

Abstract
Applications of microbial transglutaminase (mTGase) produced from Streptomyces mobarensis (S. mobarensis) were recently extended from food to pharmaceutical industry. To use mTGase for clinical applications, like generation of site specific antibody drug conjugates, it would be beneficial to manufacture mTGase in Escherichia coli (E. coli). To date, attempts to express recombinant soluble and active S. mobarensis mTGase have been largely unsuccessful. mTGase from S. mobarensis is naturally expressed as proenzyme and stepwise proteolytically processed into its active mature form outside of the bacterial cell. The pro‐domain is essential for correct folding of mTGase as well as for inhibiting activity of mTGase inside the cell. Here, we report a genetically modified mTGase that has full activity and can be expressed at high yields in the cytoplasm of E. coli. To achieve this we performed an alanine‐scan of the mTGase pro‐domain and identified mutants that maintain its chaperone function but destabilize the cleaved pro‐domain/mTGase interaction in a temperature dependent fashion. This allows proper folding of mTGase and keeps the enzyme inactive during expression at 20°C, but results in full activity when shifted to 37°C due to loosen domain interactions. The insertion of the 3C protease cleavage site together with pro‐domain alanine mutants Tyr14, Ile24, or Asn25 facilitate high yields (30–75 mg/L), and produced an enzyme with activity identical to wild type mTGase from S. mobarensis. Site‐specific antibody drug conjugates made with the E .coli produced mTGase demonstrated identical potency in an in vitro cell assay to those made with mTGase from S. mobarensis.
Keywords: microbial transglutaminase, S. mobarensis, E. coli, cloning, soluble expression, protein purification, antibody drug conjugation
Introduction
Transglutaminase (EC 2.3.2.13, protein‐glutamine γ‐glutamyltransferase, TGase) is a member of the enzyme family which catalyzes cross linking between the γ‐carboxyamide groups of glutamine residues and a variety of primary amines, including the amino group of lysine.1, 2 TGases can be found throughout all groups of organisms including prokaryotes, eukaryotes,1 and plants.3 TGases in animals (aTGase), for example, human blood coagulation factor XIII, TG2, and guinea pig liver TGase, are multi‐domain proteins and depend on calcium for regulation of enzyme function.4, 5 Microbial transglutaminases (mTGase), on the other hand, like the enzyme first discovered in S. mobarensis,6 have only one single domain and do not depend on calcium for activity.7 aTGases and mTGases utilize a catalytic triad comprised of Cys, His, and Asp residues, although, relative to the aTGase active site cysteine, in mTGase, the positions of His and Asp appear to be reversed.8, 9, 10 S. mobarensis mTGases evolved from proteases and the protein fold surrounding the catalytic triad shows similarities to the aTGases suggesting that both enzymes are related by convergent evolution. mTGase is mainly used in the food industry to modulate the texture of meat, fish, and dairy products like yogurt and cheese.7 mTGase is also used in a wide variety of applications in the pharmaceutical industry for conjugation of proteins, DNA, and peptides, as well as in tissue engineering.2 Recently, the feasibility of using mTGase for generating antibody drug conjugates (ADCs) for therapeutic applications11 and for imaging was demonstrated.12, 13 The commercially available mTGase is produced by fermentation of wild‐type S. mobarensis.6 S. mobarensis expresses mTGase as an inactive zymogen and the pro‐domain needs to be proteolytically processed in order to yield an active enzyme.14, 15 mTGase precursor is secreted into the surrounding medium together with activating proteases. The activation of mTGase occurs stepwise, and to date two endogenous enzymes, transglutaminase activating metalloprotease (TAMEP) and S. mobarensis tripeptidyl amino peptidase (SM‐TAP) have been purified and biochemically characterized.15 The pro‐domain of S. mobarensis mTGase was shown to be essential for correct folding and activity of mTGase16 and its chaperon activity was revealed when mTGase precursor was expressed as one molecule, or even when the pro‐domain and mature domain were co‐expressed as separate chains.16, 17
While S. mobarensis produced mTGase is widely used in the food industry, clinical applications of mTGase would benefit from the development of a more commonly used expression system for soluble and fully active mTGase. E. coli is a popular expression system and recently it was reported that approximately a third of currently approved recombinant therapeutic proteins are produced in E. coli.18 Large‐scale production of mTGase for pharmaceutical applications would be most convenient if accomplished in E. coli, due to available “host‐cell‐protein” assays, defined media components and cGMP documentation necessary for clinical use.18 To date, however, attempts to produce large amounts of mTGase in a soluble and fully active form in yeast and E. coli have failed.17, 19
Here, we report an mTGase expression system that allows expression of soluble S. mobarensis mTGase in the cytoplasm of E. coli yielding purified mTGase with identical enzymatic activity as S. mobarensis produced mTGase. Through pro‐domain mutations, we identified important contact residues reveling previously unknown features of the interface between pro‐domain and mature enzyme domain.
Results and Discussion
Design of mTGase precursor‐3C protease dual gene expression plasmids
Similar to previous reports,20, 21, 22 our attempts to produce mTGase directly as a soluble protein in the cytoplasm or in the periplasm of E. coli were unsuccessful. Expression of mTGase without its pro‐domain either led to significant growth retardation upon induction of mTGase or generation of inclusion bodies. These results suggest that mTGase might be toxic to E. coli, probably due to its protein crosslinking activity. Insoluble mTGase precursor and mTGase can be overexpressed in the cytoplasm of E. coli as inclusion bodies and subsequently solubilized and refolded to yield various amounts of soluble proenzyme or mTGase.21, 23 Soluble mTGase precursor can be produced in large quantities in the cytoplasm of E. coli by lowering the temperature of protein induction below 37°C.24, 25 Both soluble and inclusion body approaches to produce mTGase precursor, however, necessitate in vitro removal of the pro‐domain to yield active mTGase enzyme by proteases such as TAMEP, Dispase, or Trypsin.15, 22, 23
To produce large amounts of active mature mTGase in E. coli, it would be beneficial to have an expression system that allows soluble expression of mTGase and a straightforward purification procedure. Towards this goal, we designed bi‐cistronic E. coli expression vectors (see Materials and Methods) which facilitate expression of mTGase precursor plus 3C protease (human rhino virus 14) into E. coli cytoplasm [Fig. 1(A,B) and Supporting Information Fig. 1(A)]. To enable proteolytic processing of mTGase precursor to its active form, the recognition site of 3C protease was cloned in between mTGase pro‐domain and enzyme domain [Fig. 3(B) and Supporting Information Fig. 1(A)]. Since the two promoters utilize different reagents to induce protein expression, (IPTG for the T7 promoter and L‐arabinose for the araBAD promoter) it is possible to either co‐induce both genes or induce them sequentially and control the timing of protein expression.
Figure 1.
