

Abstract
This short article examines the usefulness of fast simulations of conformational transition paths in elucidating enzymatic mechanisms and guiding drug discovery for protein kinases. It applies the transition path method in the MOIL software package to simulate the paths of conformational transitions between six pairs of structures from the Protein Data Bank. The structures along the transition paths were found to resemble experimental structures that mimic transient structures believed to form during enzymatic catalysis or conformational transitions, or structures that have drug candidates bound. These findings suggest that such simulations could provide quick initial insights into the enzymatic mechanisms or pathways of conformational transitions of proteins kinases, or could provide structures useful for aiding structure‐based drug design.
Keywords: conformational transition paths, protein kinases, structures for ensemble docking, catalytic mechanisms of protein kinases
Introduction
In recent years, scientists have introduced many simulation techniques to identify reaction or conformational transition paths (e.g., Refs. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17). These techniques have helped to detect transient intermediate structures that are difficult to observe experimentally. Finding these short‐lived species could provide useful insights into enzymatic mechanisms or the crucial conformational changes that need to occur in order for biological molecules such as enzymes, molecular motors, or ion channels to function. Finding less populated structures can also prove useful in drug discovery as many drug candidates are known to bind to these structures. This short article focuses on examining whether an approximate but fast method can already yield useful preliminary insights into these problems, as rapid methods are essential for some applications such as practical drug discovery in which researchers call for results in days to inform new experiments.
Identifying the pathways of conformational transitions by simulations is easier when both the starting and the ending structures are known or specified at the outset. This idea was introduced in the 80′ by Pratt18 and Elber and Karplus19 with various similar approaches introduced since then.
This article focuses on estimating minimum‐energy paths using an inexpensive method developed by Ron Elber's group as it can be applied to a large number of systems more easily. The first publication in this line of work19 described a conformational transition path by a sequence of intermediate structures between two end states, such as two structures observed experimentally. By optimizing a function containing the sum of the energy of the intermediate structures and a term designed to maintain roughly equal distances between any two adjacent structures along the path, one can obtain a relatively smooth path to describe a possible pathway of transitions between the two end states. This approach often provides useful quick first estimates of conformational transitional paths that could be difficult to obtain by brute‐force molecular simulations. Elber's group has continued to refine this method and their MOIL software package provides access to this useful tool.20, 21, 22, 23
In general, the quality of a conformational transition path obtained by this method depends on the initial guess, as energy minimization usually cannot take a path too far from the initial estimate. Nevertheless, simple linear interpolation between two end structures could provide good initial estimates for some systems. Such linearly interpolated paths are also often used in graphics programs to morph one structure to another to help users appreciate the large conformational change that could occur between two stable or meta‐stable states of a protein.24, 25 The work presented in this article examined whether simply refining a linearly interpolated path by optimizing the sum of the energies of the intermediate structures subjected to the constrains of maintaining roughly equal separation distances between any adjacent structures could provide a transition path sufficiently realistic to answer some useful questions. For example, could a path constructed between the reactant and the product conformations of a protein kinase encompass structures similar to the experimental ones prepared to mimic transient states during enzymatic catalysis? If so, paths obtained this way could supplement experimental studies to provide even richer insights into the individual steps that could occur during enzymatic catalysis. And for systems for which no experimental study to probe any transient state has been carried out yet, such simulations could suggest some possibilities and propose suitable experiments to test them.
Conformational transition paths obtained from such simulations could also be useful for drug discovery. In structure‐based drug discovery, it is now well recognized that docking compounds to a single conformation of a receptor could produce many false negatives. Scientists have introduced various methods to alleviate this problem. Ensemble docking represents one such method in which a ligand is docked to multiple structures of its biomolecular receptor rather than only one.26, 27, 28 Methods such as the relaxed‐complex scheme29, 30 carry out molecular dynamics simulations on a protein receptor to generate additional structures for molecular docking. However, it takes time to run long molecular dynamics simulations. With short molecular dynamics simulations, many potentially useful structures for drug binding would still be missed. Although various enhanced sampling methods have also been introduced, (e.g., Ref. 2, 31, 32, 33, 34, 35, 36, 37, 38) they are still expensive to perform for routine use in practical drug discovery, which often calls for results within days to inform new experiments.
Another strategy takes advantage of the large number of experimental structures that have been determined for some protein families such as protein kinases. For example, Wong and Bairy28 proposed to use a large number of experimental structures that have been determined for protein kinases to build structural ensembles to account for protein flexibility in molecular docking. Many structures have been determined for some protein kinases with different ligands bound or with different modifications introduced. The variations of these structures reflect the conformational flexibility of these proteins and their use in ensemble docking could reduce the number of false negatives. For protein kinases that do not have as many experimentally structures determined yet, Wong and Bairy proposed to perform comparative modeling to use the structures from other protein kinases as well to enlarge the conformational ensemble of a protein kinase that can be built for use in molecular docking. They have also developed an automatic pipeline to ease the construction of such a structural ensemble to facilitate its update when new experimental structures become available. This short article further asks whether the structures along conformational transition paths between experimental structures could provide additional structures useful for docking. If possible, this approach will be particularly useful for proteins that do not have many experimental structures determined yet. To test this idea, this work generated conformational transition paths between pairs of experimental structures for several relatively well‐studied protein kinases, and used the intermediate structures along the paths to perform 3‐dimensional structure similarity search of the Protein Data Bank (PDB)39 to examine whether such structures could bind drug candidates. If so, this gives some confidences that higher‐energy structures generated along conformational transition paths could be found in nature and therefore provide another way to supply useful conformations of a protein receptor for ensemble docking.
Results
Six simulations showing subtle differences of conformational transition paths in protein kinases
This work chose five protein kinases and simulated six conformational transition paths. Figure 1 uses the standard deviation of pairwise distances between α carbons along the paths, calculated by chimera,40 to illustrate the substructures that underwent the most changes along the paths of transitions.
Figure 1.
