

Abstract
Even a single Gly substitution in the triple helix domain of collagen leads to pathological conditions while natural interruptions are suggested to play important functional roles. Two peptides—one mimicking a pathological Gly–Ser substitution (ERSEQ) and the other one modeling a similar natural interruption sequence (DRSER)—are designed to facilitate the comparison for elucidating the molecular basis of their different biological roles. CD and NMR investigation of peptide ERSEQ indicates a reduction of the thermal stability and disruption of hydrogen bonding at the Ser mutation site, providing a structural basis of the OI disease resulting from the Gly–Ser mutation in the highly charged RGE environment. Both CD and NMR real‐time folding results indicate that peptide ERSEQ displays a comparatively slower folding rate than peptide DRSER, suggesting that the Gly–Ser mutation may lead to a larger interference in folding than the natural interruption in a similar RSE context. Our studies suggest that unlike the rigid GPO environment, the abundant R(K)GE(D) motif may provide a more flexible sequence environment that better accommodates mutations as well as interruptions, while the electrostatic interactions contribute to its stability. These results shed insight into the molecular features of the highly charged motif and may aid the design of collagen biomimetic peptides containing important biological sites.
Keywords: NMR, CD, collagen, interruption, mutation
Introduction
Collagen plays a ubiquitous role as a biological scaffold in connective tissues such as skin, bone, tendon, ligament, cartilage, and blood vessels. The unique functional role of collagen relies on its characteristic triple helix structure.1, 2 The close packing of three polypeptide chains requires Gly as every third residue, generating the repetitive (Gly–X–Y)n amino acid sequence pattern. Twenty‐eight different types of collagens have been discovered and classified as fibril‐forming collagens (Type I, II, III, V, and XI) and nonfibrillar collagens.3, 4 The key Gly is absolutely conserved along the ∼1000 residue triple helix domain in fibrillar collagens, and even the substitution of a single Gly by a bulkier residue leads to pathological conditions.5, 6, 7 In contrast, more than 350 interruptions in the (Gly–X–Y)n pattern naturally occur in nonfibrillar collagens, and they have shown critical roles in collagen binding and degradation.8, 9, 10 It has attracted extensive attention that both Gly substitutions and natural interruptions disrupt the repetitive (Gly–X–Y)n pattern in collagen, while they cause totally different biological consequences.11, 12
Since collagen is too large to be amenable for biophysical characterizations, triple‐helical peptides have been developed as a convenient approach to investigate the structural and functional consequences of sequence variations in collagen.13, 14, 15, 16, 17, 18, 19, 20 Triple‐helical peptide studies have shown that the identity of the residue replacing Gly seriously affect the degree of peptide destabilization, and the order of stability loss was Ala ≤ Ser < Cys < Arg < Val < Glu ≤ Asp.19 X‐ray and NMR studies of peptide models of Gly–Ala and Gly–Ser mutations have shown localized perturbation in dihedral angles and hydrogen bonding at the mutation sites.14, 18 It was also observed that introduction of Gly mutations into both the (POG)10 and T1‐865 environments resulted in significant delays in the folding time.20 These peptide studies have greatly advanced our understanding of the roles of Gly mutations in triple helix stability, conformation, and folding. Meanwhile, peptide models of natural interruptions have also displayed localized alterations in conformation.8, 21, 22 However, the molecular basis underlying the contradictory functional consequences of Gly mutations and natural interruptions is still poorly understood.
Triple helical peptide studies have suggested that the local sequence environment of a Gly substitution is considered as a critical factor in defining the molecular features of the peptide.20, 23, 24 A more rigid Gly–Pro–Hyp sequence environment of a Gly–Ala substitution has been correlated with a larger drop in thermal stability (ΔT m) than a more flexible sequence.20 Host guest peptide studies have revealed that Arg, which is predominantly found in the Y position in natural collagen, forms the most stable triple helix just after Hyp.25 In addition, negatively charged amino acids in the X position have shown strong propensity to form stable triple helix structure like Pro.25 The high occurrence of charged amino acids in collagen may provide another stabilizing force of the triple helix in addition to the well‐known stabilizing Gly–Pro–Hyp triplet. However, it is barely known how a pathological mutation or a natural interruption is accommodated in a highly charged sequence environment.
Peptide models of Gly substitutions and natural interruptions suitable for comparison are needed in order to elucidate their structural and dynamic differences. The Gly–Ser mutations represent the most abundant Gly substitutions in the α(1) and α(2) chains of Type I collagen, accounting for ∼40% for all the observed OI cases resulting from Gly substitutions.26 A Gly–Ser mutation with a sequence Gly–AA1–AA2–Ser–AA4–AA5–Gly can be considered as a G5G interruption (Gly–AA1–AA2–AA3–AA4–AA5–Gly) with a central Ser at the AA3 position in terms of local amino acid sequence context. Examination of the Gly–Ser mutations and G5G natural interruptions in collagen has located two sites with similar local sequence environment: a lethal Gly–Ser mutation (ERSEQ) and a natural interruption (DRSER). Two triple helical peptides are designed to mimic these two sites, and their features such as stability, conformation, and folding are investigated by CD and NMR techniques. The two peptides show comparable stability and triple helix content as well as similar disrupted patterns in dihedral angles and hydrogen bonding. Peptide ERSEQ displays a comparatively slower folding rate than peptide DRSER, suggesting that the Gly–Ser mutation may lead to a larger interference in folding than the natural interruption in a similar RSE context. These results suggest a molecular basis of different biological roles of natural interruptions and Gly–Ser substitutions in a highly charged R(K)GE(D) sequence environment.
