

Abstract
Carnosine dipeptidase II (CN2/CNDP2) is an M20 family metallopeptidase that hydrolyses various dipeptides including β‐alanyl‐l‐histidine (carnosine). Crystallographic analysis showed that CN2 monomer is composed of one catalytic and one dimerization domains, and likely to form homodimer. In this crystal, H228 residue of the dimerization domain interacts with the substrate analogue bestatin on the active site of the dimer counterpart, indicating that H228 is involved in enzymatic reaction. In the present study, the role of intradimer interaction of CN2 in its catalytic activity was investigated using electrospray‐ionization time‐of‐flight mass spectrometry (ESI‐TOF MS). First, a dimer interface mutant I319K was prepared and shown to be present as a folded monomer in solution as examined by using ESI‐TOF MS. Since the mutant was inactive, it was suggested that dimer formation is essential to its enzymatic activity. Next, we prepared H228A and D132A mutant proteins with different N‐terminal extended sequences, which enabled us to monitor dimer exchange reaction by ESI‐TOF MS. The D132A mutant is a metal ligand mutant and also inactive. But the activity was partially recovered time‐dependently when H228A and D132A mutant proteins were incubated together. In parallel, H228A/D132A heterodimer was formed as detected by ESI‐TOF MS, indicating that interaction of a catalytic center with H228 residue of the other subunit is essential to the enzymatic reaction. These results provide evidence showing that intradimer interaction of H228 with the reaction center of the dimer counterpart is essential to the enzymatic activity of CN2.
Keywords: metallopeptidase, reaction mechanism, electrospray ionization mass spectrometry, protein complex, dimer exchange
Outline:
In order to clarify the role of dimer formation in the catalytic action of an M20 family metalloenzyme, carnosine dipeptidase II, dimer formation, and dimer exchange reaction were examined using electrospray ionization time‐of‐flight mass spectrometry. Consequently, we obtained evidence showing that intradimer interaction of H228 residue with the active center of another subunit is essential to its catalytic activity.
Introduction
Carnosine dipeptidase II (CN2 or CNDP2) is a dinuclear metallopeptidase classified into M20F family of clan MH in MEROPS peptidase database.1, 2 It is present in the cytosol of a wide variety of mammalian tissues including the brain and kidney, and is likely to have essential roles in peptide and amino acid metabolism in the tissues.3 CN2 hydrolyses β‐Ala‐l‐His (carnosine) as well as certain dipeptides such as Leu‐His and Gly‐Phe1, 2 in the presence of Mn2+, while it is inhibited by a substrate analogue, bestatin. These enzymatic properties indicated that CN2 is identical to the enzyme that was previously referred to as tissue carnosinase or cytosolic nonspecific dipeptidase.4 In mammals, carnosine is also hydrolyzed by a closely related enzyme, CN1, which has been suggested to be identical to that known as serum carnosinase.1
Crystallographic analysis of CN2 showed that it was present as a homodimeric structure in the crystal.5 Each subunit is composed of one catalytic domain and one dimerization domain, the latter of which provides the main dimer interface for homodimer formation. Each catalytic domain has one active center with two Mn2+ or Zn2+ ions, where a substrate analogue bestatin was associated in this crystal. These are consistent with basic features of M20 family member of enzymes.
The metallopeptidase M20 family contains a variety of enzymes that are expressed in a wide range of organisms from bacteria to mammals. Most of these are essential components in fundamental pathways of amino acid, peptide and nucleic acid metabolism. These include peptidases that hydrolyze dipeptides (CN1, CN2, PepV,6 PepD,7 and Sapep8), tripeptides (PepT9), and both (Dug1p10). In addition, there are several non‐peptidase enzymes such as aminoacylase‐1 (Acy‐1),11 β‐alanine synthetase,12 acetylornithine deacetylase (ArgE),13 and N‐succinyl‐l,l‐diaminopimelate desuccinylase (DapE),14 which share structural features with M20 family metallopeptidases. The majority of M20 family members including CN2 have one catalytic domain and one dimerization domain in a monomer, and possibly form a homodimer in solution, while in PepV, PepD, and Sapep, the second domain called the lid domain has a structure like a dimer of dimerization domains, and act as a monomer or form a homodimer of a different conformation of quaternary structure.6, 7, 8 These two groups of proteins show similar folding patterns in the catalytic as well as the dimerization/lid domains.
