

Abstract
The development of a modified DNA aptamer that binds HIV‐1 reverse transcriptase (RT) with ultra‐high affinity has enabled the X‐ray structure determination of an HIV‐1 RT‐DNA complex to 2.3 Å resolution without the need for an antibody Fab fragment or RT‐DNA cross‐linking. The 38‐mer hairpin‐DNA aptamer has a 15 base‐pair duplex, a three‐deoxythymidine hairpin loop, and a five‐nucleotide 5′‐overhang. The aptamer binds RT in a template‐primer configuration with the 3′‐end positioned at the polymerase active site and has 2′‐O‐methyl modifications at the second and fourth duplex template nucleotides that interact with the p66 fingers and palm subdomains. This structure represents the highest resolution RT‐nucleic acid structure to date. The RT‐aptamer complex is catalytically active and can serve as a platform for studying fundamental RT mechanisms and for development of anti‐HIV inhibitors through fragment screening and other approaches. Additionally, the structure allows for a detailed look at a unique aptamer design and provides the molecular basis for its remarkably high affinity for RT.
Keywords: reverse transcriptase, HIV, DNA aptamer, 2′‐O‐methylcytidine, p66/p51, SELEX
Abbreviations
- dsDNA
double‐stranded DNA
- HIV
human immunodeficiency virus
- RT
reverse transcriptase
- SELEX
selective evolution of ligands by exponential enrichment.
Introduction
HIV‐1 reverse transcriptase (RT) is an essential enzyme in the retroviral life cycle. RT converts the single‐stranded viral RNA genome into double‐stranded DNA via its RNA‐ and DNA‐dependent DNA polymerase and RNase H activities.1, 2, 3 The multifunctional nature of RT requires that it bind both RNA/DNA and DNA/DNA (dsDNA) substrates.
The first structure of an RT/dsDNA complex was determined in the presence of an antibody Fab fragment.4 This “binary” structure defined the nucleic acid binding track extending from the polymerase to the RNase H active sites and also defined structural elements involved in template‐primer binding, including the “primer grip” and “template grip.” A cross‐linking strategy using disulfide tethering was later used to generate a new RT/DNA crystal form that could accommodate an incoming dNTP in an RT/DNA/dNTP ternary precatalytic complex.5 Subsequently, both cross‐linking and Fab were used to study the structural basis of RNA/DNA binding and RNA degradation, the resilience of the polypurine tract (PPT) to degradation, the catalytic incorporation of nucleotides and nucleoside analog drugs (NRTIs), and the molecular mechanisms of resistance to NRTIs by different RT mutations (for reviews see Refs. 3, 6). The cross‐linking and Fab approaches require specialized reagents and additional purification steps that can hamper efficient application of currently available RT/nucleic acid complexes to structure‐based drug design efforts.
Structure‐based drug design with RT has been successful in the development of two non‐nucleoside RT drugs: etravirine/Intelence and rilpivirine/Edurant.6, 7 A protein engineering effort8 from our group generated robust crystals of RT that diffract to high resolution and enabled a crystallographic fragment‐screening campaign to find new allosteric inhibitory binding sites in RT.9 Analogous drug design and fragment screening with RT/DNA complexes will require highly reproducible and well diffracting crystals.
DNA aptamers with tight binding affinities for their target proteins that contain modified nucleotides have been developed by exploiting the higher structural diversity of these modifications.10 While aptamers typically have complex three‐dimensional structures with novel base‐pairing interactions, they can also be created with simpler topologies and still retain the signature characteristics of aptamers: the ability to bind proteins with a high degree of specificity and affinity. Previous selective evolution of ligands by exponential enrichment (SELEX) studies have demonstrated that aptamers can inhibit HIV replication through specific high affinity interactions with RT.11, 12, 13, 14, 15 Application of a modified SELEX approach has led to the development of a nucleic acid template–primer aptamer that binds RT with significantly higher affinity than a typical unselected template–primer substrate.16 These newly designed aptamers have helped explain RT's higher affinity to RNA/DNA vs. dsDNA templates by showing that even a single RNA nucleotide in the template strand near the polymerase active site can significantly increase binding affinity. The use of the more chemically stable 2′‐O‐methylation to mimic the RNA 2′‐hydroxyl retained higher binding affinity.16
The structure of a tight‐binding 38‐mer DNA aptamer complex with RT has been solved to 2.3 Å resolution, the highest resolution for an RT/nucleic acid complex so far. This crystal form should facilitate efficient fragment screening and structure‐based drug design regimes for HIV‐1 RT complexed with nucleic acid and may enable the development of novel inhibitors that bind only in the presence of nucleic acid. In addition, the structure and associated technological platform could enhance our understanding of fundamental RT polymerization mechanisms.