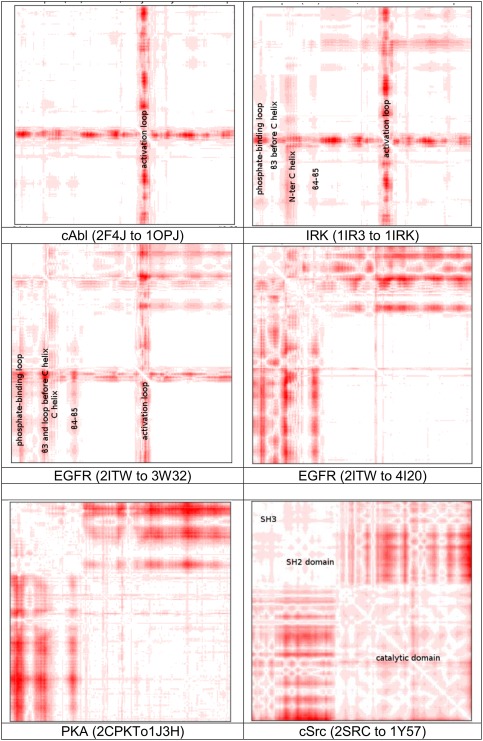
Standard deviations of distances between α carbons along six conformational transition paths for five protein kinases. Substructures were labeled in some of the figures but not in the others to allow the patterns to be seen more clearly. Redder regions carry higher standard deviations.
The simulation of cAbl (Abelson tyrosine kinase) covered the conformational transition between a structure that binds the drug Gleevec (PDB entry 1OPJ)41 and one that does not (PDB entry 2F4J42). 1OPJ is characterized by an inactive structure in which the well‐known DFG motif at the beginning of the activation loop adopts what scientists refer to as the DFG‐out conformation. 2F4J, on the other hand, represents an active structure adopting the DFG‐in conformation that cannot bind Gleevec. The DFG‐in and DFG‐out conformations flip against each other by about 180°. In spite of this large local structural change, the overall conformational change along this path was dominated only by the movement of the activation loop relative to the rest of the protein, as seen by the single thin and long band of largest standard deviations spanning along the horizontal or the vertical axis. Note that the plot is symmetric about the diagonal running from the upper left corner to the lower right corner. On the x‐axis, the N‐terminus runs towards the C‐terminus from left to right. On the y‐axis, the N‐terminus runs towards the C‐terminus from top to bottom. The thin dimension of the band on the x‐ or the y‐axis corresponds to a small segment of the protein, the activation loop in this case. The long dimension of the band from top to bottom or from left to right reflects that the whole catalytic domain of the protein experienced large relative motion with respect to the activation loop.
On the other hand, the transition between an active (PDB entry 1IR343) and an inactive (PDB entry 1IRK44) structure of the insulin receptor tyrosine kinase (IRK) gave a different picture. Although the thin and long band corresponding to the movement of the activation loop was still apparent, additional bands resulting from large movement of several regions in the N‐terminal lobe also appeared. These regions include the phosphate‐binding loop, the region just before the N‐terminus of the C helix, β3, and the β4‐loop‐β5 region, as labeled in the figure. All these regions displayed large movement with respect to the C‐terminal lobe, as seen by several thin bands spanning the length of the C‐terminal lobe. Within the N‐terminal lobe, the phosphate‐binding loop, β3, the β4‐loop‐β5 region, and the N‐terminal portion of the C helix moved somewhat as rigid bodies relative to each other, reflected by several off‐diagonal short and thin bands within the N‐terminal lobe in the upper left corner of the plot.
For the epidermal growth factor receptor (EGFR), two transition paths from the DFG‐in active conformation (PDB entry 1ITW45) to the DFG‐out inactive conformation (PDB entry 4I2046) and to the src‐like inactive conformation (PDB entry 3W3247) were simulated. Transition to the src‐like structure (2ITW to 3W32) was characterized by the large movement of the activation loop relative to the N‐terminal lobe, as in the previous two paths. Within the N‐terminal lobe, the N‐terminal region of the C helix exhibited large movement relative to the rest of the lobe, reflected by the white little square on the left upper corner (smaller movement) and the relatively red regions (larger movement) in the remaining portions of the N‐terminal lobe. On the other hand, in the transition to the DFG‐out inactive conformation (2ITW to 4I20), a smaller region of the activation loop in the C‐terminal lobe experienced large movement relative to the rest of the protein, in comparison to the above three transition paths, as reflected by the less pronounced thin long bands. Within the N‐terminal lobe, the structural change spread out more evenly throughout the lobe during the transition, as seen by the comparatively uniform red square appearing in the leftmost upper corner representing the N‐terminal lobe.
For the transition between a closed form of protein kinase A (PKA, PDB entry, 2CPK48) and an open form of PKA (PDB entry 1J3H49), the large relative movement between the N‐ and C‐terminal lobes dominated. The structural change within each lobe was significantly less noticeable in comparison. This is characterized in Figure 1 by the two relatively white squares in the upper left corner (smaller relative movement within the N‐terminal lobe) and in the lower right corner (smaller relative movement within the C‐terminal lobe) in comparison to the two relatively red off‐diagonal rectangles (large relative movement between the N‐terminal and the C‐terminal lobes).
The simulation of the cSrc kinase differed from the other five simulations in that it included a larger system containing the SH3 and the SH2 domains in addition to the catalytic domain. For the transition between the inactive (PDB entry 2SRC50) and an active conformation (PDB entry 1Y5751) of the protein, the large movement of the catalytic domain relative to the SH3 and the SH2 domains was most prominent. The smaller relative movement between the SH3 and the SH2 domains was also apparent. (The three squares corresponding to the three domains are labeled in Figure 1 along the diagonal). In addition, the structural change within the catalytic domain showed a somewhat different behavior from those observed for the other protein kinases: it spread out more evenly throughout the whole domain both in the N‐terminal and C‐terminal lobes, reflected by the relatively uniform red color scattering over the largest square occupying the lower right corner that covered the catalytic domain.
Conformational transition between the closed and the open forms of protein kinase A
Table 1 shows the crystal structures mined from the Protein Data Bank using several structures along the conformational transition path. PDBeFold52, 53, 54, 55, 56, 57 provided the search engine in this work. All the retrieved structures carried Q scores better than 0.8 with 1.0 the best possible score for the matching between a query and a retrieved structure. Because structure 1, derived from the crystal structure of the closed form (2CPK) retrieved many good hits, only the structures with the top twenty Q scores, all better than 0.8, are shown in the table. The second column of the table shows from which structure(s) on the path the PDB structure was retrieved.
Table 1.
