

Abstract
Directed evolution is a powerful tool for engineering protein function. The process of directed evolution involves iterative rounds of sequence diversification followed by assaying activity of variants and selection. The range of sequence variants and linked activities generated in the course of an evolution are a rich information source for investigating relationships between sequence and function. Key residue positions determining protein function, combinatorial contributors to activity and even potential functional mechanisms have been revealed in directed evolutions. The recent application of high throughput sequencing substantially increases the information that can be retrieved from directed evolution experiments. Combined with computational analysis this additional sequence information has allowed high‐resolution analysis of individual residue contributions to activity. These developments promise to significantly enhance the depth of insight that experimental evolution provides into mechanisms of protein function.
Keywords: directed evolution, evolution, protein function, protein sequence, next generation sequencing, high throughput sequencing, mutation
Abbreviations
- HTS
high throughput sequencing
- scFv
single chain variable fragment
Summary Statement
Understanding how proteins work will allow us to better modify and re‐design these biomolecules for use in biotechnology and medicine. Here we review the method of experimental evolution as a way to probe how the amino acid sequence of a protein determines its activity. We highlight the application of new next generation sequencing technologies, which are beginning to improve the breadth and depth of insights gained from these evolution studies.
Introduction
Directed evolution is one of the most effective strategies currently available for modifying functional activities of proteins.1, 2, 3 Although most directed evolution studies have been aimed at creating or modifying protein function, this approach can also provide significant insights into fundamental aspects of protein binding, enzyme catalysis and structure–function relationships. The key residues in the protein that determine its activity, combinatorial effects of different residues and mechanistic insights can all be revealed in a directed evolution experiment. Importantly, this does not require knowledge of the structure of the protein. Where structure is available, directed evolution provides deeper mechanistic insight and additional information. As well as contributing to understanding of fundamental aspects of protein function, the sequence–function relationships revealed by directed evolution studies have valuable practical applications. For example, the identification of activity‐determining residue positions in a protein could permit the design of more focused libraries to improve the success of evolutionary‐based engineering of that protein. Similarly, definition of stability‐determining sequence positions would be valuable for engineering more stable protein variants for therapeutic and industrial applications. Recent advances in high throughput sequencing (HTS) technologies are now set to change the quality and depth of information that we can access in an evolution and make directed evolution an even more powerful approach for probing the molecular basis of protein function and contributing to our ability to map sequence to function.
Directed Evolution
Directed evolution recapitulates features of natural evolution but on a much shorter timescale and under a selection regime designed to produce a pre‐defined outcome. Here we focus on how directed evolution can provide insights into sequence‐function relationships and therefore we only provide a brief outline of the general principle of directed evolution. Detailed discussions of directed evolution methodology are available in several excellent recent reviews, including.3, 4, 5, 6 The basic format of a directed evolution involves creating a library of random mutants encoding the protein of interest, expressing that library and selecting mutants exhibiting any enhancement towards the desired activity. These mutants are then used to generate a further mutant library, thus adding additional mutations to those already showing some desirable phenotype, and the library is again screened for improvement. Cycles of selection followed by further mutation are repeated, leading to the accumulation of combinations of mutations that result in the final phenotype3, 7 (Fig. 1). Directed evolution experiments commonly select for either quantitative improvement in an activity, such as increased binding affinity or improved enzyme stability, or a qualitative change in activity, such as the ability to catalyze conversion of a novel substrate by an enzyme. However, even an apparent qualitative change usually reflects a quantitative change in an existing activity, as evolution of a function normally requires at least some baseline level of that activity as a starting point.8, 9 Phenotypes generated in an evolution therefore generally comprise of a diverse spectrum of quantitative variations of the activity being selected for.
Figure 1.