Results
Sequence analysis
The content of charged amino acids is compared between fibrillar and nonfibrillar collagens (Supporting Information, Fig. S1). Fibrillar collagens contain ∼33% Pro at both X and Y positions, while charged amino acids account for the second largest group with ∼26% at the X position (∼70% of which is negatively charged) and ∼28% at the Y position (∼76% of which is positively charged), respectively. For nonfibrillar collagens, they contain ∼28% and ∼29% of charged amino acids at the X and Y positions, respectively. Similarly, the charged amino acids are mainly negative at the X position and positive at the Y position. There are six different combinations of positively charged Y residue and negatively charged X residue for the YGX sequence motif: RGE, RGD, KGE, KGD, HGE, and HGD. The frequency of these motifs are calculated for fibrillar (∼7.8%) as well as nonfibrillar collagens (∼9.8%), while they are predominantly R(K)GE(D) motifs (Supporting Information, Fig. S2). To conclude, both fibrillar and nonfibrillar collagens contain >1/4 charged amino acids at the X and Y positions; and particularly, both of them have a high frequency of highly charged R(K)GE(D) motifs, which have been suggested to play a stabilizing role for triple helix structure.27
Peptide design
Examination of the Gly–Ser mutations and G5G natural interruptions in collagen have located two sites with the same RSE context, where the Gly in the RGE motif is replaced by Ser. Peptide ERSEQ (with amino acid sequence: (GPO)6–GERSEQ–(GPO)6), mutation site underlined) was designed to model a lethal Gly–Ser mutation at site 454 in the α(1) chain of Type I collagen (Table 1). Peptide DRSER (with amino acid sequence: (GPO)6–GDRSER–(GPO)6), interruption site underlined) was used to model a G5G interruption at site 491–495 in the α(1) chain of Type XXIII collagen, a nonfibrillar transmembranous collagen with interrupted helices (Table 1). Peptides ERSEQ and DRSER share a very similar amino acid content, which is highly charged. The major difference of the two peptides is the change of a charged amino acid Arg in peptide DRSER to a polar residue Gln in peptide ERSEQ. Analysis of the G5G interruptions (Gly–AA1–AA2–AA3–AA4–AA5–Gly) suggested that the AA5 position contains mostly charged amino acids (∼33%), which is significantly different from that of Gly–Ser mutations (p < 0.05) (Supporting Information, Fig. S3). The two peptides exemplify the change of a charged Arg at the natural interruption site to a neutral Gln nearby a Ser mutation site from collagen sequences in a highly charged context. Compared with Gln, the charge, size, and side‐chain interaction may make Arg have a different effect on the stability, conformation, and folding of peptides.
Table 1.
Peptides Modeling a Ser Mutation and a Similar Natural Interruption, Showing Thermal Transition Temperature (T m) and Mean Residue Ellipticity at 225 nm (MRE225) at pH 7.0 and pH 3.0
| Peptide name | Peptide sequencea | pH | T m (°C) | MRE225 (deg cm2 dmol−1) |
|---|---|---|---|---|
| ERSEQ | Ac‐GPO‐GPO‐GPO‐GPO‐GPO‐GPO‐GER‐SEQ‐GPO‐GPO‐GPO‐GPO‐GPO‐GPO‐CONH2 | 7.0 | 46.5 (33.0)b | 4221 |
| 3.0 | 40.8 | 3410 | ||
| DRSER | Ac‐GPO‐GPO‐GPO‐GPO‐GPO‐GPO‐GDR‐SER‐GPO‐GPO‐GPO‐GPO‐GPO‐GPO‐CONH2 | 7.0 | 46.0 | 3539 |
| 3.0 | 41.7 | 3284 |
15N‐labeled residues are underlined.
Peptide ERSEQ shows two thermal transitions: one major transition with T m = 46.5°C and one minor transition with T m = 33.0°C.
Thermal stability
Though peptides ERSEQ and DRSER modeled totally different sites in collagen, they showed comparatively similar stabilities. At pH 7, peptide ERSEQ displayed a high molecular residue ellipticity signal at 225 nm (MRE225 = 4221 deg cm2 dmol−1) [Fig. 1(A) and Table 1]. Monitoring the ellipticity at 225 nm with increasing temperature showed two thermal transitions: one major transition with T m = 46.5°C and one minor transition with T m = 33.0 C [Fig. 1(B,C)]. It may suggest that besides the fully folded triple helix structure, peptide ERSEQ also formed a lower stability species. Similar weak species has been observed for peptide T1‐865 with a Gly–Ser mutation, which may indicate the difficulty of propagation through the mutation site.20 Peptide DRSER, in contrast, displayed a single sharp thermal transition with a melting temperature of 46.0°C [Fig. 1(D–F) and Table 1]. These results suggested that though the Ser mutation possesses similarly high stability as the interruption sequence within a similar RSE context at pH 7, the existence of a lower stability species may reflect a folding difficulty encountered by the Ser mutation but not the interruption sequence.