Structures, in the Protein Data Bank, which are Similar to One or More Structures Along the Conformational Transition Path Between the Closed Form (2CPK) and the Open Form (1J3H) of Protein Kinase A
| PDB codes | Structures identified froma | Description of structure |
|---|---|---|
| 2CPK | 1, 11 | Closed conformation of PKA |
| 2ERZ | 1, 11 | Complexed with hydroxyfasudil |
| 3MVJ | 1 | Akt inhibitor with the 3‐aminopyrrolidine scaffold |
| 3FHI | 1, 11 | Complexed with RIα regulatory subunit |
| 3AGL | 1, 11 | Complexed with the bisubstrate inhibitor ARC‐1039 |
| 2GNL | 1, 11 | 3‐mutation surrogate model of Rho kinase with H1152P inhibitor |
| 3E8E | 1, 11 | Complexed with Akt inhibitor GSK690693 |
| 1REJ | 1, 11 | Complexed with balanol analog 1 |
| 4O22 | 1, 11 | Binary complex of metal‐free PKA with substrate peptide SP20 |
| 3E8C | 1, 11 | Complexed with ATP‐competitive inhibitor: Akt inhibitor containing an imidazopyridine aminofurazan scaffold |
| 1XH8 | 1, 11 | Complexed with a protein kinase B selective inhibitor |
| 3PVB | 1 | Structure of the catalytic unit together with the 73‐244 fragment of the RIα regulatory subunit |
| 4WB6 | 1 | L205R mutant of human protein kinase A |
| 4DIN | 1 | Complexed with RIβ regulatory subunit |
| 3MVJ | 1, 11 | Complexed with an Akt inhibitor |
| 1BKX | 1, 11 | Complexed with adenosine |
| 1REK | 11 | Complexed with balanol analog 8 |
| 1STC | 11 | Complexed with staurosporine |
| 2L9L | 21 | Complexed with Akt inhibitor N(1)‐(5‐(heterocyclyl)‐thiazol‐2‐yl)−3‐(4‐trifluoromethylphenyl)−1,2‐propanediamine |
| 1SZM | 21 | Complexed with PKC inhibitor bisindolyl maleimide 2 |
| 3I9M | 21 | Triple mutant V123A, I173M, Q181K with Akt inhibitor N(1)‐(5‐(heterocyclyl)‐thiazol‐2‐yl)−3‐(4‐trifluoromethylphenyl)−1,2‐propanediamine |
| 4NTT | 21, 31 | Complexed with ADP and magnesium ion |
| 3AGM | 21 | Complexed with the bisubstrate protein kinase inhibitor ARC‐670 |
| 4C35 | 21 | PKA‐S6K1 chimera with compound 1 (NU1085) bound |
| 4NTS | 31, 41, 51 | Apo structure of the catalytic subunit of PKA |
The conformational transition path spanned between structure 1 (derived from 2CPK) and structure 51 (derived from 1J3H).
Table 1 shows that the closed form of protein kinase A retrieved many more similar structures than the open form, as there have not been as many structures determined near the open form yet. The structures retrieved include those complexed with inhibitors, those with natural substrates such as ATP or peptide mimics bound, those with the regulatory subunit associated, and those with ligands intended to mimic the intermediate states in the enzymatic mechanism. Several structures: 1REK,58 1STC,59 3L9L,60 1SZM,61 3L9M,60 4NTT,62 3AGM,63 and 4C3564 were only retrieved by the intermediate structures along the conformational transition path, not by the two end structures, the closed and the open forms derived from the experimental structures, indicating that the intermediate structures were quite different from the structures of the two end states.
Conformational transition between an active and an inactive form of the insulin receptor tyrosine kinase
Table 2 shows similar results for the insulin receptor tyrosine kinase. Not as many structures with Q score at least 0.80 were retrieved in comparison to protein kinase A that has been studied structurally for a longer time. More structures were retrieved with the active form (structure 1 derived from the PDB entry 1IR3 and structure 11 along the path) than with the inactive one (structure 51). The structures retrieved included structures with inhibitors bound, with ATP and/or peptide substrate bound, with other proteins such as the protein tyrosine phosphatase PTP1P bound, or with a mutant that mimicked a phosphorylated form of the protein.
Table 2.
Structures, in the Protein Data Bank, which are Similar to One or More Structures Along the Conformational Transition Path Between the Active Form (1IR3) and an Inactive Form (1IRK) of the Insulin Receptor Tyrosine Kinase
| PDB codes | Structures identified froma | Description of structure |
|---|---|---|
| 1IR3 | 1, 11 | Active structure with peptide substrate and ATP analog bound |
| 1GAG | 1, 11 | Complexed with a bi‐substrate inhibitor |
| 1RQQ | 1, 11 | Complexed with the SH2 domain of the adaptor protein APS |
| 3BU5 | 1, 11, 21 | Complexed with IRS2 KRLB peptide and ATP |
| 3BU3 | 1, 11, 21 | Complexed with IRS2 KRLB peptide |
| 2AUH | 1 | Complexed with the BPS region of GRB14, a member of the Grb7 adaptor protein family |
| 3BU6 | 1, 11, 21 | Complexed with IRS2 KRLB phosphopeptide |
| 2B4S | 1 | Complexed with the tyrosine phosphatase PTP1B |
| 2Z8C | 1 | Complexed with (4‐[[5‐carbamoyl‐4‐(3‐methylanilino)pyrimidin‐2‐yl]amino]phenyl)acetic acid |
| 1K3A | 11 | Structure of the insulin‐like growth factor 1 receptor kinase with an ATP analog and a peptide substrate bound |
| 1I44 | 21 | An activation loop mutant of the insulin receptor tyrosine kinase |
| 3EKK | 21, 41, 51 | Complexed with 4,6‐bis‐anilino‐1H‐pyrrolo[2,3‐d]pyrimidine |
| 3EKN | 21 | Complexed with 4,6‐bis‐anilino‐1H‐pyrrolo[2,3‐d]pyrimidine |
| 3QQU | 51 | Complexed with 2,4‐bis‐arylamino‐1,3‐pyrimidines |
The conformational transition path spanned between structure 1 (derived from PDB entry 1IR3) and structure 51 (derived from PDB entry 1IRK).
Conformational transition between an DFG‐in and an DFG‐out structure of cAbl
Although both the DFG‐in structure (PDB entry 2F4J) and the DFG‐out structure (PDB entry 1OPJ) identified many hits (>20) from the Protein Data Bank, with Q score at least 0.80, the intermediate structures identified only a few more additional hits. Structure 11 identified 11 structures in the Protein Data Bank with Q‐score 0.80, but only two were not identified by using structure 1, derived from 2F4J, as query. One structure, with PDB code 2HZI,65 represented a crystal structure between the protein and an inhibitor PD180970 related to the development of drugs to treat chronic myelogenous leukemia. The other structure, with PDB code 2G2F,66 mimics a src‐like inactive conformation lying between the DFG‐in and the DFG‐out structures.