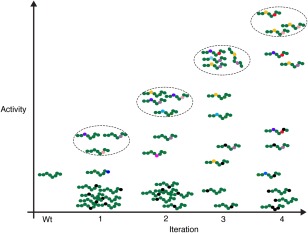
Sequence variants in a directed evolution. A schematic representation of sequence variants over four cycles of diversification and selection in a directed evolution. A simple 8‐residue sequence is depicted with each residue indicated by a coloured circle. Wild‐type residues are indicated in green and different substitute residues by other colours. At each iteration variants with the highest activity are selected (dashed ellipses) and used as a starting population for the next round of mutagenesis allowing additional mutations and further re‐sampling of substituted positions. Some mutations (black circles) will be deleterious and decrease or ablate activity. As the evolution progresses positions 3 and 6 are revealed as key activity‐determining residues. For simplicity and clarity only a very small fraction of possible variants are shown.
Practically, directed evolution experiments require some way of generating a mutant DNA or RNA library, together with an expression and assay system that permits selection for the activity of interest, whilst retaining the link between the activity and the nucleotide sequence encoding it. Error prone PCR, DNA shuffling and various gene synthesis strategies are among the methods commonly used to generate mutant libraries for evolution.2, 3, 5 Expression and assay/selection methods that provide sufficient throughput to interrogate these libraries and retain the link to encoding nucleotides, include display technologies such as phage and ribosome display10, 11 and cell surface display systems, for example yeast display that permit expression and selection of some complex proteins.12, 13 Other systems include in vitro compartmentalization which employs aqueous droplets in oil for expression and can be used together with a range of different selection methods.14, 15 Some directed evolution technologies combine mutagenesis and expression systems, by utilizing for instance immune B cells to perform in‐cell mutagenesis and expression16, 17 for facile evolution of complex mammalian proteins.18
The power of directed evolution to uncover sequence‐function relationships and mechanistic insights is rooted in the range of sequence variants and their linked activity phenotypes that are explored in the course of the evolution. Ideally, to maximize the probability of identifying sequences with improved activity each amino acid position in the sequence is individually substituted with all alternative residues. Of course, in practice experiments usually fall short of testing a full mutational spectrum at every position. However, in a well‐designed evolution most of the sequence positions that the experiment is aimed at exploring are sampled with at least some degree of amino acid diversity. Any sequence with improved activity is retained during selection and at the next iteration the sequence is re‐scanned for additional positions where substitution can improve activity. Even positions producing desirable activities are re‐sampled with alternative residues as the evolution progresses, allowing combinations of residue positions to be explored and optimized (Fig. 1). Within a directed evolution experiment there is therefore information about the effect that each sequence position has on that protein's function, how the nature of the residue at that position affects function, and the way residues work in combination to modulate function. Such information can provide deep insights into how sequence determines functional activity for a protein.
Identification of Activity‐Modulating Residues
Although most directed evolution studies have focused on modifying protein activity, these evolutions have also often revealed key activity‐determining residues. For such studies key sites of mutation are usually revealed by comparing evolved variants with wild‐type sequence. Accumulation of mutations at specific residue positions associated with the changed activity implicates these positions as important for that activity. Activity‐changing mutations may be revealed as positions where the wild‐type residue is substituted by a number of different residues across the variants, or it may be as a position where all or many of the variants have the same residue substituted in place of the one in the wild‐type protein. There are many diverse examples of key functional residues being revealed by directed evolution, including identification of the residues determining thermal stability and catalytic activity of alkaline phosphatase from the cryophile Antarctic strain TAB5,19 residue positions mediating the binding of T cell receptors to toxic shock syndrome toxin‐1,20 and residues involved in the catalytic activity of serum paraoxonases.21 Identification of key activity‐determining residues by directed evolution can allow more targeted subsequent investigation of sequence‐function relationships. For example combining previous findings from directed evolution21 with structural information and computer simulations has provided fundamental insights into mechanisms regulating activity and stability of the lipophilic lactonase paraoxonase‐1, with important wider implications for other membrane‐associated enzymes.22
Whilst directed evolution experiments reveal key activity‐determining residues there are some instances where additional non activity‐modulating positions also appear as mutated positions within the selected population, giving rise to a false positive background. An example of this is the six residue positions initially identified during a directed evolution of a lipase from Pseudomonas aeruginosa for enantioselectivity.23 Subsequent theoretical and experimental analysis of these mutations revealed that just two of these residue positions in fact contribute to the change in enantioselectivity.24, 25 Testing the effects of each of the substitutions in the finally selected variants is one way by which false positives can be identified and discounted. However, as discussed below, the availability of more comprehensive sequencing data allows true activity‐determining positions to be discriminated from false positives.
