

Abstract
β‐synuclein (βS) is a homologue of α‐synuclein (αS), the major protein component of Lewy bodies in patients with Parkinson's disease. In contrast to αS, βS does not form fibrils, mitigates αS toxicity in vivo and inhibits αS fibril formation in vitro. Previously a missense mutation of βS, P123H, was identified in patients with Dementia with Lewy Body disease. The single P123H mutation at the C‐terminus of βS is able to convert βS from a nontoxic to a toxic protein that is also able to accelerate formation of inclusions when it is in the presence of αS in vivo. To elucidate the molecular mechanisms of these processes, we compare the conformational properties of the monomer forms of αS, βS and P123H‐βS, and the effects on fibril formation of coincubation of αS with βS, and with P123H‐βS. NMR residual dipolar couplings and secondary structure propensities show that the P123H mutation of βS renders it more flexible C‐terminal to the mutation site and more αS‐like. In vitro Thioflavin T fluorescence experiments show that P123H‐βS accelerates αS fibril formation upon coincubation, as opposed to wild type βS that acts as an inhibitor of αS aggregation. When P123H‐βS becomes more αS‐like it is unable to perform the protective function of βS, which suggests that the extended polyproline II motif of βS in the C‐terminus is critical to its nontoxic nature and to inhibition of αS upon coincubation. These studies may provide a basis for understanding which regions to target for therapeutic intervention in Parkinson's disease.
Keywords: α‐synuclein, β‐synuclein, P123H‐βS, NMR, aggregation, inhibition, fibril formation, intrinsically disordered proteins, dementia with Lewy bodies
Introduction
Alpha‐synuclein (αS) is widely known for its involvement in Parkinson's disease, as Lewy Body inclusions that contain αS are found in post‐mortem diseased brains.1, 2 αS belongs to the synuclein family of proteins, which in addition to αS contains two homologs: beta and gamma synuclein.3 All of members of the synuclein family are small neuronal lipoproteins, but only αS and beta‐synuclein (βS) colocalize presynaptically in the brain.4, 5, 6 αS and βS have high sequence similarity (78%) but they differ at the point of their self‐association properties. αS self‐aggregates to pathological oligomers or fibrils, whereas βS forms oligomers more slowly and does not form fibrils on its own.7, 8, 9, 10 Interestingly, there is evidence showing that βS can inhibit αS aggregation in a dose dependent manner, and can mitigate the effects of αS toxicity in vivo.10, 11, 12, 13, 14, 15, 16, 17
Although wild type βS does not appear in pathological Lewy Body plaques or fibrils in vivo,18 two βS mutations, V70M and P123H, were identified and found in sporadic and familial dementia with Lewy Bodies (DLB), respectively.19 Studies on cell line models revealed the involvement of P123H and V70M in lysosomal pathology, and studies on mouse models for the P123H‐βS mutant proved it to be toxic. P123H‐βS exarcerbates αS pathology as the number of lysosomal inclusions increased upon coexpression of βS mutants with αS.20, 21 Furthermore, transgenic mice models of P123H‐βS exhibit extensive neuritic pathology (swelling of striatum and globus pallidus, due to formation of small spheroids), but do not result in formation of Lewy Body inclusions.20 The neuropathy of P123H‐βS is not abolished in αS knock‐out mice, but is enhanced in the P123H‐βS/αS doubly transgenic mice.20 These facts demonstrate that just a single mutation in the βS sequence is able to overcome the nonaggregating and inhibitory nature of wild type βS and that P123H‐βS is toxic by itself.19, 20, 21
αS and βS are intrinsically disordered proteins described by three regions: the N‐terminus that contains KTKXGV repeats and forms helices at membranes, the nonamyloid‐β component (NAC) region, and the highly acidic and solubilizing C‐terminus (Fig. 1).3 The N‐terminus of αS and βS are highly similar as there are only six residue differences between αS and βS, and the C‐terminus is the least conserved region with more prolines and more negatively charged residues. βS has an 11 residue deletion in the NAC region, which is in the core of the αS fibril. This suggests that the nonfibrillar nature of βS may come from this deletion; however, insertion of this region back into βS does not recover the full fibrillation potential of βS.7, 8, 9
Figure 1.
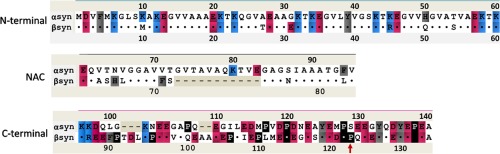
Aligned sequences of αS and βS for three regions of synucleins: N‐terminal, NAC, C‐terminal. Identical residues are shown by dots in ßS sequence and deletions are shown by dashes. The mutation site is indicated by the arrow below the sequence. Residues are color‐coded according to the scheme: blue ‐ positive charges, red ‐ negative charges, black ‐ prolines, grey‐aromatic residues.