In several M20 family proteins, it has been proposed that the dimerization/lid domain is involved in the enzymatic reaction. This possibility was first raised in PepV, in which H269 of the lid domain interacts with the active site as shown by X‐ray crystallography.6 In addition, in human aminoacylase‐1, a dimerization domain mutant, H226A, showed decreased activity, while this activity was partially recovered when it was incubated with another inactive mutant with metal‐ligand mutation, possibly due to formation of a functional reaction site as a result of dimer exchange.11, 15 In CN2, previous crystallographic analysis showed that H228 of the dimerization domain interacted with the substrate analogue bestatin at the active site of the dimer counterpart.5 In addition, H228A mutation of CN2 resulted in loss of enzymatic activity.5 These indicate that H228A of the dimer counterpart is essential to the enzymatic reaction of CN2. However, it could not exclude the possibility that the loss of enzymatic activity in the CN2 mutant protein was due to some other reason such as structural changes induced by the mutation. Therefore, further evidence should be required to clarify the role of intradimer interaction in the reaction mechanism.
In the present study, we investigated the role of intradimer interaction in the enzymatic reaction of CN2 with the help of electrospray‐ionization time‐of‐flight mass spectrometry (ESI‐TOF MS). It has been demonstrated that noncovalent protein complexes such as protein oligomers and protein‐ligand complexes can be detected by using ESI‐MS.16, 17 Because of its high mass accuracy, high throughput, and high sensitivity, mass spectrometry has great advantages in analyzing the composition and dynamics of non‐covalent protein complexes.18, 19, 20, 21 This technique has so far been applied to a variety of studies and provided valuable information on non‐covalent protein interactions in solution.22, 23, 24, 25 In this study, we used ESI‐TOF MS to analyze the oligomeric states of CN2 and to monitor its dimer exchange reaction in solution.
In the present study, first we prepared a mutant CN2 protein having a point mutation in the dimer interface (I319K), and examined its oligomeric state and enzymatic activity to test if monomeric enzyme is enzymatically active. Next, we prepared two inactive mutants, H228A and D132A with different length of N‐terminal additional sequences so that H228A/H228A, D132A/D132A, and D132A/H228A dimers could be distinguished by ESI‐TOF MS. These mutants allowed us a new approach to monitor dimer exchange reaction of homodimer proteins. Using this system, we attempted to determine if interaction of H228 to the active site of the dimer counterpart is essential to the enzymatic activity of CN2.
Results
Structure of CN2 and possible interaction of H228 with dimer counterpart
Crystal structure of CN2 in complex with a competitive inhibitor bestatin has been reported previously,5 and some structural features were summarized in Figure 1. CN2 was present as a homodimeric form in this crystal [Fig. 1(A)]. Single CN2 molecule is composed of one catalytic domain and one dimerization domain, the latter of which provides the interface for homodimer formation. In a homodimeric complex, each subunit has a dinuclear metal binding site composed of amino acids including D132 [Fig. 1(B)]. Bestatin molecules found on the metal‐binding sites indicate the position of substrate binding. Enzymatic activity of CN2 was tested at various pH using carnosine as the substrate, and confirmed that CN2 hydrolyses carnosine under neutral or weakly basic pH conditions (pH 7.5–10) in the presence of Mn2+ (Supporting Information Fig. S1).
Figure 1.