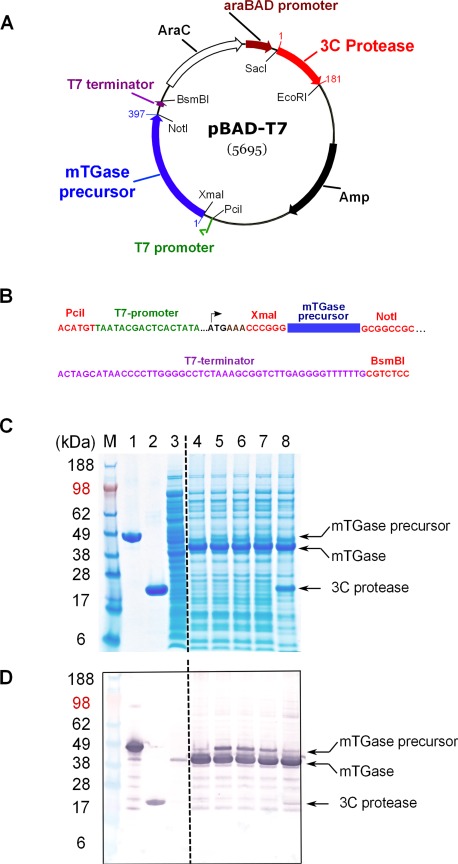
Structure of expression plasmid pBAD‐T7 and E. coli expression study. (A) Structure of the expression plasmid pBAD‐T7. The blue and red arrows show the positions of the coding regions for mTGase precursor and 3C protease, respectively. Numbers indicate first and last amino acid of each open reading frame [Supporting Information Fig. 1(A,B)]. The green arrow and brown arrow represent the positions for the T7 and araBAD promoters, respectively. Open reading frames for AraC regulator (AraC) and ampicillin resistance (Amp) are shown as open and filled black arrows, respectively. (B) DNA sequence of the T7‐promoter gene cassette. DNA sequences colored in red represent positions of restriction enzymes. Sequence color code for T7 promoter and terminator as well as the coding region for mTGase precursor matches the description in (A). (C) pBAD‐T7 E. coli small‐scale expression study. SDS‐PAGE gel: Lanes 1–3 show controls for mTGase precursor, 3C protease, and noninduced lysate, respectively. Lane 4 shows simultaneous induction of mTGase precursor and 3C protease. Lanes 5–7, separate induction of mTGase precursor, followed by induction of 3C protease, 30 min to 2 h at RT. Lane 8, induction of mTGase precursor, followed by induction of 3C protease, 30 min plus additional purified 3C protease, incubation for 2 h at RT. Arrows mark the positions for mTGase precursor, mTGase, and 3C protease. (D) Anti‐His tag western blot analysis as described in Materials and Methods section.
Figure 3.
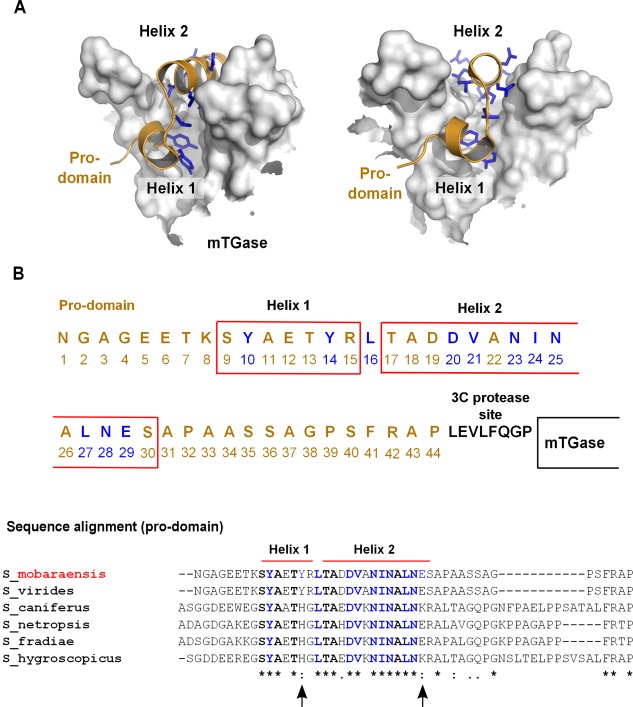
Design of mTGase pro‐domain alanine mutants. (A) mTGase precursor structural overview. Surface representation of the active site cleft of mTGase shown in gray and cartoon representation of pro‐domain colored orange (PDB 3IU0).30 Pro‐domain amino acid residues facing towards mTGase are shown in blue stick representation. (B) Pro‐domain amino acid sequence. Red boxes indicate pro‐domain alpha helices 1 and 2. Amino acids colored in blue indicate mTGase main contact residues. Amino acids colored in black indicate 3C protease cleavage site. Alignment of Streptomyces mTGase pro‐domain sequences. Red bars above the alignment indicate the region of S. mobarensis pro‐domain helix 1 and 2. Amino acids shown in bold typeface indicate conserved residues within pro‐domain helix 1 and 2. Amino acids marked blue are mutated to alanine residues in the S. mobarensis pro‐domain. Black arrows below the alignment mark amino acids of contact residues which differ among S. mobarensis species and are located within the region comprised by S. mobarensis pro‐domain helix 1 and helix 2. Stars below the alignment indicate conserved residues, two dots indicate conservative amino acid changes, and one dot indicates nonconservative changes of residues among different species.
Small scale protein expression
Expression of mTGase precursor was tested in both combinations, with mTGase precursor under the control of the araBAD or T7 promoters. The two different combinations were investigated in order to test potential differences in protein expression efficiency due to promoter characteristics (see Discussion section). In addition to the different promoter control of mTGase precursor expression, the influence of 3C protease expression by either co‐induction with mTGase precursor or delayed sequential expression was tested. The results with mTGase precursor under the control of T7 promoter and 3C protease under the control of the araBAD promoter are summarized in [Fig. 1(C)]. Co‐induction of both genes [Fig. 1(C), Lane 4] resulted in strong expression of fully processed mTGase as further demonstrated by anti His‐tag western blot analysis [Fig. 1(D)]. Sequential induction with overnight expression of mTGase precursor followed by 3C protease expression with increasing duration from 30 min up to 2 h showed gradual decrease of the amount of mTGase precursor [Fig. 1 (C,D)], Lanes 5–7). The addition of purified 3C protease to the lysate almost completely processed mTGase precursor to mTGase [Fig. 1(C,D), Lane 8].
Interestingly, the same small‐scale expression study with the alternative combination of gene expression with mTGase precursor under the control of the araBAD promoter and 3C protease expression controlled by T7 promoter only showed weak expression of soluble mTGase enzyme (data not shown). Possible reasons for the observed differences in expression levels of soluble mTGase depending on promoter combination could be related to promoter strength and tightness of promoter control. The arabinose‐inducible araBAD promoter and regulator (AraC) of E. coli is regarded as a relatively strong, yet tightly controlled promoter‐regulator system.26, 27 The phage T7‐RNA polymerase‐based promoter system on the other hand exhibits very strong expression of heterologous genes in E. coli but also high basal (leaky) expression in noninduced cells if not regulated by T7 lysozyme or Lac repressor, which are both absent in our vector construct pBAD‐T7 [Fig. 1(A)].28, 29 When 3C protease expression is controlled by T7 promoter, 3C‐enzyme concentration is presumably higher than mTGase precursor protein concentration during protein induction and quickly converts mTGase precursor to active mTGase in the cell cytoplasm. Since mTGase can be toxic to E. coli due to its crosslinking activity, significant accumulation of mTGase might be prevented. The combination of genes and promoters [Fig. 1(A)] enables higher accumulation of mTGase precursor due to lower amounts of 3C protease and results in greater overall yield of mTGase in the cytoplasm of E. coli.