From the biophysical point of view the N, NAC, and C‐terminal regions of αS display different properties. The N‐terminus has a small net charge and is best described as a polyampholite chain with more globular‐like characteristics, while the C‐terminus is highly negatively charged, has 5 proline residues and is best described as a polyelectrolye chain with more chain stiffness.22, 23 Both the N‐terminus and NAC region bind membranes and fold to a helix upon binding. The C‐terminus of αS and βS does not bind directly to the membranes and is suggested to have chaperone activity.24, 25, 26 Thus in general synucleins have an N‐terminal and NAC membrane interactive region, and a C‐terminal regulatory domain that may interact with other proteins and other factors. In the case of αS all the disease causing mutations are located in the N‐terminus, while in βS the toxic mutations are found in the NAC and C‐terminus.
In this article we compare the monomer conformations of αS, βS, and P123H‐βS and the ability of αS, βS, and P123H‐βS to accelerate or inhibit αS fibril formation upon coincubation. Our results indicate that P123H‐βS behaves more like αS both in terms of its conformation C‐terminal to the mutation site and in terms of its effect on αS fibril formation upon coincubation. Both P123H‐βS and αS have a less ordered C‐terminus, and coincubation of P123H‐βS with αS or simply doubling the concentration of αS result in identical fibril formation kinetics. Our results suggest that the single P123H mutation in the C‐terminal region of βS, which removes the double proline motif from the sequence, causes the conformational properties of the C‐terminus to be altered and to resemble the C‐terminus of αS. This renders P123H‐βS unable to perform the protective functions of the wild type βS protein and supports the view that the extended PPII motif of βS in the C‐terminus is critical to inhibition and to its nontoxic nature.20, 27
Results
Comparison of P123H‐βS and βS indicates that P123H‐βS populates a higher percentage of compact conformational ensembles due to a more compact C‐terminus
To understand the basis for the different toxicity of βS and P123H‐βS we performed their characterization via biophysical methods. αS and βS are acetylated in vivo; therefore, our studies are performed on the acetylated forms of all proteins. Acetylated P123H‐βS, similarly to wild type βS, is mostly unfolded and monomeric as shown through narrow HSQC profiles [Fig. 2(A)]. HSQC differences between these two proteins are very small and mostly located close to the mutation site. Electrospray ionization mass spectroscopy (ESI‐MS) experiments indicate that both proteins have the correct molecular weight and are pure. Additionally ESI‐MS experiments indicate that both proteins are able to probe compact and extended conformations; however the population distributions are altered for P123H‐βS with a higher percentage of compact conformation relative to wild type βS [Fig. 2(B)]. Secondary structure propensities reveal that the N‐terminus and NAC region are essentially identical while the C‐terminus displays differences particularly near the mutation site [Fig. 2(C)]. The βS C‐terminus is extended and has uniformly negative values, which is suggestive of polyproline II (PPII) secondary structure.28 The mutation at position P123H causes a discontinuity or break in the negative secondary structure propensities as shown using SSP [Fig. 2(C)].29A complementary approach using δ2D30 shows an increase in the amount of coil along with a decrease in beta‐sheet C‐ terminal to the mutation site (Supporting Information Fig. 1). Taken together, these data suggest that the conformation at the C‐terminus is not uniformly extended and that the region around the mutation site is more random coil‐like. The SSP and δ2D data, in conjunction with the ESI data suggest that the sampling of a higher population of compact conformations may be due to the more compact nature of the C‐terminus. Size exclusion chromatography was used to evaluate the existence of higher order species after five hours of incubation. Most of the protein eluted at 34 min, which is consistent with the monomer, but P123H‐βS generated more oligomers that were eluted in the void volume, suggesting that the mutant is more prone to aggregation [Fig. 2(D)].
Figure 2.
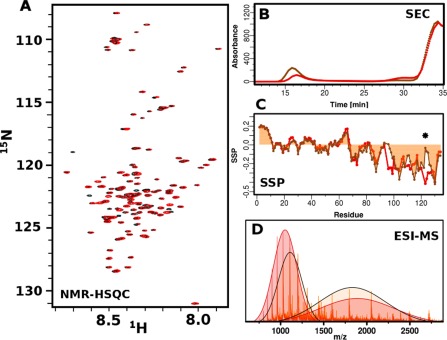
Biophysical characterization of βS and P123H‐βS mutant. (A) 1H‐15N‐HSQC spectra of βS (red) and P123H‐βS mutant (black) in 10mM MES, pH6, 100mM NaCl. (B) ESI‐MS of βS (red) and P123H‐βS (orange) in 10mM ammonium acetate buffer, pH 6. Extended and compact conformations are indicated by fitting two Gaussians. βS populates 46% compact and 54% extended conformations while P123H‐βS populates 51% compact and 49% extended conformations. (C) Comparison of secondary structure propensities (SSP) of βS (red) and P123H‐βS (orange) in 10mM MES, pH6, 100mM NaCl. Positive values indicate α‐helical secondary structure propensity, while negative values correspond to β‐Sheet or PPII propensity. The star indicates the position of the mutation. (D) Size exclusion profile using the Superose 6 column, which has a separation range of 5000 to 5000000 Da for βS (red) and P123H‐βS (orange) after 5 h of incubation at 37°C with agitation in PBS, pH 7.4. βS and P123H‐βS have similar monomer elution profiles and P123H‐βS generates oligomers that are eluted in the void volume.