Figure 1.
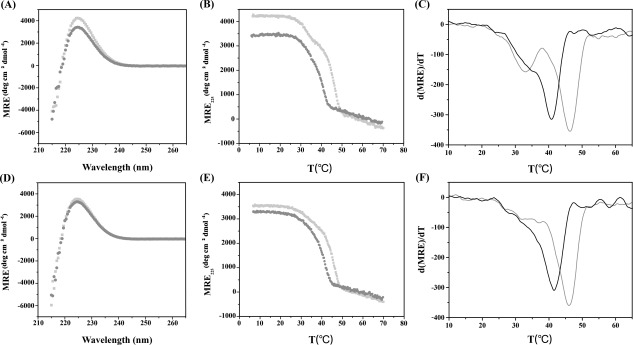
CD wavelength scans, thermal transitions and the first derivative (d(MRE)/dT) of the thermal transition curves of peptides (A–C) ERSEQ and (D–F) DRSER. Both peptides were prepared at a concentration of 1 mg/mL under two conditions: 20 mM PBS, 150 mM NaCl at PH 7 (gray), and 0.1M acetic acid, pH 3 (black). Wavelength scans were conducted at 4°C (A, D). Melting transitions were monitored by CD spectroscopy at 225 nm (B, E) and the first derivative of the thermal transition curves was calculated (C, F).
The two peptides were also characterized at acidic pH to investigate the electrostatic effect of the RSE environment. At pH 3, peptide ERSEQ still formed a nice triple helix with MRE225 = 3410 deg cm2 dmol−1, but had a decreased T m = 40.8°C, which is 5.7°C lower than that seen for this peptide at pH 7 [Fig. 1(A,B)]. These results suggested that the electrostatic interaction of the RSE sequence makes a significant contribution to the stability of the mutation site. For peptide DRSER, it showed a decreased MRE225 (3284 deg cm2 dmol−1) as well as T m (41.7°C) at pH 3 [Fig. 1(D)], suggesting the same stabilizing role of the RSE sequence at the interruption site. Peptide DRSER showed a slightly less decrease in the stability (4.3°C) than ERSEQ (5.7°C) when pH is changed from neutral to acidic, which may be due to the possible existence of favorable side‐chain interaction of Arg but not Gln at pH 3.
Triple‐helix structure
Consistent with the CD observations, the NMR HSQC spectra showed that both peptides formed complete triple helix structure (Fig. 2). For peptide ERSEQ at pH 7, Gly10 in the repetitive GPO environment showed only one trimer resonance; instead, Gly19 and Ser22 showed three trimer resonances since the three different chains displayed nonequivalent environments resulting from the one‐residue staggering within the triple helix [Fig. 2(A)]. Trimer resonances could be distinguished from monomer resonances by the 1H–15N heteronuclear NOE measurements (data not shown). Other unassigned minor resonances in the HSQC spectra resulted from cis–trans isomerization of Gly–Pro and Pro–Hyp bonds of the peptides in the unfolded states.23, 28 At pH 3, the trimer resonance of Gly10 was totally overlapped with that of Gly10 at pH 7, indicating no effect of charge on the chemical environment at the N‐terminus. However, when the pH was changed from neutral to acidic, the three trimer resonances of Gly19 and Ser22 were all shifted, while Ser22 showed a significantly larger movement, suggesting an effect of charge on the RSE sequence in the center of the triple helix.
Figure 2.

1H–15N HSQC spectra of peptides ERSEQ (A) and DRSER (B) at pH 7 (red) and pH3 (green) at 25°C. Peptide sequences are shown at the top with 15N‐labeled residues underlined. The peaks corresponding to the monomer and trimer states are denoted with a superscript M or T, respectively. The superscripted numbers 1, 2, and 3 correspond to the three different chains, respectively.
For peptide DRSER, it showed peaks indicative of fully folded triple‐helix structure at both pH 7 and pH 3 [Fig. 2(B)]. Specifically, Gly10 showed one trimer resonance, and Gly19 and Ser22 each showed three trimer resonances as expected. When the pH was changed from 7 to 3, the significant shift of the trimer resonances of Gly19 and particularly Ser22, but not Gly10, implied the electrostatic effects of the RSE sequence on the chemical environment at the interruption site in the triple helix. The similarity of both peptides changing from neutral to acidic pH suggested that the mutation and interruption share comparable electrostatic effects within the RSE context, which may indicate an alteration in local conformation around the Ser site.
Local conformation
3JHNHα‐coupling values of labeled residues in both peptides were measured using the HNHA experiments.8, 18, 29 Residues in the standard triple‐helix structure contained phi angles from −55 to −75°, giving a corresponding 3JHNHα‐coupling value of 4–6 Hz.8, 18, 29 For peptide ERSEQ at pH 7, all the labeled residues showed J coupling values between 4 and 6 Hz, supporting a standard triple‐helix conformation at the center as well as the terminal [Fig. 3(A)]. However, at pH 3, one Ser at the mutation site, 1TS22, showed a J‐coupling value larger than 6 Hz, suggesting a locally distorted triple‐helix conformation of Ser in the peptide at pH 3. A similar case was also observed for peptide DRSER [Fig. 3(B)]. When pH was changed from neutral to acidic, one and only one Ser22 (1TS22) displayed an increased J‐coupling value from 5.7 to 6.7 Hz, indicating that a standard triple‐helix conformation became a little perturbed at the interruption site.