Expression, purification, and activity of E. coli produced mTGase
Expression was scaled up to enable protein purification of soluble mTGase for protein characterization and enzyme activity assays. The cloned His‐tag at the C‐terminus of mTGase allowed for straight forward Ni‐NTA affinity purification from crude E. coli cell lysate and was followed by size exclusion chromatography (SEC). A single symmetric protein peak eluted at the expected retention time corresponding to a molecular weight of approximately 40 kDa, (Fig. 2). To our surprise, two protein bands were visible when the main peak from the SEC was run on a SDS‐page gel (Fig. 2), one prominent larger band with the expected size of mTGase and a smaller protein band with apparent molecular weight of 5–6 kDa protein size. Characterization of the Ni‐NTA and SEC purified mTGase by intact mass spectrometry revealed two masses 5,629 and 39,198 Da corresponding to prodomain and mature domain (expected mass for prodomain is 5,629 Da, and for mature domain is 39,200 Da, respectively), corresponding to cleaved mTGase pro‐domain and mature domain, respectively. The results suggest that mTGase precursor was completely processed by 3C protease during the expression, but that a significant fraction of the pro‐domain remained noncovalently attached to the enzyme domain even after sequential purification steps of Ni‐NTA and SEC chromatography.
Figure 2.
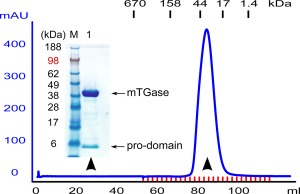
Purification of soluble mTGase from E. coli. (A) Size exclusion chromatography. Chromatogram monitored at wavelength A280 nm. Labeled dashes above the chromatogram show retention times for protein size standard. Arrowheads indicate protein elution fractions analyzed with SDS‐PAGE gel shown in inset.
mTGase secreted from its natural source (S. mobarensis), on the other hand, does not show any attached pro‐domain, determined by intact mass spectrometry (expected mass 37,862 Da, and obtained mass 37,860 Da). The complete dissociation of the pro‐domain in S. mobarensis expressed mTGase is likely due to multiple proteases acting on the pro‐domain, cleaving it in several places and yielding fully active enzyme. The E. coli expression system described here, however, contains only one engineered protease cleavage site. The selectivity of the 3C protease toward its recognition site prevents the cleavage anywhere else within the mTGase pro‐domain, and attempts to include an additional 3C cleavage site were unsuccessful (data not shown). The effect of the noncovalently associated pro‐domain on the activity of E. coli mTGase was tested with a colorimetric assay (see Materials and Methods section). The enzyme activity of the purified wild‐type mTGase from S. mobarensis was used as reference and represented 100% enzyme activity. The mTGase purified from the described E. coli system displayed about 54% enzyme activity compared to wild‐type mTGase. The data suggests that the decreased activity of E. coli mTGase is due to the presence of the noncovalently attached pro‐domain. To our knowledge, this is the first report that correlates reduced activity of heterologously expressed mTGase to the incomplete dissociation of the inhibitory pro‐domain.
Design of mTGase pro‐domain mutants
We designed a series of pro‐domain alanine mutants to test the effect of loosening the interaction between the pro‐domain and mTGase in order to facilitate their dissociation and achieve full enzymatic activity (Fig. 3). To this end, we utilized the crystal structures of the zymogen and mature form of mTGase8, 30 to select positions at the interface between pro‐domain and mTGase. Since the pro‐domain is known to also act as a folding chaperone for the mTGase,16, 17 we sought mutants that would be still able to assist in mTGase folding, but would allow the pro‐domain to dissociate more readily once the linker between pro‐domain and mTGase was cleaved by the co‐expressed 3C protease.
The crystal structure of mTGase zymogen revealed an L‐shaped pro‐domain, which completely occludes the active‐site cleft of mTGase.30 The L‐shaped pro‐domain is composed of a short α‐helix (Residues 9–15) connected by a single residue to a second α‐helix (Residues 17–30) and together they cover the active‐site cleft [Fig. 3(A,B)]. Within the first short α‐helix residues Tyr10 and Tyr14 form the majority of the interactions with the enzyme active site [Fig. 3(B)].30 Only one hydrophobic residue, Leu16, forms a short connective loop between α‐helices 1 and 2 of the mTGase pro‐domain and points deep into the active‐site cleft [Fig. 3(B)]. Eight residues in the second α‐helix face toward the active‐site cleft (Asp20, Asp21, Asn23, Ile24, Asn25, Leu27, Asn28, and Glu29) and were selected together with Tyr10, Tyr14, and Leu16 for mutagenesis [Fig. 3(B), blue‐labeled residues].
A sequence alignment of pro‐domain sequences of several species of S. mobarensis mTGases reveals high‐sequence similarity within the region covering the active site of mTGase [Fig. 3(B)]. Only two contact residues, at positions 14 and 29, relative to S. mobarensis pro‐domain, show differences between species. Position 14 can accommodate tyrosine and histidine, whereas at position 29 glutamic acid and lysine are observed [Fig. 3(B)]. Interestingly, sequences upstream and downstream of the pro‐domain region covering the active site can differ significantly in length as well as amino acid content.
Biochemical characterization of mTGase pro‐domain mutants
All of the pro‐domain alanine mutants as well as the nonmutated control of mTGase precursor were expressed as described in the method section (Fig. 4). mTGase precursor and 3C protease protein production was induced simultaneously and soluble processed mTGase was purified from crude E. coli lysate and analyzed by SDS‐polyacrylamide gel electrophoresis (PAGE) gel [Fig. 4(A)]. Nonmutated pro‐domain mTGase control [Fig. 4(A), lane 2], and all alanine mutants showed various amounts of associated pro‐domain. The amount of attached pro‐domain of mTGase mutants seemed to inversely correlate with the amount of mTGase aggregate formation (self‐crosslinking), judged by staining intensity of higher order bands on the SDS‐PAGE gel and western blot [Fig. 4(A) and Supporting Information Fig. 2(A)]. Consistent with this finding, mTGase produced from S. mobarensis lacking any pro‐domain also showed high levels of mTGase aggregates [Fig. 4(A), lane 1]. Three mutants (Tyr10, Leu16, and Val21) revealed strong formation of mTGase aggregates whereas mutants Tyr14, Asp20, Ile24, and Asn25 showed only weak formation of mTGase aggregates. All other mutants plus the control (nonmutated mTGase) did not form visible amounts of aggregates, which was also confirmed by western blot analysis [Fig. 4(A), Supporting Information Fig. 2(A)]. The appearance and amount of mTGase aggregates were closely correlated with high enzyme activity [Fig. 4(A,B)] but were inversely correlated to purified enzyme yield [Fig. 4(D)]. This correlation was especially evident with pro‐domain mutants Tyr10, Leu16, and Val21 [Fig. 4(A,B,D)] and most probably reflected the impact of active mTGase enzyme on E. coli cell viability during protein induction.
Figure 4.