Coincubation of P123H‐βS mutant with αS accelerates αS fibril formation
Coincubation of αS with βS or with P123H‐βS results in significantly different fibril formation profiles. It has been shown previously with nonacetylated protein that there is a dose dependent concentration dependence of fibril formation of αS coincubated with βS, and that αS fibril formation is delayed in the presence of βS. We have shown similar dose dependent concentration results upon coincubation of acetylated αS with acetylated βS (manuscript, submitted). While coincubation with βS alone results in delayed fibril formation [Fig. 3(A)], coincubation of αS with P123H‐βS results in increased fibril formation rates relative to αS alone [Fig. 3(B)]. More specifically, the kinetics of fibril formation resulting from doubling the original concentration of αS are almost identical to those of the coincubated αS/P123H‐βS. This supports the view that fibril formation is enhanced by P123H‐βS and that αS interacts directly with P123H‐βS. Coincubation with P123H‐βS and with twice the original concentration of αS shows a faster elongation phase as well as a doubling of Thioflavin T (ThT) intensity, suggesting that P123H‐βS behaves very similarly to αS in terms of its ability to the form fibrils [Fig. 3(B)]. P123H‐βS when incubated alone is not able to form fibrils (Fig. 3(B)] and only forms amorphous aggregates as detected by transmission electron microscopy (TEM) [Fig. 3(F)]. The differences in fibril formation are mirrored in the TEM data where αS alone forms fibrils [Fig. 3(C)], βS forms amorphous aggregates [Fig. 3(D)], αS coincubation with βS forms fibrils that are thinner and more branched [Fig. 3(E)] and coincubation of αS with P123H‐βS forms fibrils that are highly ordered [Fig. 3(G)]. These data are striking as they show that a single mutation in the C‐terminus of βS can completely reverse the inhibition properties of βS on αS and that the mutant protein behaves, in terms of fibril formation, in the same way as simply increasing the concentration of αS.
Figure 3.
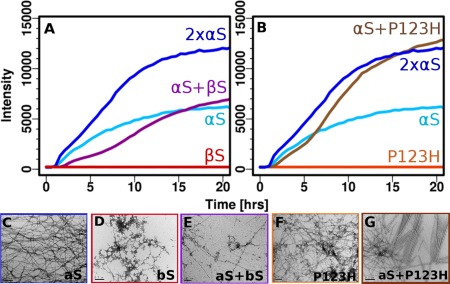
Aggregation inhibiton of αS by βS and aggregation enhancement of αS by P123H‐βS (A & B). ThT fluorescence (37°C with shaking and teflon beads, PBS) of αS coincubated with (A) βS and (B) P123H‐βS. Negatively stained electron micrographs (scale 200 nm) (C‐G) of (C) αS fibrils, (D) βS amorphous aggregates, (E) coincubated αS with βS, (F) P123H‐βS amorphous aggregates, and (G) coincubated αS with P123H‐βS.
P123H‐βS exhibits conformational characteristics of αS C‐terminal to the mutation site
A three way comparison of αS, βS and P123H‐βS is provided in order to understand, at the molecular level, why a single mutation in βS would alter its inhibitory characteristics towards αS and result in accelerated αS fibril formation kinetics. ESI‐MS experiments, SSP, and residual dipolar couplings (RDC) show clearly that there are conformational similarities between αS and P123H‐βS relative to βS. ESI‐MS experiments show that the population distribution of P123H‐βS and αS are more similar with a higher population of compact conformation relative to extended [Fig. 4(A)]. RDC profiles for all three proteins indicate that they are very similar in the N‐terminus and that major differences in conformation arise in the C‐terminus [Fig. 4(B)]. βS has the highest and most uniform RDC values suggesting that the C‐terminus from residues 95 to 134 is extended and rigid. αS shows increased RDC values in two distinct C‐terminal hydrophobic patches, in agreement with previous literature, signifying increased order in these two fragments.28, 31, 32, 33 The P123H mutation of βS exhibits characteristics of both αS and βS [Fig. 4(B)]: the RDCs are essentially identical to βS from residues 95 to 119 and are very similar to αS for residues 126 to 140. The lower RDC values of P123H‐βS from residues 126‐140 suggest a more flexible and less ordered C‐terminus than that associated with βS, for which the RDC values remain very high. SSP comparison of these three proteins additionally highlights the fact that the single mutation in the double proline motif of P123H‐βS breaks the uniformity of the beta‐like secondary structure observed for βS [Fig. 4(C)]. SSP shows two distinct C‐terminal propensities for structure for P123H‐βS with a break that is in the same position as the break in αS.
Figure 4.
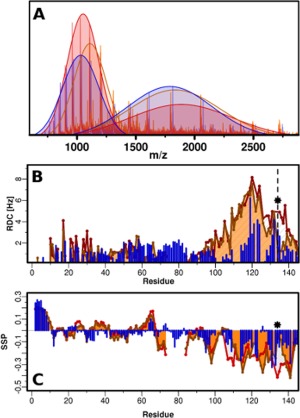
Three way comparison of αS (blue), βS (red) and P123H‐βS (orange). (A) ESI‐MS in 10mM ammonium acetate buffer, pH 6. Extended and compact conformations are indicated by fitting two Gaussians. αS: compact, 49%, extended 51%; βS: compact 46%, 54%; P123H‐βS: compact, 51%, extended 49% (B) RDC profiles of αS (blue), βS (red) and P123H‐βS (orange) in 10mM MES, pH 6, 100 mM NaCl, 250μM protein, measured in C8E5‐octanol bicelle aligning media. The star and dashed line indicate the position of the mutation. (C) SSP measured in 10mM MES, pH 6, 100 mM NaCl, 350μM proteins for αS (blue), βS (red) and P123H‐βS (orange).