In any directed evolution there are usually several different parameters that can be selected for. For an enzyme these might include catalytic activity, substrate binding, or stability, and for a binding protein on‐rate, off‐rate, affinity, or selectivity. Residues found to modulate one aspect of an activity may or may not be relevant to other aspects of that activity. Residues modifying binding off‐rates, for instance, would be expected to influence affinity,26 if they do not proportionally affect on‐rate. In contrast, because the relationship between binding affinity and selectivity is complex, residue positions affecting binding affinity for one partner could have widely differing effects on binding selectivity depending on the protein. This is illustrated in a study with calmodulin where modifying residues at the calmodulin binding interface to increase affinity for CaMKII results in both increased affinity for CamKII but dramatically decreased affinity for another calmodulin partner, calcineurin, thus increasing affinity for one partner and enhancing selectivity.27 Conversely, directed evolution of the quorum sensing protein LuxR (an acyl‐homoserine lactone‐dependent transcriptional activator) produced variants with increased affinity for cognate and some non‐cognate acyl‐homoserine lactones, simultaneously increasing affinity and decreasing selectivity.28 Residues identified as activity‐determining in an evolution experiment therefore are relevant to the specific activity selected for, but not necessarily to other, albeit related, activities.
Combinatorial Contributions to Function
Multiple residues commonly work in combination to contribute both directly and indirectly to protein activity. Combinations of key residues can participate directly in function by contributing to binding energy or catalytic activity, or indirectly by influencing either the access to or the conformation of the functional interface. Where two or more residues contribute to activity they may do this independently of each other or they may work together such that their combined effect on activity is greater than the sum of each of the individual contributions.29 Such non‐additive combinatorial effects, often called epistatic, can be synergistic or antagonistic.29 For example, mutational analysis of binding between the monoclonal antibody mAb164 and the TrpB2 subunit of E. coli tryptophan synthase reveals that the combination of residues V276 and K283 together contribute more binding energy than the sum of binding energies contributed by each of these residues individually.30 In another study, directed evolution was used to modify the binding specificity of a receptor and identified a phenylalanine at the center of the binding interface and an arginine and histidine pair at the periphery as important in selective binding to one of the receptor's ligands.18 Modifying the phenylalanine alone did not affect binding to any ligand and modifying the arginine and histidine completely abolished binding to all ligands, however, in combination the changes caused selective binding to one ligand. Understanding the mechanism of a binding interaction or catalytic activity therefore requires not only definition of the key binding positions but also how these positions work together in determining functional activity.
Where structural data are available it is possible to probe residues at the functional interface for their contribution to catalytic activity or binding. One method frequently employed for this purpose is alanine scanning, which entails replacing each residue under consideration with alanine and testing for complete or partial loss of activity indicating that the residue position contributes to activity.31 In the simplest case, where several interface residues independently contribute binding energy to an interaction or catalytic activity, and there is no synergy or antagonism between positions, alanine scanning can reveal the contribution of each residue.31 However, even with structural information alanine scanning can be very difficult where contributions to activity are non‐additive between contributing residues, and synergy or antagonism exists between positions. In such cases the number of different alanine mutants that have to be expressed, purified and assayed to test all possible combinations of residues can become unmanageable.