Results
Construction and biochemical analysis of the 38‐mer DNA aptamer (with 2′‐O‐methyl modifications at the second and fourth duplex template positions)
The aptamer used for crystallization was originally derived from selection for tight binding to HIV‐1 RT of nucleic acid duplexes in a template‐primer configuration, using a modified SELEX (Selective Evolution of Ligands by Exponential Enrichment) approach.17 The selected duplexes resembled the polypurine tract (PPT) sequence of HIV in that they had an extended run of deoxyguanosines at the 3′ end. These duplexes were further modified to produce “38NT SELEX,” a 38 nucleotide single‐stranded loop‐back aptamer with seven 3′ deoxyguanosines and a five base 5′ overhang.18 Aptamer 38NT2,4‐methyl was made from 38NT SELEX by replacing the deoxycytosine residues that were hybridized to the deoxyguanosines in the −2 and −4 (relative to the 3′ terminus) positions with 2′‐O‐methylcytosine. This modification was based on previous results showing that replacing nucleotides at these positions in the template strand of DNA–DNA template–primers with either rNTPs or 2′‐O‐methyl modified nucleotides resulted in high affinity binding with RT.16 Consistent with this, 38NT SELEX bound to HIV‐1 RT with a K D of 163 ± 24 pM and the complex has a half‐life of 47 ± 4 min. For comparison, unselected dsDNA with the same configuration bind to RT with 100 nM affinity.18 Further, 38NT2,4‐methyl DNA showed an ∼10‐fold increase in binding affinity over 38NT SELEX with a K D of 13.7 ± 1.8 pM and the half‐life of 126 ± 54 min (Supporting Information Table I).
Structure of RT‐aptamer complex
The RT‐aptamer structure was solved and refined to 2.3 Å resolution. This binary complex consists of a p66/p51 heterodimer bound to the 38‐mer DNA aptamer; the protein architecture is dominated by a large nucleic acid binding track running along the length of the heterodimer (Fig. 1). The complex crystallized in a condition and crystal form similar to those obtained earlier for RT/dsDNA and RT/RNA/DNA crosslinked complexes (Table 1).23, 24 The crystal asymmetric unit contains two copies of RT/aptamer complex. Electron density is not seen for residues beyond 544 of p66 in one heterodimer and 553 in the second copy. The heterodimers in the asymmetric unit superimpose with an RMSD of 1.34 Å for protein Cα atoms and 0.55 Å for nucleic acid atoms. The greatest variation between the two copies in the asymmetric unit occurs among RT residues 29–45 of the fingers and the RNase H domains.
Figure 1.
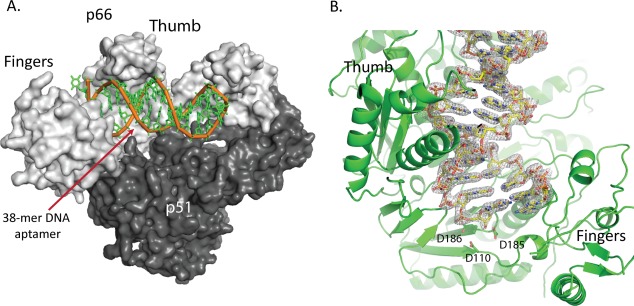
Overview of HIV‐1 RT/aptamer complex. A. HIV‐1 RT p66/p51 heterodimer (gray) shown with DNA aptamer colored in green. The first duplex DNA base pair is located at the polymerase active site and the aptamer hairpin residues are located near the RNase H active site. B. Representative electron density for the aptamer shown centered on the polymerase active site region.
Table 1.
Data Processing and Refinement Statistics Calculated Using HKL200019 and PHENIX,20, 21, 22 Respectively
| PDB ID | 5D3G |
| Wavelength (Å) | 0.9179 |
| Resolution range (Å) | 30.00–2.30 (2.34–2.30) |
| Space group | P2 1 |
| Unit cell (Å and °) | 90.02 128.91 132.62 90.0 101.3 90.0 |
| Total reflections | 886,233 (97,902) |
| Unique reflections | 133,446 (6,567) |
| Redundancy | 6.6 (5.5) |
| Completeness (%) | 99.3 (98.5) |
| Mean I/sigma (I) | 17.1 (2.0) |
| Wilson B‐factor (Å2) | 43.5 |
| R merge | 0.09 (0.76) |
| R meas | 0.11 (0.97) |
| R pim | 0.04 (0.39) |
| R work | 0.191 (0.321) |
| R free | 0.225 (0.322) |
| Number of atoms | |
| All | 17,868 |
| Protein | 15,857 |
| DNA | 1,442 |
| Glycerol/sucrose/sulfate | 122 |
| Water | 447 |
| RMS bonds (Å) | 0.004 |
| RMS angles (°) | 0.920 |
| Ramachandran favored (%) | 97.8 |
| Ramachandran allowed (%) | 2.2 |
| Ramachandran outliers (%) | 0 |
| Clashscore | 1.83 |
| Average B‐factors (Å2) | |
| All atoms | 73.9 |
| Protein | 73.8 |
| DNA | 81.7 |
| Glycerol/sucrose/sulfate | 72.0 |
| Water | 54.7 |
Data for highest resolution range shell are shown in parentheses.