Conformational transition between a DFG‐in structure (PDB entry 2ITW) and a DFG‐out inactive structure (4I20) or a src‐like inactive structure (PDB entries 3W32) of EGFR
Although the structure 2ITW identified a dozen similar structures from the Protein Data Bank, 3W32 did not identified any, and 4I20 identified only one with Q score at least 0.80. 4I20 identified 4I1Z46 as a similar structure, which represents the structure of the protein with the mutation of the gate‐keeper residue: T790M. No intermediate structures along the two paths identified structures with a Q score at least 0.80. However, structure 21 for the path between 2ITW and 4I20 identified the structure 3BCE67 with a Q score of 0.73, not far from 0.80. This represents the structure of the ERBB4 kinase, in the same family as EGFR, with the drug lapatinib bound. Structure 21 along the path between 2ITW and 3W32 identified the structure 3IKA68 with a Q‐score of 0.72. This represents the crystal structure between EGFR and the covalent inhibitor WZ4002. On the other hand, structure 31 identified the structure 2RFD69 with Q‐score = 0.72 and this represents a structure with the MIG6 peptide bound.
Conformational transition between an inactive conformation (PDB entry 2SRC) and an active conformation (PDB entry 1Y57) of cSrc
In this case, PDBeFold identified 4 structures with Q‐score at least 0.80 using structure 1 derived directly from 2SRC. It identified only itself when using the open structure 51, derived directly from 1Y57. The intermediate structures did not fish out any structure with Q‐score at least 0.80. In fact, the most similar structure gave a Q‐score of only 0.29. Removing the SH3 and SH2 domains to using only the catalytic domain in the search did not produce many more hits. Although the catalytic domain of structure 51 identified more than 20 structures with Q score better than 0.80, structure 1 retrieved only one structure (3U4W70) with Q score at least 0.80. However, structure 1 did retrieve 15 structures with Q‐score better than 0.70. Again, the intermediate structures did not identify structures with good Q‐scores. The best structure gave a Q‐score of only 0.41. This may result from the scarcity of experimental structures containing the SH3 and the SH2 domains in addition to the catalytic domain. The SH3 and SH2 domains might distort the structure of the catalytic domain significantly, especially in the more compact inactive form. This is consistent with the analysis of distance variations above in which the structural change associated with the transition between the active and inactive forms of this system spread out more extensively throughout the catalytic domain than the other systems that did not contain the SH3 and SH2 domains.
Discussion
Intermediate structures along conformational transition paths resembled known experimental structures, giving confidence that the computational approach employed here could reflect reality
As the intermediate structures along the conformational transition paths of all the protein kinases studied here, except cSrc with the SH3 and the SH2 domains included, found similar structures in the Protein Data Bank with a Q‐score of at least 0.72, it gave some confidence that the intermediate structures generated from the path simulations could exist in nature. Considering that it is difficult to observe transient intermediates experimentally and has to rely on various artificial means to trap mimics of intermediate states, fast simulations of conformational transition paths provides a useful complement to experimental techniques to hint on the conformational changes that could result from different perturbations of a protein kinase, such as by phosphorylation or de‐phosphorylation, by adding or removing ligands (e.g., inhibitors, ATP, ADP, peptide substrates, mimics of transition states, and phosphorylation products), or by mutations of the protein.
Quick simulation of paths of transitions could capture intermediates of enzymatic mechanisms or of conformational transitions in protein kinase systems
This is most evident from the study on protein kinase A for which many crystal structures have already been determined.
Typical steps in the enzymatic mechanism of a protein kinase involve the binding of ATP and a peptide or protein substrate to the catalytic domain of the protein kinase. The γ phosphate of ATP then transfers to a serine, threonine, or tyrosine residue of the substrate, followed by the release of the phosphorylated substrate and the ADP, before another cycle of catalysis occurs. Large or subtle conformational changes accompany the intermediate steps in moving from the reactants to the products. Because experimental techniques could only capture the transient intermediate states approximately and indirectly, atomistic molecular simulation could fill in additional details, especially by suitably leveraging experimental structures that have already been determined.
Structures resembling the phosphate‐transfer step were found in this study of protein kinase A. For example, 3AGL63 and 3AGM depict the structures of two compounds, ARC‐1039 and ARC‐670, which are adducts of a peptide with an ATP analog. 3AGM was mined only by using the intermediate structure 21, not by the two end states, showing the extra information that a path simulation could provide beyond existing experimental structures. In addition, intermediate structures 21 and 31 resembled the structure 4NTT (PDB code) that was intended to mimic the ADP bound form after the phosphorylated peptide or protein had dissociated from the protein kinase.
In the case of the transition between the active form of the insulin receptor tyrosine kinase and an inactive one, several conjugates between peptide and ATP analog were also found to have similar structures to the intermediate structures along the conformational transition path in this study: 1GAG,71 3BU5,72 and 3BU6.72 Intermediate structure 21 resembled the structure of a mutated protein intended to relieve the autoinhibition form of the protein, PDB entry 1I44,73 which could mimic an intermediate state during enzymatic catalysis.
For the transition between 2F4J (DFG‐in) and 1OPJ (DFG‐out) in cAbl, intermediate structure 11 identified 2G2F, an inactive conformation that Levinson et al.66 suggested to be an intermediate structure mediating the transformation from the DFG‐in conformation to the DFG‐out conformation. This suggests that the simulated path had captured key essences of the actual structural transition process.
These results show that the crude but quick simulations of transition paths performed here were capable of capturing structures that were relevant to the study of the enzymatic mechanisms or conformational transitions of these enzymes. These structures could provide useful starting points for performing even more sophisticated but more expensive simulations such as more realistic molecular dynamics simulations or quantum mechanics/molecular mechanics simulations to gain further insights into enzymatic mechanisms or conformational transitions. Without simulations such as the ones employed in this study or other enhanced sampling techniques, these higher energy states could be difficult to obtain by brute‐force simulations. For example, in studying the mechanism of phosphate transfer in the insulin receptor tyrosine kinase, Zhou and Wong74 needed to use the crystal structures of mimics of reaction intermediates from other protein kinases to build a structural model for the insulin receptor tyrosine kinase that was capable of phosphate transfer in a quantum mechanics/molecular mechanics calculation.
Structures from simulation of paths of conformational transitions are relevant for drug discovery
The fact that the three‐dimensional structure search on the Protein Data Bank with the intermediate structures along the conformational transition paths of most of the protein kinases studied here found inhibitor‐bound structures suggests that the conformations generated by quick simulations of transition paths could be useful for structure‐based drug design or virtual screening. Because these simulations can provide higher‐energy structures easier than conventional molecular dynamics simulations can and that the constraint from the two end states derived from experimental structures could reduce the possibility of approximate force fields to generate unrealistic structures, transition‐path calculations could provide a powerful tool to complement other computational techniques to generate ensemble of structures quickly to account for protein flexibility in ensemble docking or to construct dynamic pharmacophore models.75, 76, 77, 78, 79
Methods for refining paths from the chmin module in MOIL
The chmin method in MOIL is inexpensive to use and provides useful preliminary insights quickly. If needed, one can improve the paths obtained from chmin with other newer methods or to even redevelop the paths from scratch if the initial study from MOIL proves that it is worthwhile.