Figure 3.
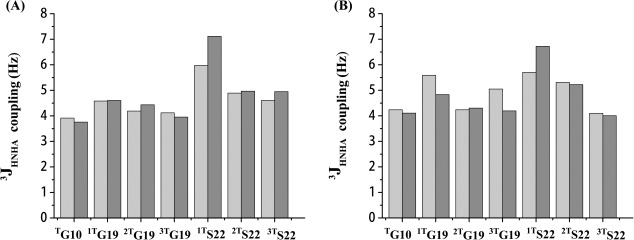
3JHNHα coupling values of model peptides ERSEQ (A) and DRSER (B). 3JHNHα‐coupling measurements were performed at pH 7 (gray) and pH 3 (black) at 25°C. Residues in the triple helical conformation typically contain phi angles from −55 to −75° and have a corresponding J coupling value of 4–6 Hz.
Hydrogen bonding
The hydrogen bonding information of both peptides was obtained by the measurements of amide proton temperature gradients, whose value was indicative of the existence of a hydrogen bond if higher than −4.6 ppb/°C (Fig. 4).18, 21 For peptide ERSEQ at pH 7, the NH gradient value for Gly10 in the trimer state was much larger than −4.6 ppb/°C, indicating the presence of typical hydrogen bonds at the N‐terminus of the triple‐helix structure [Fig. 4(A)]. The NH gradients for the three trimer resonances of Gly19 were all higher than the cut‐off value, supporting the formation of hydrogen bonds just N‐terminal to the mutation site. However, for Ser22 at the mutation site, two out of three Ser showed the NH gradient values more negative than −4.6 ppb/°C, suggesting the absence of hydrogen bonding at these two sites [Fig. 4(A)]. At pH 3, the peptide ERSEQ showed exactly the same pattern of hydrogen bonding as that of pH 7, highlighting the disruptive role of Ser at the mutation site. A similar case was also observed for peptide DRSER [Fig. 4(B)]. When pH was changed from neutral to acidic, two out of three Ser at the interruption sites remained absent of hydrogen bonding, suggesting a similarly perturbed local conformation at the interruption as well as the Ser mutation.
Figure 4.
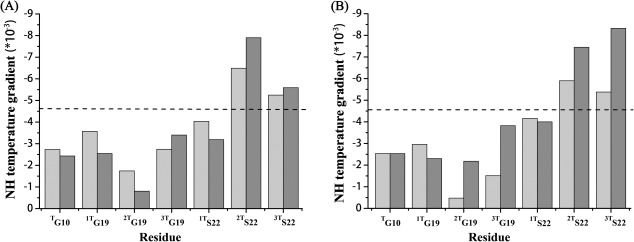
Amide proton NH temperature gradients of peptide ERSEQ (A) and DRSER (B). Amide proton NH temperature gradients were measured at pH 7 (gray) and pH 3 (black). The black dashed horizontal line corresponds to a cut‐off value for hydrogen bonding, with less negative values than −4.6 ppb/°C indicative of hydrogen bonding.
Real‐time CD and NMR monitoring of peptide folding
The folding dynamics of these two peptides was first monitored at a concentration of 1 mg/mL at pH 7 at 4°C by CD spectroscopy [Fig. 5(A)]. The empirical fraction folded half time (t 1/2) was used to compare folding since the folding profiles cannot fit simple kinetics model. For peptide ERSEQ, the folding half life (t 1/2) was 15.2 min, while peptide DRSER showed a t 1/2 of 12.9 min [Fig. 5(A)]. Compared with peptide DRSER, peptide ERSEQ showed a slightly slower folding rate at pH 7, suggesting that the mutation may reinforce more inference in the folding of the whole peptide than the interruption. The residue‐specific folding of the two peptides was further investigated by NMR at pH 7 [Fig. 5(B)]. The intensity of the monomer resonance of Ser22 in both peptides was monitored under the same conditions for ∼600 min [Fig. 5(B)]. The Ser in peptide DRSER refolded comparatively faster (gray, k = 0.055 min−1) than that in peptide ERSEQ (black, k = 0.039 min−1). The comparatively larger folding rate of Ser in peptide DRSER may imply that the interruption sequence DRSER probably has less disturbing effects on the triple helix folding than the mutation sequence ERSEQ, which is consistent with the CD observations. The faster folding of peptide DRSER than ERSEQ may be due to the complementary charge compensation between Arg and Glu, which is not available in the case of Gln and Glu.
Figure 5.

CD and NMR folding curve of peptide ERSEQ (black) and DRSER (gray). The CD folding was monitored at pH 7 at a concentration of 1 mg/mL at 4°C (A). The residue‐specific NMR folding was monitored by measuring the intensity of Ser22 in the monomer state as a function of time at pH 7 (B).