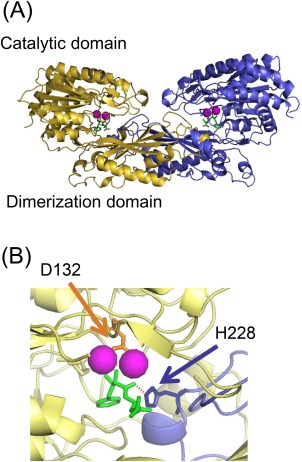
Overall structure of CN2 and its active site. (A) Overall structure of CN2 in complex with Mn2+ (magenta) and bestatin (green) (PDB: 2ZOF) (5). The yellow and blue strands represent individual subunit of a homodimer. Each subunit is composed of one catalytic and one dimerization domains. (B) Structure of the active center of CN2. The bestatin molecule on the active site of one subunit (yellow) is interacted with His 228 of the other subunit (blue) in this crystal through hydrogen bonding (red dashed line). In the active center, there is a dinuclear metal binding site, where one of the metal ligands, D132 (orange), binds to the two metal ions through carboxyl oxygen molecules. Pictures were drawn using PYMOL.26
In this crystal, a bestatin molecule interacted not only with the metal binding site of the reaction center, but also with several residues of the dimerization domain of the dimer counterpart [Fig. 1(B)]. Among them, H228 was found at the position in which the imidazole nitrogen forms a hydrogen bond with the carbonyl oxygen of the substrate. In addition, mutation of H228 to alanine resulted in loss of enzymatic activity, indicating the possibility that interaction of H228 with the reaction center of the dimer counterpart is involved in the catalytic mechanism of this enzyme.5
ESI‐TOF MS of CN2 in aqueous solutions
In the present study, in order to clarify the role of intradimer interaction of CN2, ESI‐TOF MS was used to detect CN2 dimer in aqueous solutions. First, to test if CN2 dimer can be detected by ESI‐TOF MS, wild‐type CN2 was prepared in nondenaturing or denaturing solution and applied on ESI‐TOF MS. The mass instruments were optimized so that high m/z ions from native proteins can be detected efficiently (see Materials and Methods). Expected m/z values of wild‐type CN2 dimer and monomer were shown in Supporting Information Table S1A and S1B, respectively. When CN2 was prepared in 100 mM ammonium acetate solution at neutral pH, five major peaks were observed between 4000 and 6000 m/z [Fig. 2(A)]. Since the average mass value of recombinant wild‐type CN2 monomer is 53,477, these five peaks were assigned to +24, +23, +22, +21, and +20 charged ions of CN2 dimer, respectively (Supporting Information Table S1A). Consistent with this, in size exclusion chromatography operated in the same buffer, CN2 was eluted around the elution position of 100 kDa protein [Fig. 2(D)]. Size exclusion chromatography was also carried out with lower concentrations of CN2. However, even at 100 nM, which was nearly the detection limit of this system, CN2 monomer accounted for only about 15% of the total peak area (Supporting Information Fig. S2).
Figure 2.
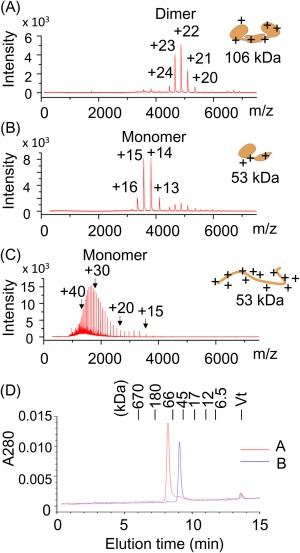
ESI‐TOF MS spectra of CN2 prepared in non‐denaturing and denaturing solutions. (A–C) Recombinant wild‐type CN2 was prepared in ammonium acetate, pH 7.5 (A), 100 mM ammonium acetate, pH 7.5/20% acetonitrile (B), or 100 mM acetic acid/20% acetonitrile, pH 2.8 (C), and subjected to ESI‐TOF MS analysis. Predicted charge numbers were shown on the top of the peaks. Predicted folding states and molecular weights were schematically drawn on the right side of the graphs. (D) The samples used in (A) and (B) (red and blue, respectively) were applied on size exclusion chromatography. The column was operated using an HPLC system at a flow rate of 0.35 mL/min, and monitored by absorbance at 280 nm. Elution positions of standard proteins were shown on top of the panel.
Next, CN2 was prepared in a neutral pH acetonitrile‐containing solution (100 mM ammonium acetate/20% acetonitrile) and analyzed by ESI‐TOF MS. In this case, major peaks were detected between 3000 and 4500 m/z [Fig. 2(B)], and were assigned to be +16, +15, +14, and +13 charged ions of CN2 monomer (Supporting Information Table S2B). Consistent with this, size‐exclusion chromatography showed that CN2 was eluted at around the position of 50 kDa protein [Fig. 2(D)].