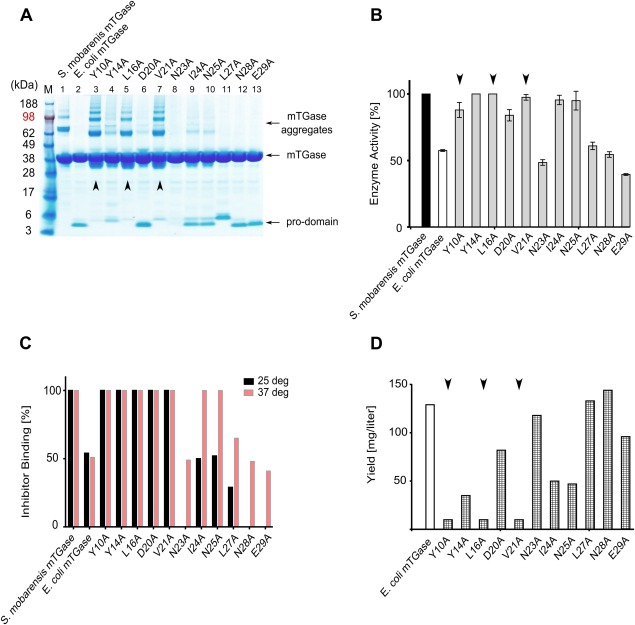
Analysis of mTGase pro‐domain alanine mutants. (A) SDS‐PAGE gel analysis of mTGase pro‐domain mutants following Ni‐NTA purification. mTGase protein runs around 38 kDa, pro‐domain around 5 kDa, and mTGase aggregates around 65–190 kDa. (B) mTGase enzyme activity assay. The black bar control represents 100% enzyme activity of wild type S. mobarensis mTGase. The white bar indicates the activity of non‐mutated but 3C protease processed mTGase. Gray bars represent activities of mTGase pro‐domain mutants. Error bars represent standard deviation of two independently performed assays. (C) mTGase inhibitor assay. Black bars indicate inhibitor binding ratios of assay performed at 25°C. Red bars indicate inhibitor binding ratios of assay performed at 37°C. Inhibitor binding to wild‐type S. mobarensis mTGase [Fig. 4(C), bar 1] was set to 100%. (D) E. coli protein yield of mTGase pro‐domain alanine mutants. The white bar represents the protein yield of nonmutated but 3C protease processed mTGase, whereas the checkered bars represent the protein yields of mTGase pro‐domain mutants. Black arrowheads in all panels highlight pro‐domain mutants Tyr10, Leu16, and Val21.
The differences in apparent pro‐domain size, most prominently displayed by mutant Leu27 [Fig. 4(A), Lane 11], is attributed to SDS page mobility change, as the correct size of mutant Leu27 pro‐domain was confirmed by mass spectroscopy (results not shown).
The level of enzyme activity of all mutants and controls was measured with a colorimetric assay at 37°C. [Fig. 4(B)]. Based on activity, the mTGase pro‐domain mutants could be grouped into two groups: the first group of mutants displayed full or nearly full activity compared to the native S. mobarensis mTGase (Tyr10, Tyr14, Leu16, Asp20, Val21, Ile24, and Asn25). The second group of mutants (Asn23, Leu27, Asn28, and Glu29) showed approximately 50% activity and was similar to the non‐mutated processed mTGase [Fig. 4(B)].
Since all mTGase pro‐domain mutants showed various levels of co‐purified and associated pro‐domain [Fig. 4(A)] it was surprising that some of the mTGase pro‐domain variants had nearly 100% activity at 37°C, but still showed the presence of the associated pro‐domain (e.g., Tyr14, Asp20, Val21) [Fig 4(A)]. To explain how some variants can display full activity with Z‐Gln‐Gly (MW 337 Da) as substrate even in the presence of presumably inhibitory pro‐domain, we tested accessibility to the binding site with a covalent inhibitor (MW 370 Da) [Fig. 4(C)]. The inhibitor binding assay was performed at 25 and 37°C. Following incubation of various mTGase pro‐domain mutants with the inhibitor, the ratio of inhibitor‐bound versus nonbound mTGase enzyme was measured by intact mass spectroscopy. When the mTGase inhibitor binding assay was done at 37°C (same temperature as the activity assay) [Fig. 4(C)], light red bars], the ratio of inhibitor bound/unbound mTGase correlated well to the measured mTGase enzyme activity [Fig. 4(B)]. The inhibitor binding experiment was repeated also at lower temperature (25°C) [Fig. 4(C), black bars]. No difference in activity was observed for pro‐domain mutants Tyr10, Tyr14, Leu16, Asp20, and Val21 tested at the two temperatures [Fig. 4(C)]. Almost all of these mutants showed identical enzyme activity as the wild‐type mTGase enzyme from S. mobarensis [Fig. 4(B)]. On the other hand, pro‐domain mutants Asn23, Ile24, Asn25, Lau27, Asn28, and Glu29 showed reduced or no inhibitor binding when the assay was performed at 25°C (see Discussion section).
In most cases, the yield of purified soluble mTGase protein per liter E. coli cell culture inversely correlated with the level of enzyme activity [Fig. 4(B,D)]. Pro‐domain mutants with high level of mTGase enzyme activity (Tyr10, Tyr14, Leu16, Val21, Ile24, and Asn25) yielded significantly less protein than mutants with low enzyme activity (Asn23, Lau27, Asn28, and Glu29). One exception was the Asp20 mutant which showed only about 20% reduced enzyme activity, but yielded relatively high expression level (75 mg/L) [Fig. 4(D)].
Previous results in the literature have shown that intact nonprocessed pro‐TGase does not have any enzymatic activity.31 3C protease clipping of the connective loop between mTGase pro‐domain and mTGase [Fig. 3(C)] loosened the interaction between the two domains enough to gain about 50% of wild‐type S. mobarensis enzyme activity [Fig. 4(B), Lane 2]. This amount of activity is likely due to the combination of pro‐domain loss during purification and structural fluctuations in the attached pro‐domain.
Pro‐domain interactions
Tyr10 and Tyr14 are located in helix1 of the L‐shaped pro‐domain while Leu16 is in the connecting loop between helix1 and helix2 [Fig. 3(B)]. All three residues protrude deeply into the active side cleft of mTGase and occlude the catalytic triad (Cys110, Aps301, and His320).30 The results of the inhibitor study shown in [Fig. 4(C)] suggest that single contact residue mutations within the short helix1, connecting loop, or the N‐terminal part of helix2 can change the strength of pro‐domain/mTGase interaction enough to allow access of substrate (or inhibitor) into the active site and can restore full activity.30
Pro‐domain mutant Asp20 displays interesting characteristics with high enzyme activity as well as relatively high protein yield. In addition, for this mutant, the amount of higher order mTGase aggregates was low compared with the apparent amount of attached pro‐domain [Fig. 4(A)]. The weak self‐crosslinking activity and relatively high yield of purified protein suggest that Asp20 mutant reaches the right balance of pro‐domain interaction strength to prevent toxic protein crosslinking in the cytoplasm of E. coli during protein expression while retaining enough flexibility at higher temperatures to have nearly full activity. Interestingly, the mutation of Val21, just adjacent to Asp20, leads to the formation of large amounts of mTGase aggregates, high enzyme activity paired with low protein yield showing the delicate balance that needs to be reached.
A pair of mutations, Ile24 and Asn25, located within the central part of pro‐domain helix 2 also showed distinct characteristics. Both mutants are as active as wild type mTGase purified from S. mobarensis and only exhibit weak self‐crosslinking [Fig. 4(A,B)]. Unlike Asp20, however, the inhibitor binding to mutants Ile24 and Asn25 shows large differences between 25 and 37°C. The more C‐terminal position within pro‐domain helix 2 of mutants Ile24 compared to Asp20 probably exhibits a more stable attachment of the pro‐domain resulting in less active site accessibility at lower temperature.