The aptamer has clear electron density for 35 of the 38 nucleotides [Supporting Information Fig. 3(A,B)]; three single‐stranded nucleotides at the 5′‐end are disordered. Numbering of the nucleotides in the aptamer (from the 5′ end) begins with −4 to 0 for the template overhang, 1–15 for the template duplex strand, 16–18 for the hairpin loop, and 19–33 for the primer duplex strand [Fig. 2(A)]. All of the 30 complementary residues in the duplex region (Supporting Information Fig. 1) form Watson–Crick base pairs. The second and fourth template nucleotides of the aptamer, referenced from the first base‐paired nucleotide residues at the primer terminus, contain the 2′‐O‐methyl modifications, which are well resolved in the electron density map [Supporting Information Fig. 3(C,D)] and are positioned near the base of the fingers and palm subdomains. Conformational analysis of the aptamer using the program x3DNA25 indicates that the first five base pairs of the duplex adopt an A‐like conformation and the remaining nucleotide base pairs conform to B‐form geometry [Fig. 2(A)], which is consistent with the conformation of normal dsDNA template–primers bound to RT.4
Figure 2.
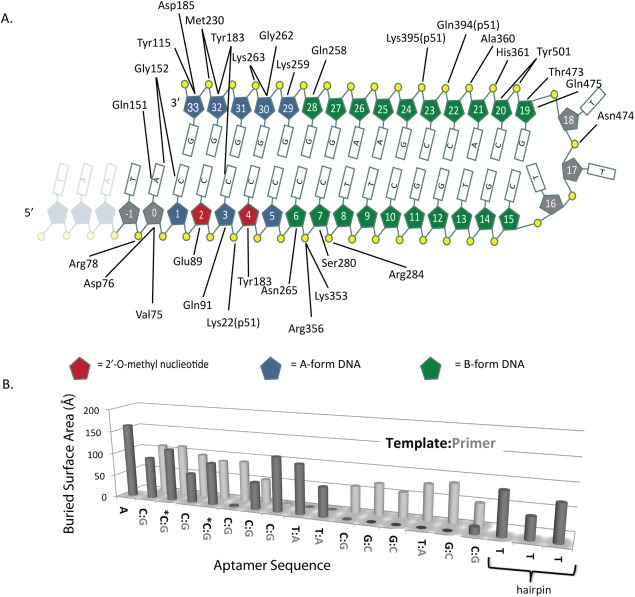
Aptamer Interactions. A. Diagram showing the protein‐aptamer interactions that are within hydrogen‐bonding distance. The first three residues are shown in gray to indicate that they are disordered in the structure. The 2′‐O‐methyl modified nucleotides are indicated in red. B. Buried surface area of each nucleotide of the aptamer shown as a bar graph.
Three deoxythymidines (nucleotides 16–18) form the hairpin loop and are bound near the RNase H primer grip.26 The nucleotide bases of the three deoxythymidines are ordered in the structure and form non‐Watson–Crick hydrogen bonds to the DNA [Fig. 5(A,B)].
Figure 5.
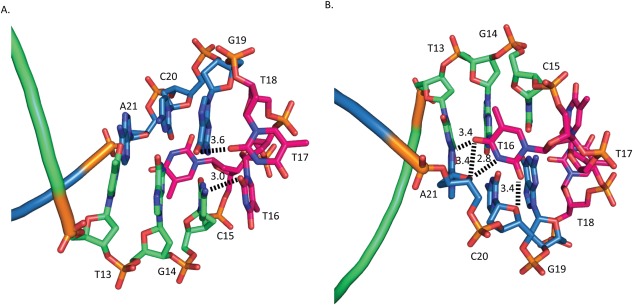
Aptamer hydrogen‐bonding interactions. Template strand is shown in blue, primer strand is shown in green, and hairpin‐loop residues in red. A. View of the aptamer looking towards the RNase H active site with bonding network created by T16, T17, and G19. B. 180° Rotation of A, showing interactions of T16.
The B‐factors of the crosslinked dsDNA (3V6D)23 range from 47 to 179. The B‐factors of the aptamer range from 43 to 178 (Supporting Information Fig. 2) with the lowest B‐factors near the polymerase and RNase H active sites, reflecting that the DNA aptamer is most ordered in the regions with the most extensive contacts with RT. The aptamer structure B‐factors were refined using a translation/libration/screw motion determination (TLSMD) model, which allows for anisotropic B‐factor refinement at lower than atomic resolution. As a result of this refinement the isotropic B‐factors increase, which explains the apparent similarity in B‐factor range between the two structures.
DNA conformation and protein–DNA interactions
The network of interactions between RT and the aptamer is shown in Figure 2(A), and the interacting surface area for individual nucleotides is shown in Figure 2(B). The distances are listed in Supporting Information Table I. With the exception of the electrostatic interactions between Gly152 to C1 and Tyr183 to C3, all of the interactions are between the protein and sugar‐phosphate backbone of the aptamer. There is one water‐mediated interaction between Glu478 and T17. There are 30 residues of p66 that interact with aptamer nucleotides through side‐chain contacts. They are clustered in three groups: dsDNA interacting with the polymerase domain (nucleotides 0–8 and 28–33) where the terminus of the aptamer interacts with key amino acid residues in the p66 fingers, palm, and thumb subdomains; the hairpin loop (nucleotides 16–18); and the remaining dsDNA near the RNase H domain (nucleotides 18–23). There is only one direct protein–DNA interaction with the three deoxythymidine residues that make up the hairpin loop, a hydrogen bond between Asn474 and the phosphate of T18 [Fig. 4(C)]. The solvent‐accessible surface area for each nucleotide buried in the context of RT is shown in Figure 2(B).
Figure 4.