Discussions
Pathological Gly substitutions and functional natural interruptions present a seemingly conflicting picture of sequence variations in the “uniform” rod‐like collagen. Understanding the structural features of these important sites will help us elucidate the etiology of collagen diseases as well as the design of biomimetic materials. Gly–Ser mutations account for about two‐fifths of all observed Gly substitutions in the OI database, and display the most complex spectrum of phenotypes among all types of Gly substitutions.5 Recent correlation of the genotypes and phenotypes of collagen by bioinformatics analysis have suggested that local amino acid environment of Ser determines its lethality.26 Earlier studies on recombinant collagen fragments showed that the presence of a KGE sequence near a Gly–Ser OI mutation was correlated with OI lethality, highlighting the crucial role of highly charged motif R(K)GE(D).30 Structural characterization of a peptide mimicking a Gly–Ser substitution within an RGE sequence context is presented here.
The destabilization effect of a Gly–Ser mutation depends on its local sequence environment. The Gly–Ser mutation in the charged RGE sequence results in a destabilization of the triple helix with the melting temperature reduced by 16.8°C from 63.3°C of the parent peptide to 46.5°C (Supporting Information, Table S1). This level of destabilization is comparable with the Gly–Ser mutation in peptide T1‐865 (T m decreased by 20.5°C from 35°C of the parent peptide to 14.5°C) and that in peptide T1‐898 (T m decreased by 19.1°C from 47.1°C of the parent peptide to 28°C), both of which contain a flexible amino acid sequence environment (Supporting Information, Table S1). However, the destabilization effect is much weaker than a Gly–Ser mutation in (POG)10(G–S), whose melting temperature is decreased by 36.5°C (from 57°C of the parent peptide to 20.5°C) (Supporting Information, Table S1). Studies have suggested that a more flexible local sequence environment possibly leads to less destabilization of triple helix structure.20 Though the RGE motif is considered as highly stabilizing, its destabilization effect is more like a flexible sequence rather than the highly rigid and stabilizing GPO context. The melting temperature of peptide ERSEQ is decreased by 5.7°C when the pH is changed from neutral to acidic, suggesting that the electrostatic interaction contributes to the stability of the peptide containing mutations. These results may suggest that unlike the rigid GPO environment, the RGE motif likely provide a more flexible sequence environment, while the electrostatic interactions greatly enhance its stability.
The conformational disruptive effect of the Gly–Ser mutation in the RGE motif is comparable to those previously reported in the context of other amino acid sequences. Similarly with peptide T1‐898, the Gly–Ser mutation in peptide ERSEQ also results in disruption of two hydrogen bonds of Ser at the mutation site (Supporting Information, Table S1). However, two out of the three Ser in peptide T1‐898 show J‐coupling values a little larger than expected for typical triple helix, while all the three Ser in peptide ERSEQ display J‐coupling values compatible with a triple‐helix structure. Both Ser and N‐terminal Gly were found to be rigid on picoseconds time scale (data not shown), suggesting any perturbations were occurring on a much longer time scale. Earlier studies of a Gly–Ala mutation in a similarly charged KGD context also showed disruption of two hydrogen bonds at the mutation site, while the dihedral angles were well maintained for canonical triple helix, which is exactly the same as peptide ERSEQ.17 These results may suggest a general role of the charged R(K)GE(D) motif in the disruption of triple‐helix structure by the introduction of Gly–Ala and Gly–Ser substitutions. The reduction of the thermal stability and the disruption of triple‐helix structure in terms of hydrogen bonding may provide a structural basis of the OI disease resulting from the Gly–Ser mutation in the RGE environment.
Peptide models of natural interruptions and Gly substitutions with a comparable sequence environment will provide a new perspective to understand the structural consequences of interruptions versus mutations. Compared with peptide ERSEQ containing a Gly–Ser mutation, peptide DRSER modeling a similar interruption sequence showed pretty similar stability, triple‐helix content, as well as local conformational features such as hydrogen bonding and dihedral angles. The effect of pH on stability and structure on both peptides was also quite similar, indicating that the electrostatic interaction of the RSE motif makes similar contribution to the mutation and interruption. Despite these similarities, both the CD and NMR real‐time folding results indicated that peptide ERSEQ displayed a comparatively slower folding rate than peptide DRSER. Considering the sequential resemblance of both peptides, the small difference in the folding rate is likely to be significant. These results are consistent with earlier observations that folding is a key factor in determining OI phenotypes.31, 32 These studies may imply that the Gly–Ser mutation likely leads to a larger interference in folding than the natural interruption, therefore resulting in OI diseases.
The highly charged R(K)GE(D) motif occurs frequently in both fibrillar and nonfibrillar collagens, and it is considered as an important stabilizing factor for triple‐helix structure.27 However, the role of this motif in OI mutations and functional interruptions is barely understood. Our studies have revealed quite different effects of the R(K)GE(D) motif on the triple helix stability and conformation as compared with the most abundant GPO context. This motif may provide a comparatively flexible environment that better accommodates mutations as well as interruptions, while the electrostatic interactions contribute to its stability. The R(K)GE(D) motif and other charged amino acid patterns have been used to create collagen heterotrimers.33, 34, 35 Our studies shed insight into the molecular features of the highly charged motif and may aid the design of collagen biomimetic peptides containing important biological sites.