The temperature‐dependent inhibitor binding pattern suggests that mutations within helix1, linker, and the N‐terminal part of helix 2 are more critical for active site access. We speculate that helix 1, which directly covers the active site Cys110, is mostly responsible for preventing premature activation of the proenzyme upon secretion of the molecule into the medium. The longer pro‐domain helix 2 seems to stabilize the interaction with the enzyme domain and might be responsible for more of the chaperone activity.
To our knowledge, this is the first study that sheds light on the residues at the pro‐domain interface that are important for mTGase chaperone, inhibitory, and activation functions and suggests that multiple cleavage sites and enzymes are necessary to facilitate complete dislodging of the pro‐domain.
Generation and in vitro potency testing of site specific antibody drug conjugates
Our inhibitor and activity data suggest that transglutaminase can catalyze the crosslinking reaction even in the presence of the associated and cleaved mutated pro‐domain. However, the mTGase substrates in these assays are much smaller than an antibody, which was our target substrate. To verify that the E. coli produced mTGase is suitable for pharmaceutical applications, we compared the drug conjugation efficiencies of commercially available mTGase (S. mobarensis mTGase) with mTGase produced and purified from E. coli (E. coli mTGase, pro‐domain mutant Y14A). Pro‐domain mutant Y14A was chosen for follow‐up studies as a representative of mutants, which yield 100% enzyme activity compared to wild‐type mTGase [Fig. 4(B)].
A schematic showing the chemical reaction catalyzed by mTGase [Fig. 5(A)] where the glutamine tag, added to the antibody [Fig. 5(B)], is the acyl donor and the linker payload [Fig. 5(C)] acts as the acyl acceptor. Previously, we and others have shown that mTGase does not recognize naturally occurring glutamines in the constant regions of glycosylated human IgG1 antibodies,12, 32 providing us with the opportunity to design a specific glutamine tag that can be engineered at desired antibody locations. Taking advantage of this finding, we utilized mTGase to conjugate the AcLys‐vc‐PABC‐Aur0101 linker‐payload (unpublished data) [Fig 5(C)] to an anti‐TROP2 (Tumor‐associated calcium signal transducer 2) antibody engineered with a glutamine tag at the C‐terminus of the heavy chain [Fig. 5(B)]. Hydrophobic interaction chromatography (HIC) revealed that both S. mobarensis TGase as well as E. coli mTGase yielded a product with an average drug‐antibody ratio (DAR) of 1.95 with a possible maximum loading of 2.0 [Fig. 5(D)]. Characterization of the conjugated anti‐TROP2 antibody by intact mass spectrometry revealed almost exclusively a mass consistent with DAR 2 (expected mass for nonconjugated anti‐TROP2 antibody is 146,380 Da; expected mass for conjugated anti‐TROP2 antibody is 148,982 Da; observed mass for conjugated anti‐TROP2 antibody is 148,983 Da; conjugated with S. mobarensis mTGase and E. coli produced mTGase, respectively).
Figure 5.
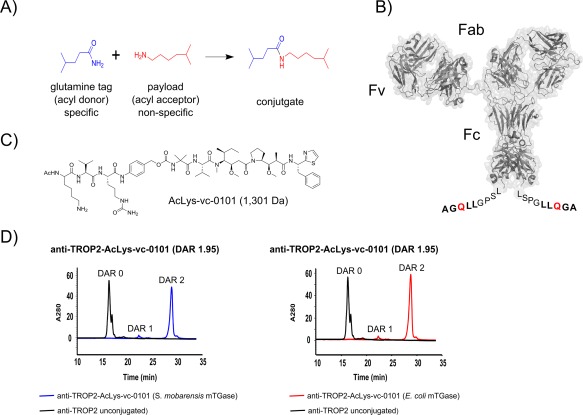
mTGase‐catalyzed generation of site specific antibody drug conjugates. (A) Chemical reaction catalyzed by mTGase. (B) Surface representation of anti‐TROP2 IgG1. Extra amino acids “LLQGA” (bold face) at the C‐terminus represent the glutamine tag, with reactive glutamine highlighted in red. Amino acids “LSPG” represent last C‐terminal residues of anti‐TROP2 antibody. (C) Chemical structure of the Linker‐payload utilized to generate the antibody drug conjugate.36 (D) Hydrophobic interaction chromatography (HIC) traces of anti‐TROP2‐AcLys‐vc‐0101 conjugated with wild type S. mobarensis. mTGase (blue) and anti‐TROP2‐AcLys‐vc‐0101 conjugated with mTGase produced and purified from E. coli (pro‐domain mutant Y14A) (red). Antibody anti‐TROP2 prior to conjugation is shown as black lines and conjugation yields [average drug‐antibody ratio (DAR)] are shown in graph titles.
We confirmed that wild‐type mTGase as well as mTGase produced in E. coli can generate highly homogenous populations of ADCs (Fig. 5). Both conjugates prepared with either the native S. mobarensis mTGase or the E. coli mTGase demonstrated equal cell killing potential with IC50 values of 0.35 and 0.31 nM, respectively (Fig. 6), while a negative control ADC showed no significant cell killing.
Figure 6.
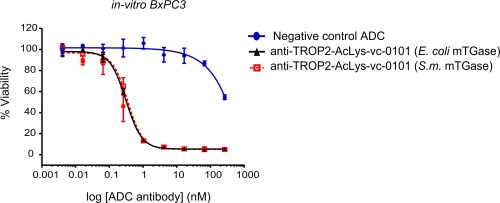
In‐vitro potency of conjugated antibody anti‐TROP2‐vc‐0101. Cytotoxicity of anti‐TROP2‐vc‐0101 conjugated with S. mobarensis mTGase ( ) and mTGase (pro‐domain mutant Y14A) produced in E. coli (
) and mTGase (pro‐domain mutant Y14A) produced in E. coli ( ) was evaluated on TROP2 target‐expressing BxPC3 cells as described in the Materials and Methods section.
) was evaluated on TROP2 target‐expressing BxPC3 cells as described in the Materials and Methods section.
For selected pro‐domain mutants and controls, we determined the EC50 values of antibody drug conjugation efficiency [Supporting Information Fig. 2(B)]. The data show that, at high enzyme concentrations, all mutants were able to achieve high conjugation yields. As expected, as the amounts of mTGase were titrated down, mutants that did not show full S. mobarensis mTGase activity also yielded worse average drug‐antibody ratios.
Conclusions
Taken together, our data show that, through engineering of the pro‐domain, it is possible to achieve good expression levels of soluble and fully active mTGase in E. coli. Alanine‐scanning of the mTGase pro‐domain amino acids revealed important residues responsible for folding chaperone activity and stability of the mTGase interaction. We identified mTGase pro‐domain residues Tyr14, Asp20, Ile24, and Asn25 as the most important residues for achieving active and soluble mTGase, while preserving high levels of protein production. Due to the detrimental crosslinking activity of mTGase to the host cell, pro‐domain chaperone function, and pro‐domain tight interaction with the enzyme, obtaining high expression yield of fully active mTGase is a delicate balance.