In contrast to alanine scanning, directed evolution reveals both additive and non‐additive contributors to activity. This ability is afforded by the iterative process of mutation and selection that acts to screen large numbers of different combinations of mutations resulting in accumulation of the mutations that act together to change activity. Residue positions contributing to activity are usually inferred by inspection of wild‐type and evolved sequences. Where there are two or more positions mutated in the evolved sequences this indicates these combinations of positions are activity‐determining, though it does not reveal whether the combinations contribute independently or in a non‐additive manner. However, during the evolution a wide range of different combinations of mutants are produced and tested through rounds of selection and further diversification. Such experiments therefore contain information on which combinations of sequence positions affect activity and any potential relationships between residue positions. As discussed below, the ability to sequence large numbers of variants, both intermediates generated during the evolution and final evolved variants, together with computational analysis tools will help to better identify combinatorial contributors to function and provide more information about how these residues work together.
Improving the Ability of Directed Evolution to Probe Sequence‐Function Relationships
A well designed directed evolution will test the activity and then select from millions of different sequence variants encompassing a wide range of substitutions at many residue positions in the sequence and many different combinations of mutated residues within sequences. This provides an unrivalled opportunity to map extensive sequence variations to functional activities. Due to sequencing limitations only a few variants, usually from later stages in the evolution process, have commonly been sequenced in the majority of directed evolution experiments to date. In such studies, mutations occurring most often in the selected variants that have been sequenced are assumed to be the key activity‐determining mutations, and these are often confirmed experimentally. The availability of HTS technologies now opens up the possibility of sequencing any number of variants selected at each iteration of the evolution to substantially enhance the depth and breadth of insight directed evolution can provide into the basis of protein function. Specifically, such data allow more accurate identification of the true activity‐determining sequence positions, investigation of covariation between residue positions involved in combinatorial contributions to activity, and helps in revealing mechanistic insight (Fig. 2).
Figure 2.
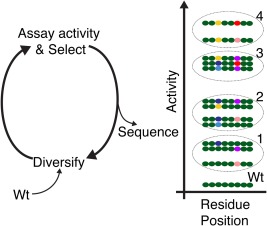
Application of HTS to directed evolution reveals sequence‐function relationships. Schematic representation of four cycles of target gene diversification, assay and selection followed by sequencing of variant populations. The dashed ellipses denote selections at each indicated iteration. Simplified sequences are shown indicating the combination of residue positions 3 and 6 emerging as activity–determining positions. Different combinations of substitutions at positions 3 and 6 produce different activities and these positions can contribute synergistically to activity. After identifying the key residue positions, sequence data can be re‐interrogated for relationships between activity and the specific combinations of substitutions at these positions. In addition, how the type of amino acid at the key positions affects activity can be retrieved, and this has the potential to suggest a functional mechanism. Wild‐type residues are indicated in green and different substitute residues by other colours. For simplicity and clarity only a very small fraction of possible variants are shown.
HTS is being applied to a growing number of mutational studies. An early example was next generation sequencing of variants selected for specific binding from a single chain variable fragment (scFv) library displayed on phage.32This allowed the evolution of scFv's to be followed as the library was enriched for binders and revealed the most abundant mutants as positive binders.32 In another study, HTS was applied to peptides selected from phage‐display libraries for binding to 22 different evolved PDZ domains, in order to identify specificity determinants in PDZ‐peptide binding.33 An important development in the HTS approach to mutational studies was introduced by Fowler et al.34 who sequenced not just the selected variants from a mutational screen but also the original starting library. This provides a means for correcting apparent preferences in mutation in the selected population for any biases in the starting population. In Fowlers study a mutant library of the WW domain of human YAP65 was created and displayed on phage allowing selection of variants that retain their ability to bind the WW domain cognate peptide ligand. HTS of starting and selected populations and analysis for enrichment or loss of variants generated a map of mutational tolerance of each residue position in the domain.34 The approach of HTS and comparative analysis in mutational studies of protein function has been developed and extended in the last few years35, 36, 37 and recently termed deep mutational scanning.38