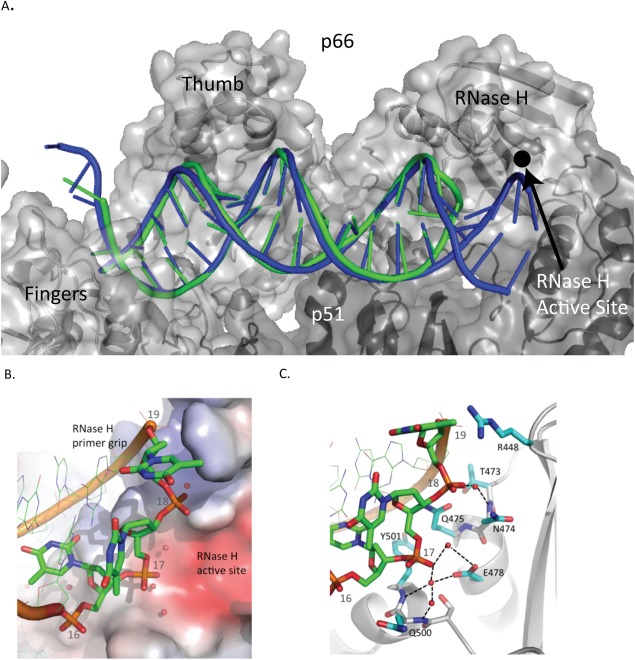
Comparison of the RT/aptamer structure (green) with the RT/cross‐linked‐dsDNA structure (3V6D) (blue). A. Nucleic acid binding cleft with the polymerase active site at the left and RNase H active site on the right. B. Aptamer hairpin (green) and cross‐linked‐dsDNA template strand (blue). Cross‐linked‐dsDNA template strand shown extending into RNase H active site. C. Aptamer binding pocket shown with selected side chains that are within 3.8 Å of nucleic acid atoms. A water molecule, shown as a red sphere, forms hydrogen bonds with Glu478 and the phosphate of T17.
Interactions of 2′‐O‐methyl groups
The 2′‐O‐methylcytidine residues at positions two and four in the duplex template strand of the aptamer experience different electrostatic environments [Fig. 3(A)]. The first 2′‐O‐methyl sits in a negatively charged environment dominated by Glu89 and Gln161 as well as the more distant Met184. The 2′‐O‐methyl at position four, however, occupies a rather hydrophobic pocket with no side chains or main‐chain carbonyl atoms within hydrogen‐bonding distance of the 2′‐O‐methyl oxygen. The nearest side chain to the 2′‐O‐methyl at position four is that of the hydrophobic residue Ile94. Separating the two 2′‐O‐methyl modified nucleotides is a loop in RT comprised of residues 91–95 of p66 with Glu91 situated beneath the third aptamer nucleotide. In addition, there is a hydrogen bond between the third aptamer residue (C3) and the phenoxyl oxygen of Tyr183.
Figure 3.
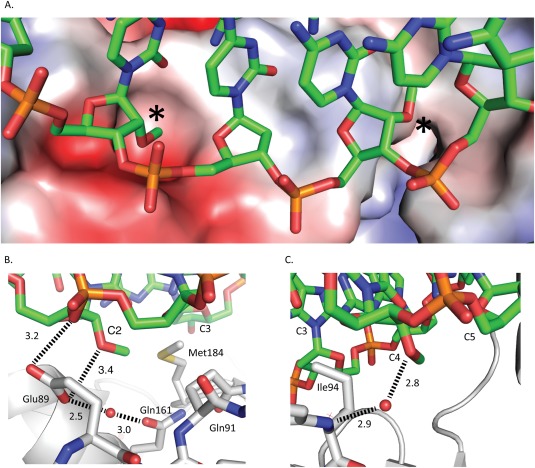
Interactions between aptamer and RT. A. RT molecular surface colored by electrostatic potential with the aptamer (colored by atom type) overlaid. The asterisks denote the locations of 2′‐O‐methyls. B. Residue two 2′‐O‐methyl shown with short electrostatic contact with Glu89. C. Residue four 2′‐O‐methyl shown in neutral pocket with water mediated interaction to Ile94.
The oxygen of the 2′‐O‐methyl in the second duplex template position forms an electrostatic interaction with RT via the side chain of Glu89 with a distance of 3.1 Å [Fig. 3(B)]; the carboxylate of the Glu89 side chain also interacts with water that forms a bridging interaction to the amide oxygen of the Gln161 side chain. The 2′‐O‐methyl modification in the fourth position lacks direct protein interactions [Fig. 3(C)], but there is a water molecule bridging from the 2′‐O‐methyl oxygen to the peptide backbone nitrogen of Ile94.