We show evidence that the pro‐domain that remains attached to the mTGase purified from E. coli facilitates higher level of protein expression without the toxic effects of protein crosslinking. Through the fine‐tuning of promoter strengths and mutagenesis of the pro‐domain, we were able to achieve 30–75 mg/L expression levels of fully active mTGase. The E. coli mTGase showed identical conjugation characteristics to the native S. mobarensis produced mTGase, and the described process can be used for generating mTGase for pharmaceutical applications.
Materials and Methods
Materials and cell strains
All chemicals used throughout the study were analytical grade and purchased from Sigma‐Aldrich, unless stated otherwise. Bacterial strains E. coli TOP10 and BL21 (DE3) were obtained from Invitrogen. All enzymes for DNA manipulation were purchased from New England Biolabs.
Bacterial strains, plasmids, and growth conditions
E. coli TOP10 and E. coli BL21 (DE3) were used as hosts for DNA manipulation and protein expression, respectively. Plasmids pET20b(+) (EMD Millipore) and pBAD‐A (Life Technologies) served as basis for expression construct generation. Luria broth (LB) and Terrific broth (TB) were used for DNA plasmid cloning and protein expression, respectively. E. coli strains were grown at 37°C for plasmid propagation and a combination of 37 and 20°C for protein expression.
Construction of expression plasmids for simultaneous expression of mTGase and 3C protease
Genes for mTGase and 3C protease were both chemically synthesized and codon‐optimized (GeneArt, Life Technologies; for gene bank accession numbers see (Supporting Information Fig. 1) for growth in E. coli. Expression plasmid pBAD‐T7 [Fig. 1(A)] was constructed by subcloning T7 promoter and T7 terminator of plasmid pET20b(+) into plasmid pBAD‐A with PciI and BsmBI restriction sites at the 5′ and 3′ end, respectively [Fig. 1(A)/(B)]. For cloning the mTGase precursor gene into plasmid pBAD‐T7 under the control of T7 promoter, restriction sites XmaI and NotI were inserted between T7 promoter and T7 terminator [Fig. 1(A)/(B)]. To optimize protein expression levels of mTGase, a codon for amino acid lysine (AAA) was placed as first amino acid following the start codon ATG.33 The six nucleotides coding for restriction site XmaI are adding amino acids proline and glycine to the N‐terminus of the mTGase pro‐domain [Fig. 1(B) and Supporting Information Fig. 1(A)]. A 3C protease cleavage site was inserted into the connecting loop of mTGase pro‐domain and mTGase between Pro44 and Asp45 [Fig. 3(B) and Supporting Information Fig. 1(A)]. Full‐length 3C protease gene was cloned into plasmid pBAD‐T7 under the control of the araBAD promoter with SacI and EcoRI restriction sites at the 5′ and 3′ end, respectively [Fig. 1(A) and Supporting Information Fig. 1(B)].
mTGase pro‐domain mutagenesis
The previously published protein crystal structure of full‐length mTGase (PDB ID 3IU0) guided us to identify contact amino acid residues between mTGase pro‐ and mTGase (Fig. 3).8 Amino acid exchange of mTGase pro‐domain residues Tyr10, Tyr14, Leu16, Asp20, Val21, Asn23, Ile24, Ala25, Leu27, Asn28, and Glu29 to alanine residues was performed using the Quick Change II site‐directed mutagenesis kit (Agilent Technologies). Mutagenesis primer and PCR conditions were designed according to manufacturer's instructions.
Fermentation and protein induction
E. coli BL21 (DE3) cells transfected with plasmid pBAD‐T7 were inoculated and grown overnight in LB broth at 37°C on a platform shaker at 250 rpm. For selection of transfected cells, Carbenicillin was added at a concentration of 100 μg/mL. A 3‐mL cell culture was added to 100 mL of TB broth, supplemented with 100 μg/mL Carbenicillin, and incubated at 37°C on a platform shaker at 250 rpm until an O.D. 600 of 1.0–1.6 was reached. The temperature was lowered to 20°C and cells were equilibrated to the lowered temperature for 30–40 min at continuous shaking. For simultaneous protein induction of genes, mPro‐TGase and 3C protease, isopropyl β‐d−1‐thiogalactopyranoside (IPTG) and L‐Arabinose were added at a final concentration of 0.4 mM and 0.2%, respectively. E. coli cells were further incubated at 20°C for 20 h. The protocol for stepwise protein induction was basically the same. Once the cells were equilibrated to 20°C, L‐Arabinose was first added at a final concentration of 0.2%. After 20 h incubation at 20°C, IPTG was added at a final concentration of 0.4 mM and protein induction was continued for various times [Fig. 1(C)/(D)].
Protein harvest and 2‐step Ni‐NTA purification
Cells were harvested by centrifugation at 6000g for 30 min. The cell pellet was frozen and stored at −80°C. The frozen cell pellet was completely thawed on ice and resuspended in 50 mL of ice‐cold 1× PBS. 30 mg of lyophilized Lysozyme (Sigma Aldrich) was dissolved in the cell suspension and incubated for 30 min on ice with careful inverting the tube every 10 min. Cells were disintegrated by sonication (Misonix; 100 W; 4000 J; 6 s/pulse; duration 1:30 min). The disintegrated cells were centrifuged at 13,000g for 30 min and the supernatant was sterile filtered through a Millex‐GP 0.2 micron syringe filter (Fisher Scientific). Imidazole, 2M stock solution at pH 8.0, and sodium chloride were added at a final concentration of 10 and 500 mM, respectively. The filtered supernatant was loaded onto a pre‐packed 5‐mL Ni‐NTA column (HisTrap FF, GE Healthcare), equilibrated in loading‐buffer (50 mM Tris‐HCl, pH 8.0, 1 M NaCl, 5 mM Imidazole) at a flow rate of 2 mL/min on an AKTA explorer system (GE Healthcare). The column was washed with 6 column volumes of wash‐buffer (50 mM Tris‐HCl, pH 8.0, 1M NaCl, 40 mM Imidazole) and protein was eluted off the column with elution‐buffer (50 mM Tris‐HCl, pH 8.0, 1M NaCl, 250 mM Imidazole). Elution fractions containing protein were pooled and concentrated to 2 mL total volume in spin concentrators (Amicon Ultra‐15, 10,000 NMWL, Millipore). As a second purification step, the protein was loaded onto a size exclusion column (Superdex 200 pg, High Load 16/600, GE Healthcare) which was equilibrated with acetate buffer (50 mM acetate, pH 5.0, 400 mM NaCl). Purified protein was flash frozen in liquid nitrogen and stored at −80°C.
SDS‐PAGE and western blot
Polyacrylamide gel electrophoresis (PAGE) was performed as described by the method of Laemmli,34 using a Novex Mini Cell system and NuPAGE 4–12% Bis‐Tris gels (Life Technologies). Each lane of the gel was loaded with 20 μL loading buffer containing equal amounts of pooled His‐tag purified protein [Fig. 1(C)], or pooled His‐tag plus SEC purified protein [Fig. 4(A)]. Protein gels were stained with Coomassie InstantBlue solution (Expedeon) according to manufacturer's instructions.