In contrast to these neutral pH conditions, CN2 prepared in acetonitrile‐containing acidic solution (20% acetonitrile/100 mM acetic acid solution, pH 2.8) exhibited more than 25 mass peaks in lower m/z region [Fig. 2(C)]. These peaks corresponded to +18 to +43 charged ions of CN2 monomer. The presence of large number of highly charged ions is a typical feature of ESI‐MS spectra of denatured proteins.21, 27 Taken together, these results indicate that subunit composition and overall folding state of CN2 can be estimated and monitored using this ESI‐TOF MS system.
Oligomeric state and enzymatic activity of dimer interface mutant, I319K
Using this system, the role of dimer formation in CN2 was examined by two different ways. First, we prepared a mutant CN2 protein having an amino acid substitution in the dimer interface (I319K), and examined its oligomeric state together with enzymatic activity. In a CN2 dimer, two subunits are interacted mainly with the hydrophobic surface of the dimerization domain [Fig. 3(A)]. This region is mainly composed of hydrophobic amino acids including Val, Leu, Ile (Kyte‐Doolittle score28 >3.0, shown in red) and Phe, Cys, Met, Ala (Kyte‐Doolittle score >1.5, shown in magenta). Among them, I319 is located at the center of the hydrophobic region, thus we prepared a mutant protein in which I319 was replaced by Lys (I319K), and analyzed it using ESI‐TOF MS [Fig. 3(B)]. The major peaks observed were those corresponded to +14 to +16 charged ions of CN2 monomer (Supporting Information Table S2). In addition, size exclusion chromatography analysis showed that I319K mutant was eluted near the position of CN2 monomer [Fig. 3(C)]. Taken together, I319K mutant protein was likely to be present as a folded monomer in solution. In this condition, the enzymatic activity was examined using carnosine as the substrate, but less than 5% of that of wild‐type CN2 was detected [Fig. 3(D)]. These results indicate that a monomeric form of CN2 is inactive even when it has a folded structure.
Figure 3.
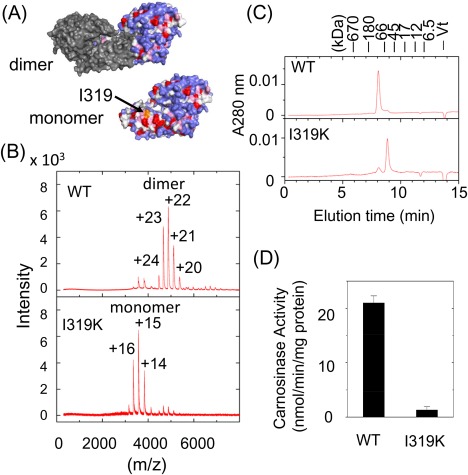
Oligomeric states and enzymatic activities of wild‐type and I319K mutant proteins of CN2. (A) Hydrophobicity of the dimer interface of CN2. The upper picture shows a CN2 dimer in which one subunit was drawn in gray, while the other subunit drawn in colors that reflect the hydrophobicity index of Kyte and Doolittle;28 red: index >3.0 (Ile, Val, Leu), magenta: index >1.5 (Phe, Cys, Met, Ala), light blue: index >−2.0 (Gly, Thr, Ser, Trp, Pro), and blue: index >3.0 (His, Glu, Gln., Asp, Asn, Lys, Arg). The lower picture shows the surface of CN2 monomer drawn in the same color. In this picture, the surface of the dimer interface is visible. The position of I319 is indicated by an arrow. (B) Mass spectrometric analysis of subunit composition of CN2. Wild‐type CN2 and I319K mutant protein were prepared in 100 mM ammonium acetate, pH 7.5, and analyzed by ESI‐TOF MS. (C) Size exclusion chromatography of wild‐type and I319K mutant proteins of CN2. Each sample was analyzed by HPLC in the same condition as described in Figure 2(D). (D) Enzymatic activities of wild‐type and I319K mutant of CN2. Activities were determined using carnosine as the substrate. Data represent mean ± SE of triplicated determinations.