C‐terminal flexibility results from loss of double proline motif
The role of the double prolines at positions 122 and 123 appears to be very important in defining the conformation of the C‐terminus as mutation of the 122P123P of βS into 122P123H of P123H‐βS results in more flexible conformations C‐terminal to the mutation as determined by RDC and SSP data. To further investigate whether the double proline motif is crucial to maintaining the extended PPII sequence we performed a Psi‐blast search to obtain the typical conformations of these motifs in the PDB. The search was performed across a 10 residue window that contained the PP motif of βS and its flanking sequences, the PS motif of αS and the PH motif of P123H‐βS. Our results show that the double proline, PP, motif of βS is the most common of the three motifs in the PDB with 117 hits, while the PS motif of αS has 49 hits, and the PH motif of P123H‐βS has only 23 hits. RMSD calculation and structure overlays of the hits were performed [Fig. 5(A–C)], and interestingly the most common motif is the least diverse (RMSD for the PP residues is the lowest with 0.35Å), while the RMSD for P123H‐βS and αS motifs are almost twice as high with 0.77 and 0.70Å, respectively. Ramachandran plots for these motifs indicate that PP is located primarily in the Phi, Psi region correpsonding to PPII [Fig. 5(F)] whereas the PS motif of αS populates the PPII conformation only approximately 1/3 of the time [Fig. 5(D)] and the PH motif in P123H‐βS does not populate the PPII conformation at all [Fig. 5(E)]. The PH motif has a tendency to be located in the forbidden region of the Ramachandran plot just outside the region of the left handed alpha helix [Fig. 5(E)]. The most varied secondary structure tendencies are sampled by the PS motif of αS with beta sheet, PPII, and the right handed alpha helix region as possibilities (but not in the left handed alpha helix region) [Fig. 5(D)]. The loss of the double proline motif clearly alters the conformational propensity around this region and explains why P123H‐βS and αS may have similar conformational propensities in the full length protein C‐terminal to the mutation site.
Figure 5.
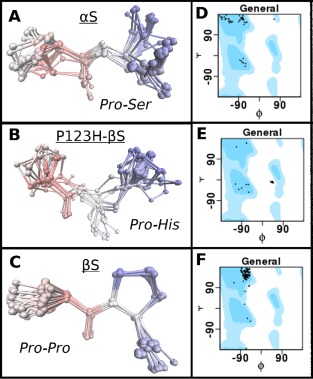
Comparison of possible conformational propensities of C‐terminal motifs of αS (PS), P123H‐βS (PH) and βS (PP) as described by Ramachandran plots. PDB database search was performed using the Psi‐Blast algorithm to obtain fragments which contain motifs PS (αS), PH (P123H‐βS) and PP (βS), but also shared similarity to the C‐terminal sequences. (A‐C) RMSD calculation and structure overlays of the hits were performed for: (A) αS (PS), (B) P123H‐βS (PH) and (C) βS (PP). (D‐E) Phi, Psi population distribution of the different hits for the respective motifs displayed as Ramachandran plots. General shows all of the allowed areas for proteins in blue and dots represent the positions of the Phi, Psi angles for the hits. Position of the Ramachandran plot shows ability of motifs to exist in certain secondary structures. Position of the secondary structures on the Ramachandran plot (Phi, Psi): helix: (−63, −43), beta‐sheet (−135, 135), PPII (−75, 150), L‐alpha helix (57, 47). (D) αS (PS), (E) P123H‐βS (PH) and (F) βS (PP.)
Discussion
A pathological mutant of βS found in DLB, P123H‐βS, shows that just a single mutation renders the normally nontoxic wild type βS into a toxic species, and that it is able to induce aggregation, overcome the inhibitory properties of βS and exarcerbate αS pathology. NMR studies reported here compare the conformational propensities of αS, βS, and P123H‐βS and indicate that the N‐terminal and NAC regions of the three proteins are very similar while the C‐terminal region of these proteins is more variable. βS exhibits the most rigid and extended structure in the C‐terminus, while αS and P123H‐βS are more similar to one another at the C‐terminus, in particular in the C‐terminal region from residues 95 to 119. The mutation at P123H in βS induces a break in the extended PPII secondary structure, suggesting the view that the double proline motif in the βS sequence is important for the extended conformation of the C‐terminus. The mutation at P123H results in a more flexible C‐terminus from residues 120‐134 and allows a wider range of conformational ensembles to be sampled, thereby making P123H‐βS more αS‐like. The fact that all three monomer conformations are similar in the N‐terminus and NAC region but that P123H‐βS and αS are similar in the C‐terminus suggests that this region is critical for the nontoxic to toxic conversion of βS to P123H‐βS. The more flexible C‐terminus may promote self‐aggregation, and the conformational heterogeneity arising from the flexible C‐terminus may increase the likelihood of sampling an aggregation prone conformation, or sampling aggregation prone inter‐chain interactions.