For applications in which single minimum‐energy paths can already provide useful insights, one can use newer methods that could provide paths closer to the minimum‐energy paths. The nudged elastic band (NEB) method80 provides one example. This method can provide smoother paths. It resembles chmin in using a sequence of connected images to create a path between two end states but differs in the methods employed in optimizing the path. During the optimization, only the interatomic forces for each image that are perpendicular to the path and the component of the constrained forces parallel to the path are included. Variations that allow the saddle point to be determined even more precisely have also been introduced.4 The string method was also introduced later as another way to obtain smooth minimum‐energy paths efficiently.81
The minimum‐energy path only reflects the most probable path at 0 K but not for finite temperatures. Methods for identifying the most probable path at a finite temperature have also been introduced. For example, Elber and Holloway used a Feynman path integral approach to find the most probable stochastic path described by Brownian dynamics at a finite temperature.82 Wong introduced a similar approach but used dynamic programming83 to find the most probable path.84 Huo and Straub,85 on the other hand, identified the most probable path at a finite temperature by maximizing the flux assuming a diffusive process of transition.
Transition‐path sampling goes beyond obtaining a single most probable path to sampling a number of significant paths.86 Once a model is chosen for calculating the probability of transition of a path, one can perform Monte Carlo simulations to evolve a starting guess path to an ensemble of significant paths for analysis. Stochastic dynamics defined the probability of each path for the first methods introduced but deterministic dynamics was presented later as well.87
It is also worthwhile to point out that although this paper focuses on finding pathways of structural transitions for qualitative analysis, methods have also been developed to provide quantitative estimates of transition rates, including some of the methods just described.
Summary
This work tested whether quick simulations of transition paths between experimental structures using the chmin module in the MOIL software package was reasonably realistic to help elucidate the enzymatic mechanisms of protein kinases, to help decipher the conformational transition paths between different populated states of these proteins, and to provide structures to aid structure‐based drug discovery. It achieved this by applying this technique to several relatively well‐studied protein kinases that have quite a few structures already determined experimentally. It expected that if the structures along the paths were reasonably realistic, similar structures would have already been determined and deposited into the Protein Data Bank for these relatively well‐studied systems. This study found this to be indeed the case for all the protein kinases studied here except for the larger system that also contained the SH2 and SH3 domains in addition to the catalytic domain for which fewer experimental structures have been determined. Experimental structures mimicking reaction intermediates were found, so were those mediating conformational transitions. In addition, many structures with inhibitor bound were mined from the Protein Data Bank by using the intermediate structures along the simulated paths, suggesting that simulation of path transitions could provide useful structures, not as easily obtained by conventional molecular simulations, for structure‐based drug design and for virtual screening.
Methods
Simulation of transition paths
The transition path method developed in the Elber laboratory was used in this work.19, 20, 21, 22, 88 The chmin module of the MOIL package23 performs such simulations. Given two structures, chmin introduces a succession of intermediate structures between them, generated by interpolation, for example, and then optimize the path by minimizing the function:
| (1) |
where gives the potential energy of structure i, and the constraint function C maintained roughly equal distance between any adjacent structures along a path:
| (2) |
where
| (3) |
Default values of η, ρ, and λ were used in this work.
In this study, N = 51 and the 49 intermediate structures between the two end structures (structure 1 and 51) were first generated by linear interpolation before path optimization was carried out. N was sufficiently large to provide a relatively smooth path in which the root‐mean‐square deviations between two adjacent structures were mostly less than 0.5 Å, although they could reach a little more than 0.6 Å between some adjacent structures, and approached 0.8 Å in one case. All energy calculations used a constant dielectric constant of 1 with no explicit water molecules included. No nonbonded cutoff was used in calculating the nonbonded interactions except in the simulation of the larger system of cSrc containing both the SH3 and the SH2 domains. In the latter case, a nonbonded cutoff of 12 Å was used.
Because each simulation of transition path needed to include the same sequence and number of residues but the two end states selected from the experimental structures could contain different sequences or number of residues—due to mutations, unresolved residues, or different constructs—the same wild‐type sequence obtained from either GenBank89 or RefSeq90 was used to build the conformations of the two end states using the corresponding experimental structures as templates. (Protein kinase A: NP_032880.1, Epidermal growth factor receptor: AAH94761.1, c‐Src kinase: NP_005408.1, Abelson tyrosine kinase: AFV09100.1, insulin receptor tyrosine kinase: AAA59452.1) MODELLER91 provides the engine to build the conformation of each end state in which residues present in the experimental structures were simply copied whereas missing or mutated residues were constructed based on MODELLER's method of satisfaction of spatial restraints. MOIL removed the uninteresting relative translation and rotation between the structures of the two end states before generating a path between them.
Mining similar structures from the protein data bank using PDBeFold
Given a structure, PDBeFold52, 53, 54, 55, 56, 57 provides an efficient means to retrieve similar structures from the Protein Data Bank. It performs a structural alignment and comparison between two structures in two stages. The first stage determines how the secondary structural elements between the two structures should be matched. Knowing roughly which parts of one protein needs to be placed with those of the second protein, the second stage completes the match by an iterative three‐dimensional structural alignment using the α carbons of the two proteins. A Q‐score measures how similar two proteins are by using both the root‐mean‐square deviation (RMSD) between the two structures and the number of residues aligned: N align instead of using either one alone. This provides a better similarity measure. Using RMSD alone, for example, could be misleading. One could obtain a smaller RMSD simply by matching a smaller portion of the two proteins. The Q‐score provides a better balance between the two quantities and is defined by:
| (4) |
where N 1and N 2 give the number of residues in sequence 1 and 2 respectively, and R 0 represents an empirical parameter that weigh the relative importance of RMSD and N align in contributing to the Q‐score. The maximum possible value of the Q‐score is 1 for two identical structures and Q decreases towards zero as the similarity between two structures declines.