Whilst inference of key functional residues by comparing wild‐type and selected sequences has been successful in numerous directed evolution studies, the availability of more comprehensive sequencing data significantly enhances the ability of such studies to identify the true activity‐determining residue positions. Sequence positions contributing to function only tolerate a limited range of residue types, dictated by the functional mechanism. Measuring the ability of each sequence position to accommodate alternative amino acids and still retain or improve activity therefore is a powerful way to identify positions important for function. These data can be accessed by sequencing of the variants in the starting library and those variants selected for a maintained or improved activity at each iteration of the evolution, or even selected and final iterations. These HTS data can be analysed for the frequency of different substitutions at each position in starting and selected populations. Sites that will tolerate only a limited range of different residues whilst retaining/improving activity will have the lowest ratio of substitution frequency in selected versus starting population, indicating these positions to be important for activity. This type of approach has been used to identify important functional residues in families of naturally evolved proteins39 and, latterly, in analysis of deep mutational scanning data34, 38. A recent directed evolution aimed at exploring the residues mediating stability and folding of human IgG‐Fc illustrates the power of HTS and quantitative analysis methods.40 By employing careful experimental design to discriminate the function under consideration from binding effects, this study used the ability of residue positions to tolerate substitution in order to generate a high‐resolution map of the involvement of each residue in determining stability and folding across a whole protein domain.41
Quantitative analysis of sequencing data from evolutions also helps discriminate true activity‐determining residue positions from false‐positives. This is possible as neutral substitutions would not be specifically enriched in selected populations to the same extent as true activity‐determining substitutions. The frequency of substitution at neutral sites therefore do not decrease in selected populations to the same extent as that seen with true activity‐determining sites, providing a quantitative means to discriminate true‐ from false‐positives.
Most analyses of HTS applied to directed evolution and other mutational studies have focused on identification of key activity‐determining residues and mapping mutational tolerance. However, this sequencing data also provides possibilities for uncovering epistatic interactions between residues and even mechanistic insight (Fig. 2). Analysis of covariation of residues between different sequences as well as within a sequence is already performed in evolutionarily related protein families and provides a way to identify functionally important residues acting in combination to influence stability, structure, binding and catalytic activities.42, 43, 44 Substitutions at functionally interacting residues are mutually constrained and this covariation pattern is evident in sequence variants.45, 46, 47, 48 Covariation analysis focused on the activity‐determining residues at each iteration of a directed evolution experiment could therefore be used to uncover mutually constrained co‐evolving residues within sequences and permit analysis of relationships between changes at each of the contributing residues and functional activities. There are a number of computational approaches used for detecting covarying residues from groups of related sequences, for example mutual information, statistical coupling analysis and direct coupling analysis scoring metrics,44, 49 which could be applied to data from experimental evolutions.
It is also possible that with more comprehensive sequencing data and appropriate analysis directed evolution experiments could more readily reveal mechanistic insights. The change in activity of a protein as it progresses from wild‐type to finally selected activity in a directed evolution can encompass a substantial range of functional variants. For example, the affinity of a protein for a partner can change over several orders of magnitude in a directed evolution for improved binding.50, 51 The availability of a range of sequential phenotypic activities can provide an opportunity to explore how the physicochemical nature (size, charge etc) of the various substitutions at each of the residue positions that have been identified as being important correlate with activity. This will require analysis of the sequence data from the evolution for how amino acid properties at the (finally identified) key positions correlate with activity. Such relationships will be valuable for constructing mechanistic models for further testing.
Conclusions
Whilst directed evolution is undoubtedly a powerful technology for generating modified protein activities, it also has enormous potential for revealing deep insights into the mechanisms by which proteins work. Interrogation of protein sequence and function using directed evolution can provide insight not available using other biochemical, biophysical or structural approaches.