We have shown here with studies of P123H‐βS that the C‐terminal conformation is important for aggregation, but the existence of the βS mutation, V70M, implicated in sporadic cases of DLB disease suggests that the central region of βS may also play an important role in aggregation inhibition. Previous studies on the central NAC region have suggested that the 11 residue deletion may be the cause of aggregation inhibition however insertion of the deletion fragment back into βS does not allow full recovery of aggregation properties.7, 8 Other studies in which existing residues in the NAC region are swapped between αS and βS increase the aggregation behavior of βS indicating that the NAC region modulates aggregation or fibrillization.9 The facts outlined here show that the molecular determinants for the aggregation of synucleins are highly complex.
The role of the C‐terminus has been extensively discussed in the αS literature and can be viewed as playing a significant role in directing aggregation versus inhibition. It has been shown that the collapse of the C‐terminus due to pH changes,34, 35, 36 the addition of polycations and metals,37, 38, 39 the substitutions of prolines to alanines,40 and C‐terminal truncations increase the aggregation propensities of αS.41, 42 Mutations of the tyrosines in the C‐terminus,43, 44 and addition of small molecules that interact with the C‐terminus such as dopamine45 and ECGC46 result in aggregation inhibition. βS, whose C‐terminus is extended, is self‐inhibitory, and P123H‐βS, whose C‐terminus is more flexible is less inhibitory. This αS literature taken together with our data suggests that a PPII extended C‐terminus, as seen in βS, or a C‐terminus that is unavailable due to interactions with small molecules, is important for inhibition and that disruption of the extended conformation makes the protein more prone to aggregation.
Our results show that the changes in the conformation of the C‐terminus of βS can alter, not only self‐aggregation properties, but also its ability to delay or inhibit αS aggregation during coincubation. Just as P123H‐βS is toxic in vivo, it also loses the ability to delay aggregation in vivo and fibril formation in vitro upon coincubation with αS. The striking difference in fibril formation of αS with βS, versus fibril formation of αS with P123H‐βS suggests that altering the extended conformational propensities of the monomer at the C‐terminus will affect inter‐chain interactions at the dimer level and beyond. We show the delicate balance in the transition from protective to pathogenic forms of βS, suggesting that the conformation of the protein at the C‐terminus may be linked to toxicity and inhibition events.
Methods
Mutagenesis, expression, and purification
P123H‐βS was prepared by site‐directed mutagenesis using AccuPrime pfx from Invitrogen. N‐terminal acetylation of all proteins was performed by coexpression with the NatB plasmid as described previously. Protein purification was performed according to previous protocols.47
NMR experiments
All NMR experiments with the exclusion of the RDC experiments were acquired on a Varian 600 MHz spectrometer at 15°C in pH 6 and 10 mM MES buffer with 100 mM salt. RDC experiments were acquired on a Bruker 700 MHz spectrometer.
NMR assignments
Assignments of P123H‐βS were performed using the protocol described elsewhere.47 NMR assignments of αS48 and βS have been performed previously (manuscript submitted). Experiments were performed on 350 µM 15N and 13C labeled sample with 10%D2O in 10 mM MES buffer pH 6 with 100 mM NaCl. Secondary structure propensities for P123H‐βS were obtained from the SSP program and δ2D30 and SSP for αS and βS were obtained previously.29
RDC experiments
C8E5‐octanol bicelle aligning medium in 100 mM NaCl, 10 mM MES buffer pH 6.49 Reagents: C8E5 and 1‐octanol were purchased from Sigma. The quadrupolar deuterium splitting constants were measured prior to the experiment. The sample was prepared by dissolving lyophilized protein in buffer and passing through 100 kD and 3kD filters. Concentration of the protein was adjusted to 250 µM. Aligning media was added to a final volume of 5%. High resolution HSQC_IPAP spectra in the absence or in the presence of an alignment medium were collected.
Kinetics of fibril formation
Kinetics of fibril formation of αS, βS and P123H‐βS were obtained along with kinetics of fibril formation of coincubation of αS with βS, αS with P123H‐βS and doubling of aS using ThT fluorescence experiments. 5−10 mg of lyophilized acetylated αS, βS, and P123H‐βS was dissolved in PBS, centrifuged for 10 min in 14,000 rpm to remove big oligomers, and purified using size exclusion chromatography (Superdex 75 GL 10/300, from GE Healthcare Life Sciences). Protein was concentrated using 3kDa centrifugal units (Millipore Inc). Final protein concentration was 70 μM with 20 µM ThT for fluorescence measurements. Measurements were recorded at 37°C with linear shaking at 600 rpm. ThT fluorescence was recorded at 30‐min intervals using a POLARstar Omega reader from BMG, as described previously.50 Each condition was repeated 4 times and data is averaged. The experimental set up was used as previously described in the presence of PTFE beads (Taylor Scientific).47
Electrospray ionization mass spectroscopy (ESI‐MS)
ESI‐MS experiments were performed as described previously.51 Samples were prepared in 10 mM Ammonium Acetate, pH 6 in final concentration 50 µM, by using 100 kDa and 3 kDa filters.