Acknowledgments
The work on using chmin in the MOIL package was started when CW was a J Tinsley Oden faculty fellow of the Institute for Computational Engineering and Sciences, University of Texas at Austin. CW thanks Professor Ron Elber, Dr. Alfredo Cardenas, and Mr. Juan Bello Rivas for useful discussions and help in using MOIL. The University of Missouri Bioinformatics Consortium provided useful computational resources for this work. As a paper for a special issue honoring Professor Ronald M Levy's 65th birthday, the author also appreciates the many insights that Professor Levy has brought to the field of biomolecular simulations.
References
- 1. Fischer S, Karplus M (1992) Conjugate peak refinement—an algorithm for finding reaction paths and accurate transition‐states in systems with many degrees of freedom. Chem Phys Lett 194:252–261. [Google Scholar]
- 2. Sugita Y, Okamoto Y (1999) Replica‐exchange molecular dynamics method for protein folding. Chem Phys Lett 314:141–151. [Google Scholar]
- 3. Bolhuis PG, Dellago C, Geissler PL, Chandler D (2000) Transition path sampling: throwing ropes over mountains in the dark. J Phys Condens Matter 12:A147–A152. [DOI] [PubMed] [Google Scholar]
- 4. Henkelman G, Uberuaga BP, Jonsson H (2000) A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J Chem Phys 113:9901–9904. [Google Scholar]
- 5. Cardenas AE, Elber R (2003) Kinetics of cytochrome C folding: atomically detailed simulations. Proteins: Struct Funct Genet 51:245–257. [DOI] [PubMed] [Google Scholar]
- 6. Cardenas AE, Elber R. (2003) Atomically detailed Simulations of helix formation with the stochastic difference equation. Biophys J 85:2919–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chu JW, Trout BL, Brooks BR (2003) A super‐linear minimization scheme for the nudged elastic band method. J Chem Phys 119:12708–12717. [Google Scholar]
- 8. Peters B, Heyden A, Bell AT, Chakraborty A (2004) A growing string method for determining transition states: comparison to the nudged elastic band and string methods. J Chem Phys 120:7877–7886. [DOI] [PubMed] [Google Scholar]
- 9. Ren W, Vanden‐Eijnden E, Maragakis PWE. (2005) Transition pathways in complex systems: application of the finite‐temperature string method to the alanine dipeptide. J Chem Phys 123:1–12. [DOI] [PubMed] [Google Scholar]
- 10. Hu J, Ma A, Dinner AR (2006) Bias annealing: a method for obtaining transition paths de novo. J Chem Phys 125:114101. [DOI] [PubMed] [Google Scholar]
- 11. Elber R (2007) A milestoning study of the kinetics of an allosteric transition: atomically detailed simulations of deoxy Scapharca hemoglobin. Biophys J 92:L85–L87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maragliano L, Vanden‐Eijnden E (2007) On‐the‐fly string method for minimum free energy paths calculation. Chem Phys Lett 446:182–190. [Google Scholar]
- 13. Pan AC, Sezer D, Roux B (2008) Finding transition pathways using the string method with swarms of trajectories. J Phys Chem B 112:3432–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bowman GR, Beauchamp KA, Boxer G, Pande VS (2009) Progress and challenges in the automated construction of Markov state models for full protein systems. J Chem Phys 131:124101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bowman GR, Huang X, Pande VS (2009) Using generalized ensemble simulations and Markov state models to identify conformational states. Methods 49:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adelman JL, Grabe M (2013) Simulating rare events using a weighted ensemble‐based string method. J Chem Phys 138:044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Donovan RM, Sedgewick AJ, Faeder JR, Zuckerman DM (2013) Efficient stochastic simulation of chemical kinetics networks using a weighted ensemble of trajectories. J Chem Phys 139:115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pratt LR (1986) A statistical method for identifying transition states in high dimensional problems. J Chem Phys 85:5045–5048. [Google Scholar]
- 19. Elber R, Karplus M (1987) A method for determining reaction paths in large molecules: application to myoglobin. Chem Phys Lett 139:375–380. [Google Scholar]
- 20. Czerminski R, Elber R (1990) Reaction path study of conformational transitions in flexible systems: Applications to peptides. J Chem Phys 92:5580–5601. [Google Scholar]
- 21. Choi C, Elber R (1991) Reaction path study of helix formation in tetrapeptides: effect of side chains. J Chem Phys 94:751–760. [Google Scholar]
- 22. Nowak W, Czerminski R, Elber R (1991) Reaction path study of ligand diffusion in proteins: application of the self penalty walk (SPW) method to calculate reaction coordinates for the motion of CO through leghemoglobin. J Am Chem Soc 113:5627–5637. [Google Scholar]
- 23. Elber R, Roitberg A, Simmerling C, Goldstein R, Li H, Verkhivker G, Keasar C, Zhang J, Ulitsky A (1995) MOIL: a program for simulations of macromolecules. Comput Phys Commun 91:159–189. [Google Scholar]
- 24. Gerstein M, Krebs W (1998) A database of macromolecular motions. Nucleic Acids Res 26:4280–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krebs WG, Gerstein M (2000) The morph server: a standardized system for analyzing and visualizing macromolecular motions in a database framework. Nucleic Acids Res 28:1665–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wong CF (2008) Flexible ligand‐flexible protein docking in protein kinase systems. Biochim Biophys Acta: Prot Proteom 1784:244–251. [DOI] [PubMed] [Google Scholar]