Structural comparison of RT‐aptamer to RT cross‐linked to dsDNA
The overall fold of RT in the RT/aptamer complex is similar to structures of RT bound to cross‐linked dsDNA (RMSD of 0.54 Å to 3V6D). The path of the crosslinked dsDNA through the RT binding cleft is similar to that of the aptamer [Fig. 4(A)] and shows similar interactions (Supporting Information Table I). The aptamer DNA and crosslinked dsDNA structures have similar conformations near the polymerase active site but diverge toward the RNase H active site [Fig. 4(A,B)]. A comparison of the averaged DNA base‐pairing parameters of the aptamer to the crosslinked DNA shows that they are similar by most metrics (Supporting Information Table III). Comparing the active site residues of the aptamer and crosslinked structures shows that the catalytic residues D110, D185, and D186 are superimposable [Supporting Information Fig. 4(A)]. In the region of the crosslinked structure where the protein is linked via a disulfide tethering to the DNA (at residue Q258C to the sixth base of the primer strand) there is no significant displacement [Supporting Information Fig. 4(B)]. While the minor groove distances are nearly identical to those of the crosslinked structure near the polymerase active site, one consequence of the hairpin is a compression of the minor groove (by >2 Å for the 10th and 11th duplex positions) near the RNase H domain (Supporting Information Fig. 5). The minor groove is ∼1 Å narrower in the aptamer DNA vs. crosslinked DNA in the vicinity of the p66 thumb subdomain.
Location and interactions of the hairpin with the RNase H domain
The polymerase and RNase H active sites of RT are separated by 18 duplex base pairs.4 The 15 base‐pair aptamer duplex spans the polymerase active site to within 9 Å of the RNase H active site [Fig. 4(A)]. Unlike conventional dsDNA template‐primers, the aptamer loops back to form the template strand [Fig. 4(C)]. The 3‐dT hairpin loop (nucleotides 16–18) interacts primarily with the RNase H primer grip. The aptamer hairpin loop makes a unique set of interactions with RT that likely facilitates the tight binding measured between RT and the aptamer. There are hydrogen bonds between the protein and the aptamer from Gln475 to the ribose of G19, Asn474 to the phosphate of T19, Tyr501 to the phosphate of A21 and the O2 of C20, and Thr473 to the phosphate of G19 [Fig. 4(C)]. There is also a water molecule bridging Glu478 to the phosphate of T17. The aptamer makes a series of intramolecular interactions, which presumably would increase the stability in a manner that is consistent with what has been determined by biophysical analysis of DNA.27, 28 Adjacent to the aptamer hairpin G19 makes Watson–Crick hydrogen bonds to C15. C15 is involved in a base‐stacking interaction with T17 of the hairpin loop [Fig. 5(A)]. Residue T16 is flipped internally to the loop and makes hydrogen bonds with the base and ribose moieties of C20 and A21 [Fig. 5(B)].
Discussion
Rationale for aptamer affinity
The direct visualization of a high‐affinity aptamer bound to RT illustrates that the key features of the aptamer are the 2′‐O‐methyl base modifications at positions two and four and the inclusion of a short hairpin loop composed of a triplet of deoxythymidine residues. In addition, the structure also serves to further validate the findings from structures of RT crosslinked to DNA, since there appears to be very little distortion of the DNA backbone as a result of the covalent tether linking RT to DNA. The aptamer with 2′‐O‐methyl modifications contains two features that may explain the 10‐fold increase in affinity as compared to the aptamer without 2′‐O‐methyl modifications. One feature is an interaction between Glu89 and the first 2′‐O‐methyl group on the second duplex template nucleotide that likely recapitulates a similar interaction with an RNA 2′‐OH. Another feature is that the second 2′‐O‐methyl group (at duplex template position four) does not make protein–nucleic acid contacts, but does occupy a pocket [Fig. 3(A)] and has a water‐mediated interaction between the 2′‐O‐methyl oxygen and the peptide backbone nitrogen of Ile94 [Fig. 3(C)].
The role of the hairpin in stabilizing the aptamer is twofold. There is a series of hydrogen bonds between the DNA backbone and RT, but these are comparable to interactions seen in the crosslinked dsDNA structure5 as well as in non‐crosslinked dsDNA and RNA/DNA structures (Supporting Information Table II).4, 26 Additional enhancement of the aptamer stability is improved by the interactions of the hairpin with RT. Previous work has shown that hairpin loops that are small and stabilized by charged ions, or in this case by charged side chains, had an increase in their T m as the number of nucleotides in the hairpin loops decreased.27 The maximum stability, as indicated by T m, was found when DNA loops were composed of deoxythymidine residues with lengths of three to five bases. This is consistent with the composition of the aptamer in this study.28
The SELEX experiments revealed that a string of seven deoxycytidine residues in the template strand led to the highest binding affinity,18 presumably by forming a highly stable G:C duplex. The role of the purine‐rich region of the aptamer may contribute to its high affinity by preferentially adopting an A‐like conformation. The conformation of dsDNA at the RT polymerase active site is A‐like and transitions to B‐form toward the RNase H site in all RT/dsDNA structures. Duplex DNA with a more random base distribution may favor B‐form in solution and adopt an A‐like conformation once bound to RT. Dickerson et al. have shown that G‐C‐rich stretches of six or more residues adopt an A‐like DNA conformation.29 We propose that the G‐C‐rich region of the aptamer is preformed as A‐like and represents a ground‐state form of the RT‐bound DNA. Polypurine DNA may have biological relevance since the HIV genome contains stretches of polypurine nucleotides that play a critical role in reverse transcription.26, 30
Finally, this structure offers a new framework for studying RT‐DNA complexes at higher resolution than previously available and may enable systematic approaches such as crystallographic fragment screening for identification of new small molecule classes as potential inhibitors or chemical probes of RT function.
