, we can only conclude that like other genetic diseases de novo mutations resulting in lipoid CAH might be equally considered in the future diagnosis of all newborn babies.
Acknowledgements
L Casas is thankful to Dr Kenneth Copeland and Dr Walter L Miller for their stimulus discussion.
Footnotes
(J Kaur and L Casas contributed equally to this work)
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
The work was supported by a grant from the National Institutes of Health (HD057876) and an intramural research grant from the Anderson Cancer Institute to HSB.
References
- 1. Miller WL & Bose HS. 2011. Early steps in steroidogenesis: intracellular cholesterol trafficking. Journal of Lipid Research 52 2111–2135. 10.1194/jlr.R016675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Metherell LA, Naville D, Halaby G, Begeot M, Huebner A, Nürnberg G, Nürnberg P, Green J, Tomlinson JW, Krone KP et al. 2009. Nonclassic lipoid congenital adrenal hyperplasia masquerading as familial glucocorticoid deficiency. Journal of Clinical Endocrinology and Metabolism 94 3865–3871. 10.1210/jc.2009-0467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller WL & Auchus RJ. 2011. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocrine Reviews 32 81–151. 10.1210/er.2010-0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Helgason A, Sigurðardóttir S, Nicholson J, Sykes B, Hill EW, Bradley DG, Bosnes V, Gulcher JR, Ward R & Stefánsson K. 2000. Estimating Scandinavian and Gaelic ancestry in the male settlers of iceland. American Journal of Human Genetics 67 697–717. 10.1086/303046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pawlak KJ, Prasad M, McKenzie KA, Wiebe JP, Gairola CG, Whittal RM & Bose HS. 2011. Decreased cytochrome c oxidase IV expression reduces steroidogenesis. Journal of Pharmacology and Experimental Therapeutics 338 598–604. 10.1124/jpet.111.182634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Prasad M, Kaur J, Pawlak KJ, Bose M, Whittal RM, Bose HS & Mitochondria-Associated ER. 2015. membrane regulates steroidogenic activity via StAR-VDAC2 interaction. Journal of Biological Chemistry 290 2604–2616. 10.1074/jbc.M114.605808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bens S, Mohn A, Yüksel B, Kulle AE, Michalek M, Chiarelli F, Özbek MN, Leuschner I, Grötzinger J et al. 2010. Congenital lipoid adrenal hyperplasia: functional characterization of three novel mutations in the StAR gene. Journal of Clinical Endocrinology and Metabolism 95 1301–1308. 10.1210/jc.2009-1176 [DOI] [PubMed] [Google Scholar]
- 8. Bose HS. 2011. Mechanistic sequence of mitochondrial cholesterol transport by StAR proteins. Journal of Proteins and Proteomics 2 1–9. [Google Scholar]
- 9. Gauthier J & Rouleau GA. 2012. De novo mutations in neurological and psychiatric disorders: effects, diagnosis and prevention. Genome Medicine 4 1–11. 10.1186/gm372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A et al. 2012. Rate of de novo mutations and the importance of father's age to disease risk. Nature 488 471–475. 10.1038/nature11396 [DOI] [PMC free article] [PubMed] [Google Scholar]