Role of H228 in enzymatic reaction of CN2
In order to clarify the role of H228 in the enzymatic reaction, we next prepared D132A in addition to H228A mutant proteins with different N‐terminal additional sequences so that different homo‐ and heterodimers could be clearly distinguished on ESI‐TOF MS (Fig. 4). The mutant proteins were prepared by cloning the corresponding cDNAs into pGEX‐6P3 and pGEX‐4T3 vectors, respectively. The resultant H228A protein is calculated to be 245 Da larger than D132A protein. The calculated average mass values of D132A/D132A, D132A/H228A, and H228A/H228A dimers are 106,331 Da, 106,577 Da and 106,822 Da, respectively.
Figure 4.
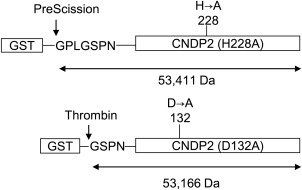
Preparation of H228A and D132A mutant proteins of CN2 with different N‐terminal additional sequences. cDNAs encoding inactive CN2 mutants, H228A, and D132A, were cloned into pGEX‐6P3 and pGEX‐4T3 vectors, respectively (Supporting Information Fig. S2), and expressed in E. coli as GST‐fusion proteins. The recombinant proteins were then trapped on glutathione‐Sepharose beads and digested with PreScission™ or thrombin protease, respectively. The yielded proteins have expected average molecular weights of 53,411 and 53,166 Da, respectively.
The D132A mutant protein had no enzymatic activity [Fig. 5(A)], possibly because the D132 residue is coordinated with two metal ions in the catalytic center in wild‐type CN2 [Fig. 1(B)], and thus the D132A mutant protein would not have metal binding activity. On the other hand, H228A mutant protein was also inactive [Fig. 5(A)], but was expected to retain metal‐binding activity. The Mn2+‐binding activities of the two mutants were experimentally tested using ESI‐TOF MS (Supporting Information Fig. S3). When wild‐type apo‐CN2 was incubated with 50 μM Mn2+, each ion peak showed a mass shift that nearly corresponded to four Mn2+ ions per dimer. In H228A mutant, incubation with Mn2+ also induced a peak shift to similar extent. In contrast, D132A mutant did not show a peak shift after Mn2+‐addition. These results confirmed that H228A protein, but not D132A protein, has Mn2+‐binding activity.
Figure 5.
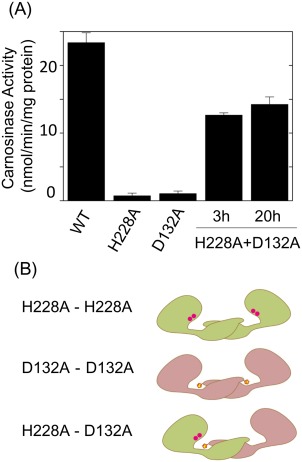
Compensation of enzymatic activities of two inactive mutant proteins, H228A and D132A. (A) Recovery of enzymatic activity in H228A and D132A mixture. Enzymatic activity of wild‐type, H228A, D132A, and H228A/D132 mixture of CN2 were determined using carnosine as the substrate. (B) Schematic representation of H228A (green) homodimer, D132A (pink) homodimer and H228A/D132A heterodimer. In H228A/D132A heterodimer, enzymatic activity could be recovered when the metal binding site of H228A subunit was interacted with H228 of the D132A subunit.
Although H228A and D132A mutant proteins were both inactive as mentioned, enzymatic activity was partially recovered in at least 3 hours after mixing the two mutants [Fig. 5(A)]. This can be explained if the catalytic domain of H228A mutant had a functional conformation and that subunit exchange reaction resulted in formation of D132A/H228 heterodimer in which the active center of the H228A subunit interacted with the H228 residue of D132A subunit [Fig. 5(B)].
To examine if H228A/D132A heterodimer was actually formed during the incubation period, time‐dependent formation of heterodimer was monitored by ESI‐TOF MS. The spectral patterns showed that, before mixing, each protein was present as folded homodimer in solution [Fig. 6(A), left panel]. The highest peak of H228A homodimer was the +21 charged ion whose observed m/z value at the peak apex was 5090.5 [Fig. 6(A), right panel]. This is in good agreement with the calculated value of 5087.8 (Supporting Information Table S3). Similar spectral pattern was observed with D132A protein solution [Fig. 6(B)]. The m/z value of its +21 ion at the apex was 5065.3, which is also in good agreement with the calculated value, 5064.4 (Supporting Information Table S3). In both cases, m/z values of the peak apexes were slightly higher than the calculated values. In addition, these peaks had a tailing toward the higher mass region. These could be ascribed to the adduct of solvent molecules due to incomplete desolvation,18 and are commonly seen in ESI‐MS spectra of proteins in aqueous solutions.