Materials and Methods
Aptamer design and selection: End‐labeling of 38NT2,4‐methyl aptamer with T4 polynucleotide kinase (PNK)
Aptamers (synthesized without 5′ phosphate) were 5′ 32P end‐labeled in a 50‐µL volume containing 10 pmole of aptamer, 1X T4 polynucleotide kinase reaction buffer (New England Biolabs), 10 U of T4 PNK enzyme, and 10 µL of (γ‐32P) ATP (3000 Ci mmol−1, 10 µCi µL−1). The labeling reaction was done at 37°C for 30 min according to manufacturer's protocol. PNK enzyme was heat‐inactivated by incubating the reaction at 75°C for 15 min. Excess radiolabeled nucleotides were removed by centrifugation using a Sephadex G‐25 column (Harvard Apparatus).
Dissociation rate constant (K D) and half‐life (t 1/2) determinations using nitrocellulose filter binding assay
Reactions for K D determinations were performed in 1 mL of buffer containing (final concentrations): 2 pM 5′ end‐labeled aptamer, 50 mM Tris‐HCl pH 8, 80 mM KCl, 1 mM DTT, 2 mM MgCl2, and 0.1 µg mL−1 BSA. Increasing amounts of HIV‐1 RT (prepared as described in Hou et al.31) diluted in the above reaction buffer were added at the following final concentrations for analysis of 38NT2, 4‐methyl DNA: 0, 1 pM, 2 pM, 4 pM, 8 pM, 16 pM, 32 pM, 64 pM, 128 pM, 256 pM, and 10 nM; and for 38NT SELEX: 0, 12.5 pM, 25 pM, 50 pM, 100 pM, 150 pM, 200 pM, 250 pM, 400 pM, 800 pM, 1.6 nM, and 10 nM. After 10 min at room temperature, the reactions were applied to a 25‐mm nitrocellulose disk (0.45‐µm pore, Protran BA 85, Whatman™) presoaked in filter wash buffer (25 mM Tris‐HCl pH 7.5, 10 mM KCl) under vacuum. The filter was washed three times with 1 mL of wash buffer at a flow rate of ∼0.5 mL sec−1. Filters were then counted in a scintillation counter. The concentration of bound aptamer was determined for each RT concentration using the 10 nM saturating RT level as a reference for the maximum amount of material that could be bound in the assay. A plot of bound aptamer vs. RT concentration was fit to the following equation using KaleidaGraph in order to determine the K D: [ED] = 0.5([E]t + [D]t + K D) – 0.5(([E]t + [D]t + K D)2 – 4[E]t[D]t)1/2, where [E]t is the total enzyme concentration and [D]t is the total primer–template concentration.32 Half‐life determinations were performed in the same buffer with 5 nM (final concentration) of 5′ end‐labeled aptamer and 5 nM of HIV RT in 98 µL. The reaction was incubated at room temperature for 5 min. Two microliters of nonradioactive aptamer (1 µM final concentration) was added to the reaction at time “0.” This was used as a “trap” to sequester RT molecules as they dissociated from the labeled aptamer. Aliquots of 10 µL were vacuum filtered as above at times 0 min, 15 min, 30 min, 60 min, 90 min, 120 min, 180 min, and 240 min. The nitrocellulose filter disks were washed and processed as described above. Off‐rates (k off) were determined by plotting the retained counts vs. time, and fitting the data to an equation for single, two parameter exponential decay using SigmaPlot. Half‐life values were calculated from k off using the equation t 1/2= 0.693/k off. Experiments were repeated at least three times and averages ± standard deviations are reported.
RT expression and purification
HIV‐1 RT plasmid RT152A allows for the co‐expression of p66Δ555 and p51Δ428. The RT152A construct produces protein mutations in p66:C280S,D498N; and in p51: C280S. The protein was cloned, expressed, and purified following the previously published protocol.8 The 38‐nucleotide DNA aptamer, 38NT2,4‐methyl, (Supporting Information Fig. 1) with 2′‐O‐methyl at the second and fourth duplex positions of the template strand, was custom synthesized by Integrated DNA Technologies and Midland Certified Reagent Company. The complex of RT and aptamer was generated by mixing RT at a concentration of 85 µM (10 mg mL−1) with a 1.2‐fold excess of 38NT2,4‐methyl. Gel filtration analysis indicated a single peak corresponding to a 1:1 RT/aptamer complex in addition to a small amount of unbound DNA (data not shown).
Crystallization
The RT/38NT2,4‐methyl DNA complex was subjected to crystallization condition screening using Hampton Research Index Screen HT and Crystal Screen HT, and Rigaku Wizard Classic Screens I and II. Drops with a final volume of 0.4 µL were setup in a 1:1 ratio of well solution to protein complex solution in 96 well Intelli‐Plates using an Art Robbins Instruments Gryphon Crystallization Robot. The plates were incubated at 4°C and were monitored for crystal growth. The final crystals used for diffraction experiments were crystallized using the hanging drop method utilizing conditions previously identified.24 The well solution was composed of 7–10% (w/v) PEG 8000, 25 mM Bis‐tris Propane pH 6.8, 75 mM Bis‐tris Propane pH 7.4, 50 mM ammonium sulfate, 5% (v/v) glycerol, and 5% (w/v) sucrose. A 1:1 ratio of protein‐to‐well solution was suspended in a 2 µL drop over a 1‐mL well. Plates were incubated for 7–14 days at 4°C.