- 27. Amaro RE, Li WW (2010) Emerging methods for ensemble‐based virtual screening. Curr Top Med Chem 10:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wong CF, Bairy S (2013) Drug design for protein kinases and phosphatases: flexible‐receptor docking, binding affinity and specificity, and drug‐binding kinetics. Curr Pharm Des 19:4739–4754. [DOI] [PubMed] [Google Scholar]
- 29. Lin JH, Perryman AL, Schames JR, McCammon JA (2002) Computational drug design accommodating receptor flexibility: the relaxed complex scheme. J Am Chem Soc 124:5632–5633. [DOI] [PubMed] [Google Scholar]
- 30. Lin JH, Perryman A, Schames J, McCammon JA (2003) The relaxed complex method: accommodating receptor flexibility for drug design with an improved scoring scheme. Biopolymers 68:47–62. [DOI] [PubMed] [Google Scholar]
- 31. Voter A (1997) Hyperdynamics: accelerated molecular dynamics of infrequent events. Phys Rev Lett 78:3908–3911. [Google Scholar]
- 32. Sugita Y, Kitao A, Okamoto Y (2000) Multidimensional replica‐exchange method for free‐energy calculations. J Chem Phys 113:6042–6051. [Google Scholar]
- 33. Sugita Y, Okamoto Y (2000) Replica‐exchange multicanonical algorithm and multicanonical replica‐exchange method for simulating systems with rough energy landscape. Chem Phys Lett 329:261–270. [Google Scholar]
- 34. Laio A, Parrinello M (2002) Escaping free‐energy minima. Proc Natl Acad Sci U S A 99:12562–12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu XW, Brooks BR (2003) Self‐guided Langevin dynamics simulation method. Chem Phys Lett 381:512–518. [Google Scholar]
- 36. Hamelberg D, Mongan J, McCammon J (2004) Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J Chem Phys 120:11919–11929. [DOI] [PubMed] [Google Scholar]
- 37. Laio A, Rodriguez‐Fortea A, Gervasio FL, Ceccarelli M, Parrinello M (2005) Assessing the accuracy of metadynamics. J Phys Chem B 109:6714–6721. [DOI] [PubMed] [Google Scholar]
- 38. Huang Z, Wong CF, Wheeler RA (2008) Flexible protein‐flexible ligand docking with disrupted velocity simulated annealing. Proteins 71:440–454. [DOI] [PubMed] [Google Scholar]
- 39. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 41. Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W, Clarkson B, Superti‐Furga G, Kuriyan J (2003) Structural basis for the autoinhibition of c‐Abl tyrosine kinase. Cell 112:859–871. [DOI] [PubMed] [Google Scholar]
- 42. Young MA, Shah NP, Chao LH, Seeliger M, Milanov ZV, Biggs Iii WH, Treiber DK, Patel HK, Zarrinkar PP, Lockhart DJ, Sawyers CL, Kuriyan J (2006) Structure of the kinase domain of an imatinib‐resistant Abl mutant in complex with the aurora kinase inhibitor VX‐680. Cancer Res 66:1007–1014. [DOI] [PubMed] [Google Scholar]
- 43. Hubbard SR (1997) Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J 16:5572–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hubbard SR, Wei L, Elis L, Hendrickson WA (1994) Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 372:746–754. [DOI] [PubMed] [Google Scholar]
- 45. Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ (2007) Structures of lung cancer‐derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 11:217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gajiwala KS, Feng J, Ferre R, Ryan K, Brodsky O, Weinrich S, Kath JC, Stewart A (2013) Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure 21:209–219. [DOI] [PubMed] [Google Scholar]
- 47. Kawakita Y, Seto M, Ohashi T, Tamura T, Yusa T, Miki H, Iwata H, Kamiguchi H, Tanaka T, Sogabe S, Ohta Y, Ishikawa T (2013) Design and synthesis of novel pyrimido[4,5‐b]azepine derivatives as HER2/EGFR dual inhibitors. Bioorg Med Chem 21:2250–2261. [DOI] [PubMed] [Google Scholar]
- 48. Knighton DR, Zheng J, Ten Eyck LF, Ashford VA, Xuong N, Taylor SS, Sowadski JM (1991) Crystal structure of the catalytic subunit of cyclic adenosine monophosphate‐dependent protein kinase. Science 253:407–414. [DOI] [PubMed] [Google Scholar]
- 49. Akamine P, Madhusudan Wu J, Xuong NH, Ten Eyck LF, Taylor SS (2003) Dynamic features of cAMP‐dependent protein kinase revealed by apoenzyme crystal structure. J Mol Biol 327:159–171. [DOI] [PubMed] [Google Scholar]
- 50. Xu W, Doshi A, Lei M, Eck MJ, Harrison SC (1999) Crystal structures of c‐Src reveal features of its autoinhibitory mechanism. Mol Cell 3:629–638. [DOI] [PubMed] [Google Scholar]
- 51. Cowan‐Jacob SW, Fendrich G, Manley PW, Jahnke W, Fabbro D, Liebetanz J, Meyer T (2005) The crystal structure of a c‐Src complex in an active conformation suggests possible steps in c‐Src activation. Structure 13:861–871. [DOI] [PubMed] [Google Scholar]
- 52. Krissinel E, Henrick K (2004) Secondary‐structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr Sect D: Biol Crystallogr 60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 53. Krissinel EB, Henrick K (2004) Common subgraph isomorphism detection by backtracking search. Software—Pract Exp 34:591–607. [Google Scholar]
- 54. Krissinel EB, Winn MD, Ballard CC, Ashton AW, Patel P, Potterton EA, McNicholas SJ, Cowtan KD, Emsley P (2004) The new CCP4 Coordinate Library as a toolkit for the design of coordinate‐related applications in protein crystallography. Acta Crystallogr Sect D: Biol Crystallogr 60:2250–2255. [DOI] [PubMed] [Google Scholar]
- 55. Krissinel E, Henrick K. Multiple alignment of protein structures in three dimensions. (2005) Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics). pp. 67–78.