Figure 6.
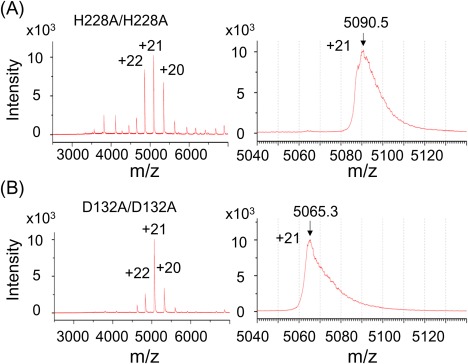
Representative ESI‐TOF MS spectra of H228A and D132A mutant protein. The H228A and D132A mutant proteins of CN2 were prepared in 100 mM ammonium acetate, pH 7.5, and subjected to ESI‐TOF MS analysis. The overall spectra were shown in the left panel. Right panels are magnified graphs around +21 charged ions. Arrows indicate the positions of calculated m/z values.
Next, in order to monitor the subunit exchange reaction, the two mutant proteins were incubated together and analyzed by ESI‐TOF MS [Fig. 7(A)]. The overall spectral patterns did not change throughout the incubation period, but each charge‐state of the peak envelopes showed that, in addition to the peaks of H228A/H228A and D132A/D132A homodimers, a third peak appeared between these two peaks and was increased in a time‐dependent manner [Fig. 7(A)]. The mass value of the third peak was in agreement with the predicted value of H228A/D132A heterodimer.
Figure 7.
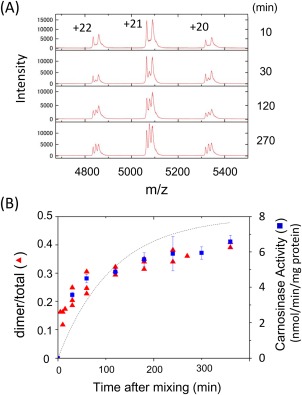
Time‐dependent homodimer exchange of CN2 analyzed by ESI‐TOF MS. (A) Detection of dimer exchange reaction. The H228A and D132A mutant proteins of CN2 prepared in 100 mM ammonium acetate, pH 7.5, were mixed at equimolar concentration and incubated for indicated time periods. Then, aliquots of samples were applied on ESI‐TOF MS. This figure shows the mass region around the +20, +21, and +22 ions of CN2 dimer. Time course of H228A/D132A heterodimer formation and enzymatic activity. Relative amounts of three CN2 dimers (H228A/H228A, H228A/D132A, and D132A/D132A) were estimated from the ESI‐TOF MS spectra using a peak‐fitting program, Peakfit (Seasolv, Framingham). The ratio of heterodimer relative to the whole dimer peak area was plotted (red triangle). The enzymatic activity was determined using carnosine as the substrate (blue square). Enzymatic activities were represented as mean ± SEM of triplicated determinations.
The increase in the peak intensity of H228A/D132A heterodimer was compared with carnosine‐hydrolyzing activity [Fig. 7(B)]. Heterodimer peak were detected within 30 min after mixing, and further increased over 6 h. In parallel with this, enzymatic activity was increased in a similar time course [Fig. 7(B)]. The rate of dimer exchange reaction depends on dissociation and association rates of CN2. If monomer concentrations are constant during incubation, heterodimer formation rate would be constant, while heterodimer dissociation rate would depend on the heterodimer concentration. Under the conditions, a simple exponential equation can be built as described in the Materials and Methods section, and a nonlinear regression analysis was carried out using this equation [Fig. 7(B)]. The regression line reflected the time course of heterodimer formation, although a few residual difference between the regression line and actual data was observed. The rate constant for dimer dissociation calculated from the regression analysis was 0.0081 (min−1). On the other hand, the rate constant for dissociation calculated from enzymatic activity was 0.012 (min−1), which was in good agreement with that from ESI‐TOF MS analysis. These results suggest that the time‐dependent recovery of enzymatic activity was due to the formation of H228A/D132A heterodimer.