Indeed, even relatively simple laboratory evolutions have already revealed key residues important in binding, combinatorial contributions to binding and have provided mechanistic models for testing for a number of proteins. However, there is an enormous information content inherent in the range of mutant variants and functional activities generated in a directed evolution experiment. Access to this information has previously been restricted by factors such as limitations in sequencing ability. High‐throughput sequencing technologies and coupling of sequences to functional activities now allows researchers to use directed evolution directly to probe sequence‐function relationships and functional mechanisms more comprehensively and to a much deeper level than previously possible. In order to maximize the utility of the data generated in such experiments it will be necessary to adapt and develop computational tools for analysis of relationships between residue positions, residue properties and functional activities.
Acknowledgment
The authors thank the British Heart Foundation (PG/13/43/30312) for support.
References
- 1. Dougherty MJ, Arnold FH (2009) Directed evolution: new parts and optimized function. Curr Opin Biotechnol 20:486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bershtein S, Tawfik DS (2008) Advances in laboratory evolution of enzymes. Curr Opin Chem Biol 12:151–158. [DOI] [PubMed] [Google Scholar]
- 3. Jäckel C, Kast P, Hilvert D (2008) Protein design by directed evolution. Ann Rev Biophys 37:153–173. [DOI] [PubMed] [Google Scholar]
- 4. Lane MD, Seelig B (2014) Advances in the directed evolution of proteins. Curr Opin Chem Biol 22C:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Currin A, Swainston N, Day PJ, Kell DB (2015) Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently. Chem Soc Rev 44:1172–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Packer MS, Liu DR (2015) Methods for the directed evolution of proteins. Nat Rev Genet 16:379–394. [DOI] [PubMed] [Google Scholar]
- 7. Tracewell CA, Arnold FH (2009) Directed enzyme evolution: climbing fitness peaks one amino acid at a time. Curr Opin Chem Biol 13:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arnold FH (1996) Directed evolution: creating biocatalysts for the future. Chem Eng Sci 51:5091–5102. [Google Scholar]
- 9. Romero PA, Arnold FH (2009) Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol 10:866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith GP (1991) Surface presentation of protein epitopes using bacteriophage expression systems. Curr Opin Biotechnol 2:668–673. [DOI] [PubMed] [Google Scholar]
- 11. Hanes J, Pluckthun A (1997) In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci U S A 94:4937–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boder ET, Wittrup KD (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol 15:553–557. [DOI] [PubMed] [Google Scholar]
- 13. Georgiou G, Stathopoulos C, Daugherty PS, Nayak AR, Iverson BL Curtiss R, 3rd (1997) Display of heterologous proteins on the surface of microorganisms: from the screening of combinatorial libraries to live recombinant vaccines. Nat Biotechnol 15:29–34. [DOI] [PubMed] [Google Scholar]
- 14. Tawfik DS, Griffiths AD (1998) Man‐made cell‐like compartments for molecular evolution. Nat Biotechnol 16:652–656. [DOI] [PubMed] [Google Scholar]
- 15. Kaltenbach M, Hollfelder F (2012) Snap display: in vitro protein evolution in microdroplets. Methods Mol Biol 805:101–111. [DOI] [PubMed] [Google Scholar]
- 16. Wang CL, Yang DC, Wabl M (2004) Directed molecular evolution by somatic hypermutation. Protein Eng Des Sel 17:659–664. [DOI] [PubMed] [Google Scholar]