- 56. Krissinel E (2007) On the relationship between sequence and structure similarities in proteomics. Bioinformatics 23:717–723. [DOI] [PubMed] [Google Scholar]
- 57. Read RJ, Adams PD, Arendall Iii WB, Brunger AT, Emsley P, Joosten RP, Kleywegt GJ, Krissinel EB, Lütteke T, Otwinowski Z, Perrakis A, Richardson JS, Sheffler WH, Smith JL, Tickle IJ, Vriend G, Zwart PH (2011) A new generation of crystallographic validation tools for the Protein Data Bank. Structure 19:1395–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Akamine P, Madhusudan, Brunton LL, Ou HD, Canaves JM, Xuong NH, Taylor SS (2004) Balanol analogues probe specificity determinants and the conformational malleability of the cyclic 3′,5′‐adenosine monophosphate‐dependent protein kinase catalytic subunit. Biochemistry (Moscow) 43:85–96. [DOI] [PubMed] [Google Scholar]
- 59. Prade L, Engh RA, Girod A, Kinzel V, Huber R, Bossemeyer D (1997) Staurosporine‐induced conformational changes of cAMP‐dependent protein kinase catalytic subunit explain inhibitory potential. Structure 5:1627–1637. [DOI] [PubMed] [Google Scholar]
- 60. Zeng Q, Allen JG, Bourbeau MP, Wang X, Yao G, Tadesse S, Rider JT, Yuan CC, Hong FT, Lee MR, Zhang S, Lofgren JA, Freeman DJ, Yang S, Li C, Tominey E, Huang X, Hoffman D, Yamane HK, Fotsch C, Dominguez C, Hungate R, Zhang X (2010) Azole‐based inhibitors of AKT/PKB for the treatment of cancer. Bioorg Med Chem Lett 20:1559–1564. [DOI] [PubMed] [Google Scholar]
- 61. Gassel M, Breitenlechner CB, König N, Huber R, Engh RA, Bossemeyer D (2004) The protein kinase C inhibitor bisindolyl maleimide 2 binds with reversed orientations to different conformations of protein kinase A. J Biol Chem 279:23679–23690. [DOI] [PubMed] [Google Scholar]
- 62. Bastidas AC, Wu J, Taylor SS (2015) Molecular features of product release for the PKA catalytic cycle. Biochemistry (Moscow) 54:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pflug A, Rogozina J, Lavogina D, Enkvist E, Uri A, Engh RA, Bossemeyer D (2010) Diversity of bisubstrate binding modes of adenosine analogue‐oligoarginine conjugates in protein kinase A and implications for protein substrate interactions. J Mol Biol 403:66–77. [DOI] [PubMed] [Google Scholar]
- 64. Couty S, Westwood IM, Kalusa A, Cano C, Travers J, Boxall K, Chow CL, Burns S, Schmitt J, Pickard L, Barillari C, McAndrew PC, Clarke PA, Linardopoulos S, Griffin RJ, Aherne GW, Raynaud FI, Workman P, Jones K, Van Montfort RML (2013) The discovery of potent ribosomal S6 kinase inhibitors by high throughput screening and structure‐guided drug design. Oncotarget 4:1647–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cowan‐Jacob SW, Fendrich G, Floersheimer A, Furet P, Liebetanz J, Rummel G, Rheinberger P, Centeleghe M, Fabbro D, Manley PW (2009) Structural biology contributions to the discovery of drugs to treat chronic myelogenous leukemia. NATO Science for Peace and Security Series A: Chemistry and Biology. pp. 37–61. [DOI] [PMC free article] [PubMed]
- 66. Levinson NM, Kuchment O, Shen K, Young MA, Koldobskiy M, Karplus M, Cole PA, Kuriyan J (2006) A Src‐like inactive conformation in the Abl tyrosine kinase domain. PLoS Biol 4:753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Qiu C, Tarrant MK, Choi SH, Sathyamurthy A, Bose R, Banjade S, Pal A, Bornmann WG, Lemmon MA, Cole PA, Leahy DJ (2008) Mechanism of activation and Inhibition of the HER4/ErbB4 kinase. Structure 16:460–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Janne PA (2009) Novel mutant‐selective EGFR kinase inhibitors against EGFR T790M. Nature 462:1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang XW, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J (2007) Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature 450:741 U713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Georghiou G, Kleiner RE, Pulkoski‐Gross M, Liu DR, Seeliger MA (2012) Highly specific, bisubstrate‐competitive Src inhibitors from DNA‐templated macrocycles. Nat Chem Biol 8:366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Parang K, Till JH, Ablooglu AJ, Kohanski RA, Hubbard SR, Cole PA (2001) Mechanism‐based design of a protein kinase inhibitor. Nat Struct Biol 8:37–41. [DOI] [PubMed] [Google Scholar]
- 72. Wu J, Tseng YD, Xu CF, Neubert TA, White MF, Hubbard SR (2008) Structural and biochemical characterization of the KRLB region in insulin receptor substrate‐2. Nat Struct Mol Biol 15:251–258. [DOI] [PubMed] [Google Scholar]
- 73. Till JH, Ablooglu AJ, Frankel M, Bishop SM, Kohanski RA, Hubbard SR (2001) Crystallographic and solution studies of an activation loop mutant of the insulin receptor tyrosine kinase: insights into kinase mechanism. J Biol Chem 276:10049–10055. [DOI] [PubMed] [Google Scholar]
- 74. Zhou B, Wong CF (2009) A computational study of the phosphorylation mechanism of the insulin receptor tyrosine kinase. J Phys Chem A 113:5144–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Carlson HA, Masukawa KM, McCammon JA (1999) Method for including the dynamic fluctuations of a protein in computer‐aided drug design. J Phys Chem A 103:10213–10219. [Google Scholar]
- 76. Carlson HA, Masukawa KM, Rubins K, Bushman FD, Jorgensen WL, Lins RD, Briggs JM, McCammon JA (2000) Developing a dynamic pharmacophore model for HIV‐1 integrase. J Med Chem 43:2100–2114. [DOI] [PubMed] [Google Scholar]
- 77. Carlson HA, McCammon JA (2000) Accommodating protein flexibility in computational drug design. Mol Pharmacol 57:213–218. [PubMed] [Google Scholar]
- 78. Carlson HA (2002) Protein flexibility is an important component of structure‐based drug discovery. Curr Pharm Des 8:1571–1578. [DOI] [PubMed] [Google Scholar]
- 79.Carlson A (2002) Protein flexibility and drug design: how to hit a moving target. Curr Opin Chem Biol 6:447–452. [DOI] [PubMed] [Google Scholar]
- 80. Henkelman G, Jonsson H (2000) Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J Chem Phys 113:9978–9985. [Google Scholar]
- 81. Weinan E, Ren WQ, Vanden‐Eijnden E (2002) String method for the study of rare events. Phys Rev B 66:052301. [DOI] [PubMed] [Google Scholar]
- 82. Elber R, Shalloway D (2000) Temperature dependent reaction coordinates. J Chem Phys 112:5539–5545. [Google Scholar]
- 83. Bellman R (1957) Dynamic programming. Princeton University Press. [Google Scholar]
- 84. Wong CF, Incorporating drug‐binding kinetics in drug design In: Cavasotto CN, Ed. (2015) Silico drug discovery and design: theory, methods, challenges and applications. Boca Raton, FL: CRC Press. [Google Scholar]
- 85. Huo S, Straub JE (1997) The MaxFlux algorithm for calculating variationally optimized reaction paths for conformational transitions in many body systems at finite temperature. J Chem Phys 107:5000–5006. [Google Scholar]
- 86. Dellago C, Bolhuis PG, Chandler D (1998) Efficient transition path sampling: application to Lennard‐Jones cluster rearrangements. J Chem Phys 108:9236–9245. [Google Scholar]
- 87. Bolhuis PG, Dellago C, Chandler D (1998) Sampling ensembles of deterministic transition pathways. Faraday Discuss 110:421–436. [Google Scholar]
- 88. Czerminski R, Elber R (1989) Reaction path study of conformational transitions and helix formation in a tetrapeptide. Proc Natl Acad Sci U S A 86:6963–6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Benson DA, Clark K, Karsch‐Mizrachi I, Lipman DJ, Ostell J, Sayers EW (2015) GenBank. Nucleic Acids Res 43:D30–D35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tatusova T, Ciufo S, Fedorov B, O'Neill K, Tolstoy I (2014) RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res 42:D553–D559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815. [DOI] [PubMed] [Google Scholar]