Conservation of H228 of CN2 among M20 family metalloenzymes
The H228 residue of CN2 is located in a loop region between β11 and α11 strands of the dimerization domain [Fig. 8(A)]. To examine if this residue is conserved among the M20 family, M20 family members whose three‐dimensional (3D) structures are available in the PDB database were subjected to sequence alignment analysis [Fig. 8(B)]. These included CN2 from mouse5 (PDB: 2ZOF), CN1 from human (3DLJ), PepV from Lactobacillus delbrueckii 6 (1LFW), Xaa‐His dipeptidase from Vibrio alginolyticus 7 (3MRU), Sepap from Staphylococcus aureus (3KI9),8 PepT from Escherichia coli 9 (1VIX), acetylornithine deacetylase, ArgE, from Bacteroides thetaiotaomicron (3CT9), succinyl‐diaminopimelate desuccinylase, DapE, from Neisseria meningitidis 14 (1VGY), and β‐alanine synthetase from Saccharomyces kluyveri 30 (2V8H). To compare the sequences around the His residue, the 3D structure of each enzyme was browsed to identify the amino acids corresponding to the β11‐loop‐α11 region of CN2. Then the sequences of this region were subjected to sequence alignment analysis. Consequently, it was found that a histidine residue was present in the loop regions of the M20 family member proteins examined.
Figure 8.
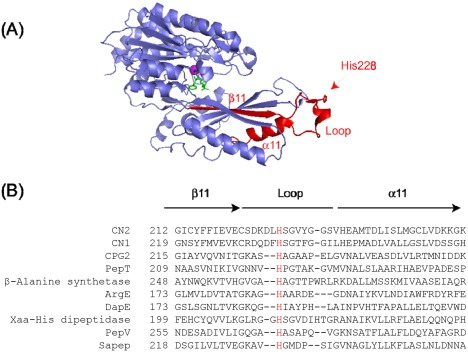
Conservation of histidine residues corresponding to H228 of CN2 among M20 family metalloproteins. (A) One subunit of a CN2 dimer was drawn as a ribbon model. Location of the loop containing His228, together with neighboring β11 and α11 strands were shown in red. The side chain of His228 was drawn in red sticks. (B) The amino acid sequence of β11‐loop‐α11 region of CN2 was subjected to sequence alignment analysis with eight other members of M20 family proteins whose structures have been reported in a published paper or deposited to PDB database. Sequence alignment was carried out using the Clustal omega program.29 Histidine residues corresponding to H228 of CN2 were shown in red.
The conserved histidine residues were mapped in representative crystal structures of M20 family enzymes (Supporting Information Fig. S4). As mentioned in the introduction section, there are two types of enzymes with different quaternary structures in this family. The majority of M20 family proteins are composed of one catalytic and one dimerization domains as in the case with CN2, and are likely to be present as a dimer. On the other hand, PepV, Sapep, and Xaa‐His dipeptidase are composed of one catalytic domain and one lid domain that is unlikely to form CN2‐like structures of homodimeric complex. In all these structures, the histidine residue was located in the loop region corresponding to the loop between β11 and α11 of CN2.
Discussion
In the present study, we examined the reaction mechanism of metallopeptidase CN2 using native ESI‐TOF MS, which enabled us to distinguish folded dimer, folded monomer and denatured monomer in solution. We then developed a method to monitor dimer exchange reaction of CN2 using ESI‐TOF MS in combination with recombinant DNA technology, and obtained evidence showing that intradimer interaction of H228 of the dimerization domain with the active site of the dimer counterpart is essential to the catalytic activity of CN2.
It has now been considered that ESI‐TOF MS is one of the strategies to analyze non‐covalent protein complexes in aqueous solution.18, 19, 20, 21, 25 To analyze a protein complex with ESI‐TOF MS, however, the interaction should be strong enough so that the complex is kept during measurement. Thus we first tested if CN2 homodimer, which is formed mainly through hydrophobic interaction, is detected actually as a homodimer in ESI‐TOF MS analysis. As the result, CN2 prepared in a neutral pH non‐denaturing solution was detected predominantly as a dimer in ESI‐TOF MS as well as in size exclusion chromatography (Fig. 2). These indicated that ESI‐TOF MS spectra of CN2 reflects its oligomeric states in an aqueous solution.