Materials and Methods
Sequence analysis
Sequences of all fibrillar and nonfibrillar collagens were obtained from Genbank.36 The amino acids were grouped into hydrophobic (Val, Ile, Leu, Met, Phe, Tyr, and Trp), charged (His, Lys, Arg, Glu, and Asp), small (Gly, Ala, Cys, and Ser), polar (Asn, Gln, and Thr), and pro (Pro and Hyp). To analyze the frequency of amino acid types in the X and Y positions, charged amino acids were further divided into two subgroups positively charged (His, Lys, and Arg) and negatively charged (Glu and Asp). The frequency of highly charged motifs RGE, RGD, KGE, KGD, he, and HGD was calculated as the total occurrences of each motif divided by the total number of the repetitive Gly–X–Y triplets in fibrillar (Type I, II, III, V, and XI) and nonfibrillar collagens, respectively. Fisher's exact test was used to test whether the types of residues at position AA5 in the sequence of Gly–AA1–AA2–AA3–AA4–AA5–Gly of G5G interruptions were different from those found adjacent to Gly–Ser mutations of the α1(I) chain of Type I collagen. All Gly–Ser substitutions in the α1(I) chain of Type I collagen were collected from the Database of Collagen Mutations (www.le.ac.uk/genetics/collagen) at the University of Leicester.37 The analyses were performed by the software SPSS (Statistical Product and Service Solutions, IBM), and a p‐value of <0.05 was considered significant.
Sample preparation
Peptides ERSEQ and DRSER were synthesized by Chinese Peptide Company (Hangzhou, China). Both peptides were selectively 15N labeled at positions G10, G19, and S22 (Table 1). NMR samples of both peptides were prepared in 10% D2O/90% PBS (10 mM, pH 7.0) with a concentration of 2.8 mM.
Circular dichroism spectroscopy
CD spectra were recorded on a Chirascan CD spectrometer (Applied Photophysics, UK). Cells with a path length of 2 mm were used, and the temperature of the cells was controlled using a Peltier temperature controller. The CD samples were prepared at a concentration of 1 mg/mL in 20 mM PBS at PH 7. Wavelength scans were conducted from 215 to 260 nm with a 0.5 nm increment per step and a 5 s averaging time at 4°C, and each scan was repeated three times. CD was applied to determine the thermal stability by monitoring the amplitude of the peak at 225 nm as a function of increasing temperature with an average heating rate of 0.2°C/min.38 The peptides were equilibrated for at least 24 h at 4°C prior to the melting experiments. The first derivative of the thermal transition curves was calculated and smoothed, and the melting temperature (T m) was assigned as the extrema of the first derivative. The CD folding experiments were performed by denaturing the sample at 80°C for 20 min and then being rapidly quenched in an ice water bath and transferred to a pre‐equilibrated CD cell at 4°C. The dead time was on the order of 40 s. The ellipticity at 225 nm was monitored for ∼10 h, with a time interval of 10 s and time constant of 2 s. The half time of refolding, t 1/2, was calculated as the time at which the fraction folded reached 0.5.
NMR spectroscopy
All NMR experiments were performed on a Varian INOVA 600 MHz spectrometer equipped with High‐Filed Indirect Detection NMR Probe Installation (Varian). 1H–15N heteronuclear single‐quantum coherence (HSQC) spectra39 and 3D 15N‐edited NOESY‐HSQC experiments40, 41, 42 with a mixing time of 50 ms were carried out for assignments of NMR resonances at 25°C. HNHA experiments were performed with an H–H coupling period of 25 ms at 25°C to measure 3JHNHα coupling constants.43 The correction factor for the 3JHNHα coupling constants was obtained as described.21 Relaxation R1, R2, and heteronuclear NOE measurements44, 45, 46 were performed at 25°C. For measurements of amide proton temperature gradients, a series of 1H–15N HSQC spectra were recorded from 25°C to 50°C. The samples were equilibrated at each temperature for at least 1 h. Amide proton temperature gradients were obtained by linear regression analysis of the amide proton chemical shift versus temperature. All data were processed using NMRPipe47 and analyzed with NMRView.48
The NMR folding experiments were performed as previously described.31, 49 The samples were denatured by heating to 80°C for 15 min and quickly transferred to the spectrometer that was equilibrated at 25°C. A series of HSQC spectra were acquired every 5.2 min immediately after the sample was placed in the probe. The kinetics of folding was monitored by measuring cross‐peak intensities as a function of time. The rate constant k was obtained by exponential fitting and the intensities of the monomer peaks were normalized as previously described.31, 49
Supporting information
Supporting Information
References
- 1. Ramachandran GN, Kartha G (1955) Structure of collagen. Nature 176:593–595. [DOI] [PubMed] [Google Scholar]
- 2. Rich A, Crick FH (1955) The structure of collagen. Nature 176:915–916. [DOI] [PubMed] [Google Scholar]
- 3. van der Rest M, Garrone R (1991) Collagen family of proteins. FASEB J 5:2814–2823. [PubMed] [Google Scholar]