Negative straining transmission electron microscopy (TEM)
Samples were incubated for 14 h and after this time aliquots were taken for imaging. Fibrils were visualized using a JEM‐100CXII manufactured by JEOL. Negative staining TEM was performed using the single droplet procedure52 at ambient temperature. Micrographs were recorded at a magnification of 100,000. All of the chemicals were purchased from Sigma.
Size exclusion chromatography
Samples of βS and P123H‐βS were prepared by dissolving 12 mg/mL of protein in PBS buffer, spinning down for 1 h at 14,000 rpm and incubating with orbital shaking for 5 h at 37°C degrees. After that time samples were spun down for 10 min and injected into a Superpose 6 (GE Healthcare) column with a flow of 0.5 mL/min.
PSI‐BLAST analysis
PSI‐BLAST53 analysis was performed to obtain the typical conformations of PP, PS and PH motifs in the PDB. The search was performed across a 10 residue window that contained the PP motif of βS and its flanking sequences, the PS motif of αS and the PH motif of P123H‐βS. Alignment to the average structure, and RMSD was calculated for all structures from the set using VMD visualization program.54 Phi and psi values for all the residues were calculated using AMBER cpptraj analysis tool55 and represented in Ramachandran plots.56
Supporting information
Supporting Information
Acknowledgments
The authors would like to thank Ron Levy for a very productive and stimulating collaboration on α‐synuclein in the early stages of the project. They thank Pawel Janowski for assistance with the bioinformatics analysis, Dr. Valentin Starovoytov for assistance with the TEM experiments and Gina Moriarty for helpful discussions. This work was supported by grant GM110577 from the National Institutes of Health.
References
- 1. Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M (1997) alpha‐synuclein in Lewy bodies. Nature 388:839–840. [DOI] [PubMed] [Google Scholar]
- 2. Goedert M (2001) Parkinson's disease and other alpha‐synucleinopathies. Clin Chem Lab Med 39:308–312. [DOI] [PubMed] [Google Scholar]
- 3. George JM (2002) The synucleins. Genome Biol 3:REVIEWS3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T (1995) The precursor protein of non‐A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron 14:467–475. [DOI] [PubMed] [Google Scholar]
- 5. Nakajo S, Shioda S, Nakai Y, Nakaya K (1994) Localization of phosphoneuroprotein 14 (PNP 14) and its mRNA expression in rat brain determined by immunocytochemistry and in situ hybridization. Brain Res Mol Brain Res 27:81–86. [DOI] [PubMed] [Google Scholar]
- 6. Murphy DD, Rueter SM, Trojanowski JQ, Lee VMY (2000) Synucleins are developmentally expressed, and alpha‐synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci 20:3214–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rivers RC, Kumita JR, Tartaglia GG, Dedmon MM, Pawar A, Vendruscolo M, Dobson CM, Christodoulou J (2008) Molecular determinants of the aggregation behavior of alpha‐ and beta‐synuclein. Protein Sci 17:887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zibaee S, Jakes R, Fraser G, Serpell LC, Crowther RA, Goedert M (2007) Sequence determinants for amyloid fibrillogenesis of human alpha‐synuclein. J Mol Biol 374:454–464. [DOI] [PubMed] [Google Scholar]
- 9. Roodveldt C, Andersson A, De Genst EJ, Labrador‐Garrido A, Buell AK, Dobson CM, Tartaglia GG, Vendruscolo M (2012) A rationally designed six‐residue swap generates comparability in the aggregation behavior of alpha‐synuclein and beta‐synuclein. Biochemistry 51:8771–8778. [DOI] [PubMed] [Google Scholar]
- 10. Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, Goedert M, Fink AL (2002) Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha‐synuclein assembly by beta‐ and gamma‐synucleins. J Biol Chem 277:11970–11978. [DOI] [PubMed] [Google Scholar]
- 11. Fujita M, Sekigawa A, Sekiyama K, Sugama S, Hashimoto M (2009) Neurotoxic conversion of beta‐synuclein: a novel approach to generate a transgenic mouse model of synucleinopathies? J Neurol 256:286–292. [DOI] [PubMed] [Google Scholar]
- 12. Masliah E, Hashimoto M (2002) Development of new treatments for Parkinson's disease in transgenic animal models: A role for beta‐synuclein. Neurotoxicology 23:461–468. [DOI] [PubMed] [Google Scholar]
- 13. Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E (2001) beta‐Synuclein inhibits alpha‐synuclein aggregation: a possible role as an anti‐parkinsonian factor. Neuron 32:213–223. [DOI] [PubMed] [Google Scholar]
- 14. Hashimoto M, Rockenstein E, Mante M, Crews L, Bar‐On P, Gage FH, Marr R, Masliah E (2004) An antiaggregation gene therapy strategy for Lewy body disease utilizing beta‐synuclein lentivirus in a transgenic model. Gene Therapy 11:1713–1723. [DOI] [PubMed] [Google Scholar]