Crystals were prepared for flash cooling in liquid nitrogen by transferring them step‐wise into solutions containing the crystallization buffers supplemented with increasing amounts of glycerol until the final concentration was 25% (v/v).
Data collection, processing, and structure determination
Data were collected on flash‐cooled crystals at 100 K on the F1 beamline at the Cornell High Energy Synchrotron Source. A 360° dataset was collected on an ADSC Q270 at a wavelength of 0.9179 Å with 1° oscillations. The data were integrated and scaled using HKL2000.19 Phasing was performed by molecular replacement using phenix.phaser20, 21 with the PDB entry 3DLK and refinement was completed with phenix.refine. 21, 22, 33 There were two complexes in the asymmetric unit, therefore non‐crystallographic symmetry (NCS) restraints were used between the two copies of p66, p51, and aptamer. A combination of NCS restraints and TLS refinement produced a final model with significantly improved agreement indices and stereochemical quality vs. other strategies of refinement. Inclusion of TLS parameterization led to a lowering of R free from 0.24 to 0.225, but also led to a substantial increase of the average underlying isotropic B factors (74 Å2 vs. 69 Å2). Manual rebuilding was performed using Coot.34 Data and refinement statistics are shown in Table 1. Figures were generated using PyMol.35 Structure analysis was performed using x3DNA,25 PyMol,35 cealign,36 and NCONT.37 The coordinates were deposited in the PDB under accession number 5D3G.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
The authors contribute this article in honor of their friend and colleague Professor Ron Levy on the occasion of his 65th birthday. They acknowledge the staff at the Cornell High Energy Synchrotron Source (CHESS), and also Samantha Wadsworth, Mena Issa, and Sergio Martinez for assistance and helpful discussions.
References
- 1. Coffin JM, Hughes SH, Varmus HE, editors (1997) Retroviruses. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. [PubMed] [Google Scholar]
- 2. Hu WS, Hughes SH (2012) HIV‐1 reverse transcription. Cold Spring Harb Perspect Med 2:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA, Hughes SH, Arnold E (2009) Structure and function of HIV‐1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J Mol Biol 385:693–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jacobo‐Molina A, Ding J, Nanni RG, Clark AD Jr, Lu X, Tantillo C, Williams RL, Kamer G, Ferris AL, Clark P, Hizi A, Hughes SH, Arnold E (1993) Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double‐stranded DNA at 3.0 Å resolution shows bent DNA. Proc Natl Acad Sci USA 90:6320–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang H, Chopra R, Verdine GL, Harrison SC (1998) Structure of a covalently trapped catalytic complex of HIV‐1 reverse transcriptase: implications for drug resistance. Science 282:1669–1675. [DOI] [PubMed] [Google Scholar]
- 6. Das K, Clark AD Jr, Lewi PJ, Heeres J, de Jonge MR, Koymans LMH, Vinkers HM, Daeyaert F, Ludovici DW, Kukla MJ, De Corte B, Kavash RW, Ho CY, Ye H, Lichtenstein MA, Andries K, Pauwels R, de Béthune M, Boyer PL, Clark P, Hughes SH, Janssen PAJ, Arnold E (2004) Roles of conformational and positional adaptability in structure‐based design of TMC125‐R165335 (Etravirine) and related non‐nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild‐type and drug‐resistant HIV‐1 variants. J Med Chem 47:2550–2560. [DOI] [PubMed] [Google Scholar]
- 7. Janssen PA, Lewi PJ, Arnold E, Daeyaert F, de Jonge M, Heeres J, Koymans L, Vinkers M, Guillemont J, Pasquier E, Kukla M, Ludovici D, Andries K, de Bethune MP, Pauwels R, Das K, Clark AD Jr, Frenkel YV, Hughes SH, Medaer B, De Knaep F, Bohets H, De Clerck F, Lampo A, Williams P, Stoffels P (2005) In search of a novel anti‐HIV drug: multidisciplinary coordination in the discovery of 4‐[[4‐[[4‐[(1E)−2‐cyanoethenyl]‐2,6‐dimethylphenyl]amino]‐2‐ pyrimidinyl]amino]benzonitrile (R278474, rilpivirine). J Med Chem 48:1901–1909. [DOI] [PubMed] [Google Scholar]
- 8. Bauman JD, Das K, Ho WC, Baweja M, Himmel DM, Clark AD, Jr , Oren DA, Boyer PL, Hughes SH, Shatkin AJ, Arnold E (2008) Crystal engineering of HIV‐1 reverse transcriptase for structure‐based drug design. Nucleic Acids Res 36:5083–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bauman JD, Patel D, Dharia C, Fromer MW, Ahmed S, Frenkel Y, Vijayan RS, Eck JT, Ho WC, Das K, Shatkin AJ, Arnold E (2013) Detecting allosteric sites of HIV‐1 reverse transcriptase by X‐ray crystallographic fragment screening. J Med Chem 56:2738–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davies DR, Gelinas AD, Zhang C, Rohloff JC, Carter JD, O'Connell D, Waugh SM, Wolk SK, Mayfield WS, Burgin AB, Edwards TE, Stewart LJ, Gold L, Janjic N, Jarvis TC (2012) Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc Natl Acad Sci USA 109:19971–19976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Joshi P, Prasad VR (2002) Potent inhibition of human immunodeficiency virus type 1 replication by template analog reverse transcriptase inhibitors derived by SELEX (systematic evolution of ligands by exponential enrichment). J Virol 76:6545–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Whatley AS, Ditzler MA, Lange MJ, Biondi E, Sawyer AW, Chang JL, Franken JD, Burke DH (2013) Potent inhibition of HIV‐1 reverse transcriptase and replication by Nonpseudoknot, “UCAA‐motif” RNA aptamers. Mol Ther Nucleic Acids 2:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Joshi PJ, North TW, Prasad VR (2005) Aptamers directed to HIV‐1 reverse transcriptase display greater efficacy over small hairpin RNAs targeted to viral RNA in blocking HIV‐1 replication. Mol Ther 11:677–686. [DOI] [PubMed] [Google Scholar]