For Western blot analysis, proteins were transferred to nitrocellulose membrane using the iBlot system (Live Technologies) following manufactures instructions. The membrane was incubated in blocking solution, 50 mM Tris‐HCl, pH 8.0, 150 mM NaCl, 0.01% Tween 20, 1.5% BSA, for 2 h at 4°C. His‐tagged proteins on the membrane were detected with mouse anti‐His tag antibody (GenScript) for 1 h at RT in binding buffer, 50 mM Tris‐HCl, pH 8.0, 150 mM NaCl, 0.01% Tween20, 1% BSA. Nonbound antibody was washed away with washing buffer, 50 mM Tris‐HCl, 150 mM NaCl, 0.01% Tween20, three times 10 min on orbital shaker. Bound antibody was probed with secondary goat anti mouse alkaline phosphatase conjugated F(ab′)2 fragment (Jackson Immuno) in binding buffer for 1 h at RT. Blotted protein was detected on the membrane with 1‐Step™ NBT/BCIP solution (Thermo Scientific). The reaction was stopped by washing with ddH2O three times.
Transglutaminase activity assay
mTGase enzyme activity was measured with a commercially available kit, which is based on a previously described colorimetric hydroxamate procedure using N‐carbobenzoxy‐L‐glutaminyl‐glycine (Zedira, product number Z009, Germany).35 mTGase protein concentration was measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific) assuming a calculated protein molar extinction coefficient of 70,410 cm−1 M−1 at A280 [A280/1 cm (1 mg/mL): 1.796, assuming all Cysteines are reduces, as calculated with online available software Pepstats, Emboss Bioinformatics] . mTGase enzyme protein concentration of wild type and all mutants was adjusted to 0.125 mg/mL (3 μM) to assure activity measurements within the linear range of enzyme assay.
mTGase inhibitor binding assay
A DMSO stock solution of mTGase inhibitor (Zedira, product number C102, chemical formula not disclosed, Germany) with concentration of 5 mM was prepared. mTGase wild type control protein and mTGase pro‐domain mutants were prepared at protein concentrations of about 25 μM in 20 μL Tris‐HCl buffer at pH 8.0. About 2 μL of mTGase inhibitor stock solution were added to the mTGase protein solution and samples were incubated for 1 h at 25 or 37°C.
Mass spectroscopy
Prior to LC/MS analysis, ADCs were deglycosylated with PNGase F (NEB, cat#P0704L) under nondenaturing conditions at 37°C overnight. ADCs or TGase (500 ng) were loaded into a reverse phase column packed with a polymeric material (Michrom‐Bruker, cat# CM8/00920/00). LC/MS analysis was performed using Agilent 1100 series HPLC system, comprising binary HPLC pump, degasser, temperature controlled auto sampler, column heater and diode‐array detector, coupled to an Orbitrap Velos Pro (Thermo Scientific) mass spectrometer with electrospray ion source. The mass ranges acquired were m/z 1000–4000 and m/z 500–3000 for the detection of ADC and TGase, respectively. The resulting mass spectra were deconvoluted using ProMass software (Thermo Fisher Scientific).
Antibody conjugation
Anti‐TROP2 antibody with C‐terminal glutamine tag [Fig. 5(B)] was produced in‐house as previously described.36 For the conjugation of anti‐TROP2 antibody to AcLys‐vc‐0101,36 (unpublished data) antibody was adjusted to 5 mg/mL in buffer containing 25 mM Tris‐HCl at pH 8.0, and 150 mM NaCl, AcLys‐vc‐0101 was added in a 10‐fold molar excess over antibody and the enzymatic reaction initiated by addition of mTGase.11 Following incubation with gentle shaking at 22°C for 16 h, the ADC was purified using MabSelect SuRe (GE Healthcare) using standard procedures. The linker‐payload AcLys‐vc‐0101 was synthesized in‐house as previously described.36
HIC chromatography
Antibody‐drug conjugates with zero, one or two drugs per antibody were separated using a TSK‐GEL® Butyl‐NPR column (4.6 mm × 3.5 cm) (Tosoh Bioscience, King of Prussia, PA) on an Agilent HP 1100 HPLC (Agilent, Santa Clara, CA). The HIC method utilized a mobile phase of 1.5M ammonium sulfate, 50 mM potassium phosphate at pH 7 for Buffer A, and 50 mM potassium phosphate, 15% 2‐propanol at pH 7 for Buffer B. Using a flow rate of 0.8 mL/min, 40 μg of ADC in 0.75M ammonium sulfate was loaded onto the column and eluted with a gradient consisting of a 2.5 min hold at 0% B, followed by a 35‐min linear gradient into 100% B. The column was washed with 100% Buffer B for 2.5 min and re‐equilibrated with initial conditions for 5 minutes.
In vitro cytotoxicity assay
In vitro cytotoxicity of anti‐TROP2 ADCs was tested on the pancreas adenocarcinoma cell line BxPC3. Cells were seeded in white walled clear bottom plates at 2000 cells per well 24 h before treatment. Cells were treated with 4‐fold serially diluted antibody‐drug conjugates in triplicates. Cell viability was determined by CellTiter‐Glo® Luminescent Cell Viability Assay 96 (Promega, Madison, WI) 96 h after treatment. Relative cell viability was determined as percentage of untreated control. EC50s were calculated by GraphPad Prism 5 software.
Supporting information
Supporting Information Figure 1.
Supporting Information Figure 2.
References
- 1. Yokoyama K, Nio N, Kikuchi Y (2004) Properties and applications of microbial transglutaminase. Appl Microbiol Biotechnol 64:447–454. [DOI] [PubMed] [Google Scholar]
- 2. Strop P (2014) Versatility of microbial transglutaminase. Bioconjug Chem 25:855–862. [DOI] [PubMed] [Google Scholar]
- 3. Di Sandro A, Del Duca S, Verderio E, Hargreaves AJ, Scarpellini A, Cai G, Cresti M, Faleri C, Iorio RA, Hirose S, Furutani Y, Coutts IG, Griffin M, Bonner PL, Serafini‐Fracassini D (2010) An extracellular transglutaminase is required for apple pollen tube growth. Biochem J 429:261–271. [DOI] [PubMed] [Google Scholar]
- 4. Folk JE (1980) Transglutaminases. Annu Rev Biochem 49:517–531. [DOI] [PubMed] [Google Scholar]
- 5. Klock C, Khosla C (2012) Regulation of the activities of the mammalian transglutaminase family of enzymes. Protein Sci 21:1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ando HAM, Umeda K, Matsuura A, Nonaka M, Uchio R, Tanaka H, Motoki M (1989) Purification and characterization of a novel transglutaminase derived from microorganism. Agric Biol Chem 53:2613–2617. [Google Scholar]
- 7. Zhang D, Zhu Y, Chen J (2010) Microbial transglutaminase production: understanding the mechanism. Biotechnol Genet Eng Rev 26:205–222. [DOI] [PubMed] [Google Scholar]
- 8. Kashiwagi T, Yokoyama K, Ishikawa K, Ono K, Ejima D, Matsui H, Suzuki E (2002) Crystal structure of microbial transglutaminase from Streptoverticillium mobaraense. J Biol Chem 277:44252–44260. [DOI] [PubMed] [Google Scholar]