- 15. Fan Y, Limprasert P, Murray IV, Smith AC, Lee VM, Trojanowski JQ, Sopher BL, La Spada AR (2006) Beta‐synuclein modulates alpha‐synuclein neurotoxicity by reducing alpha‐synuclein protein expression. Human Mol Genet 15:3002–3011. [DOI] [PubMed] [Google Scholar]
- 16. Park JY, Lansbury PTJ (2003) Beta‐synuclein inhibits formation of alpha‐synuclein protofibrils: a possible therapeutic strategy against Parkinson's disease. Biochemistry 42:3696–3700. [DOI] [PubMed] [Google Scholar]
- 17. Tsigelny IF, Bar‐On P, Sharikov Y, Crews L, Hashimoto M, Miller MA, Keller SH, Platoshyn O, Yuan JX, Masliah E (2007) Dynamics of alpha‐synuclein aggregation and inhibition of pore‐like oligomer development by beta‐synuclein. Febs J 274:1862–1877. [DOI] [PubMed] [Google Scholar]
- 18. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) alpha‐synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ohtake H, Limprasert P, Fan Y, Onodera O, Kakita A, Takahashi H, Bonner LT, Tsuang DW, Murray IV, Lee VM, Trojanowski JQ, Ishikawa A, Idezuka J, Murata M, Toda T, Bird TD, Leverenz JB, Tsuji S, La Spada AR (2004) Beta‐synuclein gene alterations in dementia with Lewy bodies. Neurology 63:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fujita M, Sugama S, Sekiyama K, Sekigawa A, Tsukui T, Nakai M, Waragai M, Takenouchi T, Takamatsu Y, Wei J, Rockenstein E, Laspada AR, Masliah E, Inoue S, Hashimoto M (2010) A beta‐synuclein mutation linked to dementia produces neurodegeneration when expressed in mouse brain. Nature Commun 1:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wei J, Fujita M, Nakai M, Waragai M, Watabe K, Akatsu H, Rockenstein E, Masliah E, Hashimoto M (2007) Enhanced lysosomal pathology caused by beta‐synuclein mutants linked to dementia with Lewy bodies. J Biol Chem 282:28904–28914. [DOI] [PubMed] [Google Scholar]
- 22. Narayanan C, Weinstock DS, Wu KP, Baum J, Levy RM (2012) Investigation of the polymeric properties of alpha‐synuclein and comparison with NMR experiments: a replica exchange molecular dynamics study. J Chem Theory Comput 8:3929–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferreon ACM, Moosa MM, Gambin Y, Deniz AA (2012) Counteracting chemical chaperone effects on the single‐molecule alpha‐synuclein structural landscape. Proc Natl Acad Sci USA 109:17826–17831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park SM, Jung HY, Kim TD, Park JH, Yang CH, Kim J (2002) Distinct roles of the N‐terminal‐binding domain and the C‐terminal‐solubilizing domain of alpha‐synuclein, a molecular chaperone. J Biol Chem 277:28512–28520. [DOI] [PubMed] [Google Scholar]
- 25. Ulmer TS, Bax A, Cole NB, Nussbaum RL (2005) Structure and dynamics of micelle‐bound human alpha‐synuclein. J Biol Chem 280:9595–9603. [DOI] [PubMed] [Google Scholar]
- 26. Sung YH, Eliezer D (2006) Secondary structure and dynamics of micelle bound beta‐ and gamma‐synuclein. Protein Sci 15:1162–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hashimoto M, La Spada AR (2012) beta‐synuclein in the pathogenesis of Parkinson's disease and related alpha‐synucleinopathies:emerging roles and new directions. Future Neurol 7:155–163. [Google Scholar]
- 28. Bertoncini CW, Rasia RM, Lamberto GR, Binolfi A, Zweckstetter M, Griesinger C, Fernandez CO (2007) Structural characterization of the intrinsically unfolded protein beta‐synuclein, a natural negative regulator of alpha‐synuclein aggregation. J Mol Biol 372:708–722. [DOI] [PubMed] [Google Scholar]
- 29. Marsh JA, Singh VK, Jia ZC, Forman‐Kay JD (2006) Sensitivity of secondary structure propensities to sequence differences between alpha‐ and gamma‐synuclein: implications for fibrillation. Protein Sci 15:2795–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Camilloni C, De Simone A, Vranken WF, Vendruscolo M (2012) Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry 51:2224–2231. [DOI] [PubMed] [Google Scholar]
- 31. Bernado P, Bertoncini CW, Griesinger C, Zweckstetter M, Blackledge M (2005) Defining long‐range order and local disorder in native alpha‐synuclein using residual dipolar couplings. J Am Chem Soc 127:17968–17969. [DOI] [PubMed] [Google Scholar]
- 32. Cho MK, Kim HY, Bernado P, Fernandez CO, Blackledge M, Zweckstetter M (2007) Amino acid bulkiness defines the local conformations and dynamics of natively unfolded alpha‐synuclein and tau. J Am Chem Soc 129:3032+. [DOI] [PubMed] [Google Scholar]
- 33. Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, Zweckstetter M (2005) Release of long‐range tertiary interactions potentiates aggregation of natively unstructured alpha‐synuclein. Proc Natl Acad Sci USA 102:1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McClendon S, Rospigliosi CC, Eliezer D (2009) Charge neutralization and collapse of the C‐terminal tail of alpha‐synuclein at low pH. Protein Sci 18:1531–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu KP, Weinstock DS, Narayanan C, Levy RM, Baum J (2009) Structural reorganization of alpha‐synuclein at low pH observed by NMR and REMD simulations. J Mol Biol 391:784–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Uversky VN, Li J, Fink AL (2001) Evidence for a partially folded intermediate in alpha‐synuclein fibril formation. J Biol Chem 276:10737–10744. [DOI] [PubMed] [Google Scholar]