- 17. Wang L, Jackson WC, Steinbach PA, Tsien RY (2004) Evolution of new nonantibody proteins via iterative somatic hypermutation. Proc Natl Acad Sci USA 101:16745–16749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brindle NP, Sale JE, Arakawa H, Buerstedde JM, Nuamchit T, Sharma S, Steele KH (2013) Directed evolution of an angiopoietin‐2 ligand trap by somatic hypermutation and cell surface display. J Biol Chem 288:33205–33212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koutsioulis D, Wang E, Tzanodaskalaki M, Nikiforaki D, Deli A, Feller G, Heikinheimo P, Bouriotis V (2008) Directed evolution on the cold adapted properties of tab5 alkaline phosphatase. Protein Eng Des Sel 21:319–327. [DOI] [PubMed] [Google Scholar]
- 20. Buonpane RA, Moza B, Sundberg EJ, Kranz DM (2005) Characterization of t cell receptors engineered for high affinity against toxic shock syndrome toxin‐1. J Mol Biol 353:308–321. [DOI] [PubMed] [Google Scholar]
- 21. Harel M, Aharoni A, Gaidukov L, Brumshtein B, Khersonsky O, Meged R, Dvir H, Ravelli RB, McCarthy A, Toker L, Silman I, Sussman JL, Tawfik DS (2004) Structure and evolution of the serum paraoxonase family of detoxifying and anti‐atherosclerotic enzymes. Nat Struct Mol Biol 11:412–419. [DOI] [PubMed] [Google Scholar]
- 22. Ben‐David M, Sussman JL, Maxwell CI, Szeler K, Kamerlin SCL, Tawfik DS (2015) Catalytic stimulation by restrained active‐site floppiness—the case of high density lipoprotein‐bound serum paraoxonase‐1. J Mol Biol 427:1359–1374. [DOI] [PubMed] [Google Scholar]
- 23. Reetz MT, Wilensek S, Zha D, Jaeger K‐E (2001) Directed evolution of an enantioselective enzyme through combinatorial multiple‐cassette mutagenesis. Angew Chem Int Ed Engl 40:3589–3591. [DOI] [PubMed] [Google Scholar]
- 24. Reetz MT, Puls M, Carballeira JD, Vogel A, Jaeger K‐E, Eggert T, Thiel W, Bocola M, Otte N (2007) Learning from directed evolution: further lessons from theoretical investigations into cooperative mutations in lipase enantioselectivity. Chembiochem 8:106–112. [DOI] [PubMed] [Google Scholar]
- 25. Bocola M, Otte N, Jaeger K‐E, Reetz MT, Thiel W (2004) Learning from directed evolution: theoretical investigations into cooperative mutations in lipase enantioselectivity. Chembiochem 5:214–223. [DOI] [PubMed] [Google Scholar]
- 26. Graff CP, Chester K, Begent R, Wittrup KD (2004) Directed evolution of an anti‐carcinoembryonic antigen scfv with a 4‐day monovalent dissociation half‐time at 37 degrees c. Protein Eng Des Sel 17:293–304. [DOI] [PubMed] [Google Scholar]
- 27. Yosef E, Politi R, Choi MH, Shifman JM (2009) Computational design of calmodulin mutants with up to 900‐fold increase in binding specificity. J Mol Biol 385:1470–1480. [DOI] [PubMed] [Google Scholar]
- 28. Collins CH, Arnold FH, Leadbetter JR (2005) Directed evolution of vibrio fischeri luxr for increased sensitivity to a broad spectrum of acyl‐homoserine lactones. Mol Microbiol 55:712–723. [DOI] [PubMed] [Google Scholar]
- 29. Reetz MT (2013) The importance of additive and non‐additive mutational effects in protein engineering. Angew Chem Int Ed Engl 52:2658–2666. [DOI] [PubMed] [Google Scholar]
- 30. Rondard P, Bedouelle H (1998) A mutational approach shows similar mechanisms of recognition for the isolated and integrated versions of a protein epitope. J Biol Chem 273:34753–34759. [DOI] [PubMed] [Google Scholar]
- 31. Cunningham BC, Wells JA (1989) High‐resolution epitope mapping of hgh‐receptor interactions by alanine‐scanning mutagenesis. Science 244:1081–1085. [DOI] [PubMed] [Google Scholar]
- 32. Ravn U, Gueneau F, Baerlocher L, Osteras M, Desmurs M, Malinge P, Magistrelli G, Farinelli L, Kosco‐Vilbois MH, Fischer N (2010) By‐passing in vitro screening–next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res 38:e193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ernst A, Gfeller D, Kan Z, Seshagiri S, Kim PM, Bader GD, Sidhu SS (2010) Coevolution of pdz domain‐ligand interactions analyzed by high‐throughput phage display and deep sequencing. Mol Biosyst 6:1782–1790. [DOI] [PubMed] [Google Scholar]