- 9. Liu S, Cerione RA, Clardy J (2002) Structural basis for the guanine nucleotide‐binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci U S A 99:2743–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pinkas DM, Strop P, Brunger AT, Khosla C (2007) Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol 5:e327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Strop P, Liu SH, Dorywalska M, Delaria K, Dushin RG, Tran TT, Ho WH, Farias S, Casas MG, Abdiche Y, Zhou D, Chandrasekaran R, Samain C, Loo C, Rossi A, Rickert M, Krimm S, Wong T, Chin SM, Yu J, Dilley J, Chaparro‐Riggers J, Filzen GF, O'Donnell CJ, Wang F, Myers JS, Pons J, Shelton DL, Rajpal A (2013) Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol 20:161–167. [DOI] [PubMed] [Google Scholar]
- 12. Jeger S, Zimmermann K, Blanc A, Grunberg J, Honer M, Hunziker P, Struthers H, Schibli R (2010) Site‐specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew Chem Int Ed Engl 49:9995–9997. [DOI] [PubMed] [Google Scholar]
- 13. Lhospice F, Bregeon D, Belmant C, Dennler P, Chiotellis A, Fischer E, Gauthier L, Boedec A, Rispaud H, Savard‐Chambard S, Represa A, Schneider N, Paturel C, Sapet M, Delcambre C, Ingoure S, Viaud N, Bonnafous C, Schibli R, Romagne F (2015) Site‐specific conjugation of monomethyl auristatin E to anti‐CD30 antibodies improves their pharmacokinetics and therapeutic index in rodent models. Mol Pharm 12:1863–1871. [DOI] [PubMed] [Google Scholar]
- 14. Zotzel J, Keller P, Fuchsbauer HL (2003) Transglutaminase from Streptomyces mobaraensis is activated by an endogenous metalloprotease. Eur J Biochem 270:3214–3222. [DOI] [PubMed] [Google Scholar]
- 15. Zotzel J, Pasternack R, Pelzer C, Ziegert D, Mainusch M, Fuchsbauer HL (2003) Activated transglutaminase from Streptomyces mobaraensis is processed by a tripeptidyl aminopeptidase in the final step. Eur J Biochem 270:4149–4155. [DOI] [PubMed] [Google Scholar]
- 16. Yurimoto H, Yamane M, Kikuchi Y, Matsui H, Kato N, Sakai Y (2004) The pro‐peptide of Streptomyces mobaraensis transglutaminase functions in cis and in trans to mediate efficient secretion of active enzyme from methylotrophic yeasts. Biosci Biotechnol Biochem 68:2058–2069. [DOI] [PubMed] [Google Scholar]
- 17. Liu S, Zhang D, Wang M, Cui W, Chen K, Du G, Chen J, Zhou Z (2011) The order of expression is a key factor in the production of active transglutaminase in Escherichia coli by co‐expression with its pro‐peptide. Microb Cell Fact 10:112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang CJ, Lin H, Yang X (2012) Industrial production of recombinant therapeutics in Escherichia coli and its recent advancements. J Ind Microbiol Biotechnol 39:383–399. [DOI] [PubMed] [Google Scholar]
- 19. Takehana S, Washizu K, Ando K, Koikeda S, Takeuchi K, Matsui H, Motoki M, Takagi H (1994) Chemical synthesis of the gene for microbial transglutaminase from Streptoverticillium and its expression in Escherichia coli. Biosci Biotechnol Biochem 58:88–92. [DOI] [PubMed] [Google Scholar]
- 20. Kawai M, Takehana S, Takagi H (1997) High‐level expression of the chemically synthesized gene for microbial transglutaminase from Streptoverticillium in Escherichia coli. Biosci Biotechnol Biochem 61:830–835. [DOI] [PubMed] [Google Scholar]
- 21. Yokoyama KI, Nakamura N, Seguro K, Kubota K (2000) Overproduction of microbial transglutaminase in Escherichia coli, in vitro refolding, and characterization of the refolded form. Biosci Biotechnol Biochem 64:1263–1270. [DOI] [PubMed] [Google Scholar]
- 22. Marx CKHTC, Pietzsch M (2007) Soluble expression of a pro‐transglutaminase from Streptomyces mobaraensis in Escherichia coli. Enzyme & Microb Technol 40:1543–1550. [Google Scholar]
- 23. Yang HL, Pan L, Lin Y (2009) Purification and on‐column activation of a recombinant histidine‐tagged pro‐transglutaminase after soluble expression in Escherichia coli. Biosci Biotechnol Biochem 73:2531–2534. [DOI] [PubMed] [Google Scholar]
- 24. Marx CK, Hertel TC, Pietzsch M (2008) Random mutagenesis of a recombinant microbial transglutaminase for the generation of thermostable and heat‐sensitive variants. J Biotechnol 136:156–162. [DOI] [PubMed] [Google Scholar]
- 25. Sommer C, Volk N, Pietzsch M (2011) Model based optimization of the fed‐batch production of a highly active transglutaminase variant in Escherichia coli. Protein Expr Purif 77:9–19. [DOI] [PubMed] [Google Scholar]
- 26. Khlebnikov A, Datsenko KA, Skaug T, Wanner BL, Keasling JD (2001) Homogeneous expression of the P(BAD) promoter in Escherichia coli by constitutive expression of the low‐affinity high‐capacity AraE transporter. Microbiology 147:3241–3247. [DOI] [PubMed] [Google Scholar]
- 27. Terpe K (2006) Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol 72:211–222. [DOI] [PubMed] [Google Scholar]
- 28. Studier FW (1991) Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J Mol Biol 219:37–44. [DOI] [PubMed] [Google Scholar]
- 29. Spehr V, Frahm D, Meyer TF (2000) Improvement of the T7 expression system by the use of T7 lysozyme. Gene 257:259–267. [DOI] [PubMed] [Google Scholar]
- 30. Yang MT, Chang CH, Wang JM, Wu TK, Wang YK, Chang CY, Li TT (2011) Crystal structure and inhibition studies of transglutaminase from Streptomyces mobaraense. J Biol Chem 286:7301–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pasternack R, Dorsch S, Otterbach JT, Robenek IR, Wolf S, Fuchsbauer HL (1998) Bacterial pro‐transglutaminase from Streptoverticillium mobaraense–purification, characterisation and sequence of the zymogen. Eur J Biochem 257:570–576. [DOI] [PubMed] [Google Scholar]
- 32. Strop P, Dorywalska M, Rajpal A, Shelton DL, Liu S‐H, Pons J, Dushin R. Engineered polypeptide conjugates and methods for making thereof using transglutaminase. (2012). Patent WO2012059882.
- 33. Looman AC, Bodlaender J, Comstock LJ, Eaton D, Jhurani P, de Boer HA, van Knippenberg PH (1987) Influence of the codon following the AUG initiation codon on the expression of a modified lacZ gene in Escherichia coli. Embo J 6:2489–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. [DOI] [PubMed] [Google Scholar]
- 35. Folk JE, Cole PW (1966) Mechanism of action of guinea pig liver transglutaminase. I. Purification and properties of the enzyme: identification of a functional cysteine essential for activity. J Biol Chem 241:5518–5525. [PubMed] [Google Scholar]
- 36. Dorywalska M, Strop P, Melton‐Witt JA, Hasa‐Moreno A, Farias SE, Galindo Casas M, Delaria K, Lui V, Poulsen K, Loo C, Krimm S, Bolton G, Moine L, Dushin R, Tran TT, Liu SH, Rickert M, Foletti D, Shelton DL, Pons J, Rajpal A (2015) Effect of attachment site on stability of cleavable antibody drug conjugates. Bioconjug Chem 26:650–659. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.