- 37. Munishkina LA, Fink AL, Uversky VN (2009) Accelerated fibrillation of alpha‐synuclein induced by the combined action of macromolecular crowding and factors inducing partial golding. Curr Alzheimer Res 6:252–260. [DOI] [PubMed] [Google Scholar]
- 38. Fernandez CO, Hoyer W, Zweckstetter M, Jares‐Erijman EA, Subramaniam V, Griesinger C, Jovin TM (2004) NMR of alpha‐synuclein‐polyamine complexes elucidates the mechanism and kinetics of induced aggregation. Embo J 23:2039–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Uversky VN, Li J, Fink AL (2001) Metal‐triggered structural transformations, aggregation, and fibrillation of human alpha‐synuclein—a possible molecular link between Parkinson's disease and heavy metal exposure. J Biol Chem 276:44284–44296. [DOI] [PubMed] [Google Scholar]
- 40. Meuvis J, Gerard M, Desender L, Baekelandt V, Engelborghs Y (2010) The conformation and the aggregation kinetics of alpha‐synuclein depend on the proline residues in its C‐terminal region. Biochemistry 49:9345–9352. [DOI] [PubMed] [Google Scholar]
- 41. Hoyer W, Cherny D, Subramaniam V, Jovin TM (2004) Impact of the acidic C‐terminal region comprising amino acids 109‐140 on alpha‐synuclein aggregation in vitro. Biochemistry 43:16233–16242. [DOI] [PubMed] [Google Scholar]
- 42. McLean PJ, Hyman BT (2002) An alternatively spliced form of rodent alpha‐synuclein forms intracellular inclusions in vitro: role of the carboxy‐terminus in alpha‐synuclein aggregation. Neurosci Lett 323:219–223. [DOI] [PubMed] [Google Scholar]
- 43. Izawa Y, Tateno H, Kameda H, Hirakawa K, Hato K, Yagi H, Hongo K, Mizobata T, Kawata Y (2012) Role of C‐terminal negative charges and tyrosine residues in fibril formation of alpha‐synuclein. Brain Behav 2:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ulrih NP, Barry CH, Fink AL (2008) Impact of Tyr to Ala mutations on alpha‐synuclein fibrillation and structural properties. Biochim Biophys Acta 1782:581–585. [DOI] [PubMed] [Google Scholar]
- 45. Norris EH, Giasson BI, Hodara R, Xu SH, Trojanowski JQ, Ischiropoulos H, Lee VMY (2005) Reversible inhibition of alpha‐synuclein fibrillization by dopaminochrome‐mediated conformational alterations. J Biol Chem 280:21212–21219. [DOI] [PubMed] [Google Scholar]
- 46. Lorenzen N, Nielsen SB, Yoshimura Y, Vad BS, Andersen CB, Betzer C, Kaspersen JD, Christiansen G, Pedersen JS, Jensen PH, Mulder FAA, Otzen DE (2014) How epigallocatechin gallate can inhibit alpha‐synuclein oligomer toxicity in vitro. J Biol Chem 289:21299–21310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang LJ, Wu KP, Vendruscolo M, Baum J (2011) The A53T mutation is key in defining the differences in the aggregation kinetics of human and mouse alpha‐synuclein. J Am Chem Soc 133:13465–13470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kang LJ, Moriarty GM, Woods LA, Ashcroft AE, Radford SE, Baum J (2012) N‐terminal acetylation of alpha‐synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci 21:911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ruckert M, Otting G (2000) Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J Am Chem Soc 122:7793–7797. [Google Scholar]
- 50. Kang L, Janowska MK, Moriarty GM, Baum J (2013) Mechanistic insight into the relationship between N‐terminal acetylation of alpha‐synuclein and fibril formation rates by NMR and fluorescence. Plos One 8:e75018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moriarty GM, Minetti CA, Remeta DP, Baum J (2014) A revised picture of the Cu(II)‐alpha‐synuclein complex: the role of N‐terminal acetylation. Biochemistry 53:2815–2817. [DOI] [PubMed] [Google Scholar]
- 52. Horne RW, Cockayne DJH (1991) Negative staining. Micron Microscop Acta 22:319‐319 [Google Scholar]
- 53. Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38, 27‐38. [DOI] [PubMed] [Google Scholar]
- 55. Case DA, Berryman JT, Betz RM, Cerutti DS, Cheatham ITE, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AH, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Luchko T, Luo R, Madej B, Merz KM, Monard G, Needham P, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Salomon‐Ferrer R, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, YD M., Kollman PM (2015) AMBER 2015. University of California, San Francisco. [Google Scholar]
- 56. Lovell SC, Davis IW, Adrendall WB, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC (2003) Structure validation by C alpha geometry: phi,psi and C beta deviation. Proteins 50:437–450. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information