- 14. Ditzler MA, Bose D, Shkriabai N, Marchand B, Sarafianos SG, Kvaratskhelia M, Burke DH (2011) Broad‐spectrum aptamer inhibitors of HIV reverse transcriptase closely mimic natural substrates. Nucleic Acids Res 39:8237–8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kissel JD, Held DM, Hardy RW, Burke DH (2007) Active site binding and sequence requirements for inhibition of HIV‐1 reverse transcriptase by the RT1 family of single‐stranded DNA aptamers. Nucleic Acids Res 35:5039–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Olimpo JT, DeStefano JJ (2010) Duplex structural differences and not 2′‐hydroxyls explain the more stable binding of HIV‐reverse transcriptase to RNA‐DNA versus DNA‐DNA. Nucleic Acids Res 38:4426–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. DeStefano JJ, Cristofaro JV (2006) Selection of primer‐template sequences that bind human immunodeficiency virus reverse transcriptase with high affinity. Nucleic Acids Res 34:130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DeStefano JJ, Nair GR (2008) Novel aptamer inhibitors of human immunodeficiency virus reverse transcriptase. Oligonucleotides 18:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Otwinowski Z, Minor W (2001) DENZO and SCALEPACK. Boston: Kluwer Academic Publishers, pp 226–235. [Google Scholar]
- 20. Terwilliger TC, Dimaio F, Read RJ, Baker D, Bunkoczi G, Adams PD, Grosse‐Kunstleve RW, Afonine PV, Echols N (2012) phenix.mr_rosetta: molecular replacement and model rebuilding with Phenix and Rosetta. J Struct Funct Genom 13:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Cryst D 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Echols N, Headd JJ, Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner RD, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2011) The Phenix software for automated determination of macromolecular structures. Methods 55:94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Das K, Martinez SE, Bauman JD, Arnold E (2012) HIV‐1 reverse transcriptase complex with DNA and nevirapine reveals non‐nucleoside inhibition mechanism. Nat Struct Mol Biol 19:253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Das K, Martinez SE, Bandwar RP, Arnold E (2014) Structures of HIV‐1 RT‐RNA/DNA ternary complexes with dATP and nevirapine reveal conformational flexibility of RNA/DNA: insights into requirements for RNase H cleavage. Nucleic Acids Res 42:8125–8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng G, Lu XJ, Olson WK (2009) Web 3DNA—a web server for the analysis, reconstruction, and visualization of three‐dimensional nucleic‐acid structures. Nucleic Acids Res 37:W240–W246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sarafianos SG, Das K, Tantillo C, Clark AD Jr, Ding J, Whitcomb JM, Boyer PL, Hughes SH, Arnold E (2001) Crystal structure of HIV‐1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J 20:1449–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kuznetsov SV, Ren CC, Woodson SA, Ansari A (2008) Loop dependence of the stability and dynamics of nucleic acid hairpins. Nucleic Acids Res 36:1098–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuznetsov SV, Shen Y, Benight AS, Ansari A (2001) A semiflexible polymer model applied to loop formation in DNA hairpins. Biophys J 81:2864–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ng HL, Kopka ML, Dickerson RE (2000) The structure of a stable intermediate in the A B DNA helix transition. Proc Natl Acad Sci USA 97:2035–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rausch JW, Le Grice SF (2004) ‘Binding, bending and bonding’: polypurine tract‐primed initiation of plus‐strand DNA synthesis in human immunodeficiency virus. Int J Biochem Cell Biol 36:1752–1766. [DOI] [PubMed] [Google Scholar]
- 31. Hou EW, Prasad R, Beard WA, Wilson SH (2004) High‐level expression and purification of untagged and histidine‐tagged HIV‐1 reverse transcriptase. Protein Expr Purif 34:75–86. [DOI] [PubMed] [Google Scholar]
- 32. Hsieh JC, Zinnen S, Modrich P (1993) Kinetic mechanism of the DNA‐dependent DNA polymerase activity of human immunodeficiency virus reverse transcriptase. J Biol Chem 268:24607–24613. [PubMed] [Google Scholar]
- 33. Afonine PV, Grosse‐Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst D68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 35. DeLano WL (2002) The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific. [Google Scholar]
- 36. Shindyalov IN, Bourne PE (1998) Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng 11:739–747. [DOI] [PubMed] [Google Scholar]
- 37. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Cryst D 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information