- 4. Gordon MK, Hahn RA (2010) Collagens. Cell Tissue Res 339:247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, Hyland JC, Korkko J, Prockop DJ, De Paepe A, Coucke P, Symoens S, Glorieux FH, Roughley PJ, Lund AM, Kuurila‐Svahn K, Hartikka H, Cohn DH, Krakow D, Mottes M, Schwarze U, Chen D, Yang K, Kuslich C, Troendle J, Dalgleish R, Byers PH (2007) Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat 28:209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Myllyharju J, Kivirikko KI (2001) Collagens and collagen‐related diseases. Ann Med 33:7–21. [DOI] [PubMed] [Google Scholar]
- 7. Byers PH, Cole WG, Osteogenesis imperfecta In: Royce PM, Steinmann B, Eds. (2002) Connective tissue and its hereditable disorders. New York: Wiley‐Liss, pp 385–430. [Google Scholar]
- 8. Thiagarajan G, Li Y, Mohs A, Strafaci C, Popiel M, Baum J, Brodsky B (2008) Common interruptions in the repeating tripeptide sequence of non‐fibrillar collagens: sequence analysis and structural studies on triple‐helix peptide models. J Mol Biol 376:736–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miles AJ, Skubitz AP, Furcht LT, Fields GB (1994) Promotion of cell adhesion by single‐stranded and triple‐helical peptide models of basement membrane collagen alpha 1(IV)531‐543. Evidence for conformationally dependent and conformationally independent type IV collagen cell adhesion sites. J Biol Chem 269:30939–30945. [PubMed] [Google Scholar]
- 10. Parkin JD, San Antonio JD, Pedchenko V, Hudson B, Jensen ST, Savige J (2011) Mapping structural landmarks, ligand binding sites, and missense mutations to the collagen IV heterotrimers predicts major functional domains, novel interactions, and variation in phenotypes in inherited diseases affecting basement membranes. Human Mut 32:127–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bella J (2014) A first census of collagen interruptions: collagen's own stutters and stammers. J Struct Biol 186:438–450. [DOI] [PubMed] [Google Scholar]
- 12. Myllyharju J, Kivirikko KI (2004) Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet 20:33–43. [DOI] [PubMed] [Google Scholar]
- 13. Brodsky B, Thiagarajan G, Madhan B, Kar K (2008) Triple‐helical peptides: an approach to collagen conformation, stability, and self‐association. Biopolymers 89:345–353. [DOI] [PubMed] [Google Scholar]
- 14. Bella J, Eaton M, Brodsky B, Berman HM (1994) Crystal and molecular structure of a collagen‐like peptide at 1.9 A resolution. Science 266:75–81. [DOI] [PubMed] [Google Scholar]
- 15. Baum J, Brodsky B (1999) Folding of peptide models of collagen and misfolding in disease. Curr Opin Struct Biol 9:122–128. [DOI] [PubMed] [Google Scholar]
- 16. Xiao J, Addabbo RM, Lauer JL, Fields GB, Baum J (2010) Local conformation and dynamics of isoleucine in the collagenase cleavage site provide a recognition signal for matrix metalloproteinases. J Biol Chem 285:34181–34190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiao J, Cheng H, Silva T, Baum J, Brodsky B (2011) Osteogenesis imperfecta missense mutations in collagen: structural consequences of a glycine to alanine replacement at a highly charged site. Biochemistry 50:10771–10780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li Y, Brodsky B, Baum J (2009) NMR conformational and dynamic consequences of a Gly to Ser substitution in an osteogenesis imperfecta collagen model Peptide. J Biol Chem 284:20660–20667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beck K, Chan VC, Shenoy N, Kirkpatrick A, Ramshaw JA, Brodsky B (2000) Destabilization of osteogenesis imperfecta collagen‐like model peptides correlates with the identity of the residue replacing glycine. Proc Natl Acad Sci USA 97:4273–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bryan MA, Cheng H, Brodsky B (2011) Sequence environment of mutation affects stability and folding in collagen model peptides of osteogenesis imperfecta. Biopolymers 96:4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Y, Brodsky B, Baum J (2007) NMR shows hydrophobic interactions replace glycine packing in the triple helix at a natural break in the (Gly‐X‐Y)n repeat. J Biol Chem 282:22699–22706. [DOI] [PubMed] [Google Scholar]
- 22. Xiao J, Sun X, Balaram M, Brodsky B, Baum J (2015) NMR studies demonstrate a unique AAB composition and chain register for a heterotrimeric type IV collagen model peptide containing a natural interruption site. J Biol Chem 290:24201–24209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xiao J, Madhan B, Li Y, Brodsky B, Baum J (2011) Osteogenesis imperfecta model peptides: incorporation of residues replacing Gly within a triple helix achieved by renucleation and local flexibility. Biophys J 101:449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Persikov AV, Ramshaw JA, Brodsky B (2000) Collagen model peptides: sequence dependence of triple‐helix stability. Biopolymers 55:436–450. [DOI] [PubMed] [Google Scholar]