- 34. Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D, Fields S (2010) High‐resolution mapping of protein sequence‐function relationships. Nat Methods 7:741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hietpas RT, Jensen JD, Bolon DN (2011) Experimental illumination of a fitness landscape. Proc Natl Acad Sci U S A 108:7896–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Araya CL, Fowler DM, Chen W, Muniez I, Kelly JW, Fields S (2012) A fundamental protein property, thermodynamic stability, revealed solely from large‐scale measurements of protein function. Proc Natl Acad Sci U S A 109:16858–16863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Whitehead TA, Chevalier A, Song Y, Dreyfus C, Fleishman SJ, De Mattos C, Myers CA, Kamisetty H, Blair P, Wilson IA, Baker D (2012) Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol 30:543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fowler DM, Fields S (2014) Deep mutational scanning: a new style of protein science. Nat Methods 11:801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Valdar WS (2002) Scoring residue conservation. Proteins 48:227–241. [DOI] [PubMed] [Google Scholar]
- 40. Traxlmayr MW, Hasenhindl C, Hackl M, Stadlmayr G, Rybka JD, Borth N, Grillari J, Ruker F, Obinger C (2012) Construction of a stability landscape of the ch3 domain of human igg1 by combining directed evolution with high throughput sequencing. J Mol Biol 423:397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Traxlmayr MW, Obinger C (2012) Directed evolution of proteins for increased stability and expression using yeast display. Arch Biochem Biophys 526:174–180. [DOI] [PubMed] [Google Scholar]
- 42. Atchley WR, Wollenberg KR, Fitch WM, Terhalle W, Dress AW (2000) Correlations among amino acid sites in bhlh protein domains: an information theoretic analysis. Mol Biol E 17:164–178. [DOI] [PubMed] [Google Scholar]
- 43. Gloor GB, Martin LC, Wahl LM, Dunn SD (2005) Mutual information in protein multiple sequence alignments reveals two classes of coevolving positions. Biochemistry 44:7156–7165. [DOI] [PubMed] [Google Scholar]
- 44. Ashenberg O, Laub MT (2013) Using analyses of amino acid coevolution to understand protein structure and function. Methods Enzymol 523:191–212. [DOI] [PubMed] [Google Scholar]
- 45. Vernet T, Tessier DC, Khouri HE, Altschuh D (1992) Correlation of co‐ordinated amino acid changes at the two‐domain interface of cysteine proteases with protein stability. J Mol Biol 224:501–509. [DOI] [PubMed] [Google Scholar]
- 46. Gobel U, Sander C, Schneider R, Valencia A (1994) Correlated mutations and residue contacts in proteins. Proteins 18:309–317. [DOI] [PubMed] [Google Scholar]
- 47. Lockless SW, Ranganathan R (1999) Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286:295–299. [DOI] [PubMed] [Google Scholar]
- 48. Mateu MG, Fersht AR (1999) Mutually compensatory mutations during evolution of the tetramerization domain of tumor suppressor p53 lead to impaired hetero‐oligomerization. Proc Natl Acad Sci U S A 96:3595–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. de Juan D, Pazos F, Valencia A (2013) Emerging methods in protein co‐evolution. Nat Rev Genet 14:249–261. [DOI] [PubMed] [Google Scholar]
- 50. Boder ET, Midelfort KS, Wittrup KD (2000) Directed evolution of antibody fragments with monovalent femtomolar antigen‐binding affinity. Proc Natl Acad Sci U S A 97:10701–10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li Y, Moysey R, Molloy PE, Vuidepot A‐L, Mahon T, Baston E, Dunn S, Liddy N, Jacob J, Jakobsen BK, Boulter JM (2005) Directed evolution of human t‐cell receptors with picomolar affinities by phage display. Nat Biotechnol 23:349–354. [DOI] [PubMed] [Google Scholar]