- 25. Persikov AV, Ramshaw JA, Kirkpatrick A, Brodsky B (2000) Amino acid propensities for the collagen triple‐helix. Biochemistry 39:14960–14967. [DOI] [PubMed] [Google Scholar]
- 26. Xiao J, Yang Z, Sun X, Addabbo R, Baum J (2015) Local amino acid sequence patterns dominate the heterogeneous phenotype for the collagen connective tissue disease Osteogenesis Imperfecta resulting from Gly mutations. J Struct Biol 192:127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Persikov AV, Ramshaw JA, Kirkpatrick A, Brodsky B (2005) Electrostatic interactions involving lysine make major contributions to collagen triple‐helix stability. Biochemistry 44:1414–1422. [DOI] [PubMed] [Google Scholar]
- 28. Buevich AV, Dai QH, Liu X, Brodsky B, Baum J (2000) Site‐specific NMR monitoring of cis–trans isomerization in the folding of the proline‐rich collagen triple helix. Biochemistry 39:4299–4308. [DOI] [PubMed] [Google Scholar]
- 29. Kramer RZ, Bella J, Mayville P, Brodsky B, Berman HM (1999) Sequence dependent conformational variations of collagen triple‐helical structure. Nat Struct Biol 6:454–457. [DOI] [PubMed] [Google Scholar]
- 30. Xu K, Nowak I, Kirchner M, Xu Y (2008) Recombinant collagen studies link the severe conformational changes induced by osteogenesis imperfecta mutations to the disruption of a set of interchain salt bridges. J Biol Chem 283:34337–34344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Buevich AV, Silva T, Brodsky B, Baum J (2004) Transformation of the mechanism of triple‐helix peptide folding in the absence of a C‐terminal nucleation domain and its implications for mutations in collagen disorders. J Biol Chem 279:46890–46895. [DOI] [PubMed] [Google Scholar]
- 32. Hyde TJ, Bryan MA, Brodsky B, Baum J (2006) Sequence dependence of renucleation after a Gly mutation in model collagen peptides. J Biol Chem 281:36937–36943. [DOI] [PubMed] [Google Scholar]
- 33. Jalan AA, Hartgerink JD (2013) Pairwise interactions in collagen and the design of heterotrimeric helices. Curr Opin Chem Biol 17:960–967. [DOI] [PubMed] [Google Scholar]
- 34. Fallas JA, Lee MA, Jalan AA, Hartgerink JD (2012) Rational design of single‐composition ABC collagen heterotrimers. J Am Chem Soc 134:1430–1433. [DOI] [PubMed] [Google Scholar]
- 35. Xu F, Li J, Jain V, Tu RS, Huang Q, Nanda V (2012) Compositional control of higher order assembly using synthetic collagen peptides. J Am Chem Soc 134:47–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Benson DA, Cavanaugh M, Clark K, Karsch‐Mizrachi I, Lipman DJ, Ostell J, Sayers EW (2013) GenBank. Nucleic Acids Res 41:D36–D42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dalgleish R (1997) The human type I collagen mutation database. Nucleic Acids Res 25:181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Persikov AV, Pillitteri RJ, Amin P, Schwarze U, Byers PH, Brodsky B (2004) Stability related bias in residues replacing glycines within the collagen triple helix (Gly‐Xaa‐Yaa) in inherited connective tissue disorders. Hum Mutat 24:330–337. [DOI] [PubMed] [Google Scholar]
- 39. Kay LE, Keifer P, Saarinen T (1992) Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc 114:10663–10665. [Google Scholar]
- 40. Messerle BA, Wider G, Otting G, Weber C, Wüthrich K (1989) Solvent suppression using a spin‐lock in 2D and 3D NMR spectroscopy with H2O solutions. J Magn Reson 85:608–613. [Google Scholar]
- 41. Marion D, Kay LE, Sparks SW, Torchia DA, Bax A (1989) Three‐dimensional heteronuclear NMR of 15N labeled proteins. J Am Chem Soc 111:1515–1517. [Google Scholar]
- 42. Fesik SW, Zuiderweg ER (1988) Heteronuclear three‐dimensional nmr spectroscopy. A strategy for the simplification of homonuclear two‐dimensional NMR spectra. J Magn Reson 78:588–593. [Google Scholar]
- 43. Vuister GW, Bax A (1993) Quantitative J correlation: a new approach for measuring homonuclear three‐bond J(HNHA) coupling constants in 15N‐enriched proteins. J Am Chem Soc 115:7772–7777. [Google Scholar]
- 44. Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman‐Kay JD, Kay LE (1994) Backbone dynamics of a free and phosphopeptide‐complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33:5984–6003. [DOI] [PubMed] [Google Scholar]
- 45. Palmer AG 3rd (1993) Dynamic properties of proteins from NMR spectroscopy. Curr Opin Biotechnol 4:385–391. [DOI] [PubMed] [Google Scholar]
- 46. Fan P, Li MH, Brodsky B, Baum J (1993) Backbone dynamics of (Pro‐Hyp‐Gly)10 and a designed collagen‐like triple‐helical peptide by 15N NMR relaxation and hydrogen‐exchange measurements. Biochemistry 32:13299–13309. [DOI] [PubMed] [Google Scholar]
- 47. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293. [DOI] [PubMed] [Google Scholar]
- 48. Johnson BA, Blevins RA (1994) NMR View ‐ a computer‐program for the visualization and analysis of NMR data. J Biomol NMR 4:603–614. [DOI] [PubMed] [Google Scholar]
- 49. Liu X, Siegel DL, Fan P, Brodsky B, Baum J (1996) Direct NMR measurement of folding kinetics of a trimeric peptide. Biochemistry 35:4306–4313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information