CN2 dimer seems to be a stable complex in solution. At 10 µM, which is the concentration of samples used for ESI‐TOF MS analysis, CN2 was detected almost exclusively as a dimer in size exclusion chromatography [Fig. 2(D)]. We tried to analyze lower concentrations of CN2, but even at 0.1 µM, CN2 monomer accounted for about 15% of the total peak area (Supporting Information Fig. S2). Therefore, more sensitive method would be required to determine the binding constant of the homodimer precisely. However, from the present result, a rough estimation of K d value was calculated to be 2.6 nM.
It has previously been shown that, in a CN2 crystal, H228 of the dimerization domain physically interacts with the substrate analogue on the catalytic domain.5 Such interaction of a dimerization/lid domain residue with the active site was first found in PepV‐inhibitor complex (6), and then also shown in several other M20 family proteins including β‐alanine synthetase (28) and Dug1p (10). We confirmed in this study that a his residue corresponding to H228 of CN2 is conserved in most other known structures of M20 family proteins including CPG2, PepT, ArgE, and DapE (Fig. 8). M20 family proteins are present either as “closed” or “open” form, possibly due to domain motion upon substrate binding.9, 19 Crystal structures of closed forms of various enzymes show that the histidine residue could interact with the active site (Supporting Information Fig. S4), indicating that this is a feature shared by many M20 family enzymes. In some proteins, however, structure of the closed form has not been available yet, and thus are waited to be solved.
Involvement of dimerization/lid domain in enzymatic reaction was also suggested by biochemical analyses of aminoacylase‐1 (Acy‐1), where activity of a dimerization domain mutant H206N was significantly reduced, and was partially recovered by incubation with a metal ligand mutant such as E147A.11, 15 Although the 3D structure of dimerization domain was not available, sequence similarity between Acy‐1 and PepV, together with the crystal structure of PepV induced a conclusion that H206 is essential to the enzymatic reaction.11, 15
In CN2, H228A mutant protein showed no enzymatic activity, indicating that H228 is essential to its activity.5 However, it could not exclude the possibility that the loss of enzymatic activity was due to some changes in three dimensional structure in the catalytic domain or the overall structure induced by the mutation. To clarify this problem, in the present study, H228A mutant of CN2 was incubated with another inactive mutant, D132A, and shown that this resulted in partial recovery of enzymatic activity (Fig. 5). This can be explained by the formation of H228A/D132A heterodimer, in which a metal binding site from H228A mutant and the His228 residue from D132A mutant interacted to form a catalytically active reaction center. In other words, the catalytic domain of H228A mutant has a functional conformation, and its reaction center is enzymatically active when H228 residue from the dimer counterpart is available.
Although the recovery of activity could be explained by H228A/D132A heterodimer formation, it is still to be clarified if the heterodimer was actually formed in the solution. So far, several methods have been reported to prepare proteins with which dimer exchange reaction could be monitored by mass spectrometry. For example, protein preparations of an enzyme from two different bacterial species were used for monitoring dimer exchange reaction by mass spectrometry.31 Another study showed that preparation of normal and N15‐labeled proteins were applicable to mass spectrometric analysis of dimers.32 In this study, we have prepared H228A and D132A mutant proteins with different N‐terminal additional sequences. This was achieved by subcloning cDNAs into pGEX‐6P3 and pGEX‐4T3 vectors, whose final products after protease digestion had an N‐terminal additional sequences of GPLGSPN and GSPN, respectively (Fig. 4). Therefore, the mass value of H228A monomer was 245.3 Da larger than D132A monomer. Since adding different N‐ or C‐terminal sequences is applicable to any recombinant proteins, and the short chain added to the terminus might scarcely affect the folding of a native form, this method could be a general strategy to monitor subunit exchange reaction of homodimeric proteins.
The mass spectrometric analysis showed that dimer exchange reaction of CN2 was a slow event, taking at least several hours to reach near‐equilibrium (Fig. 7). Since this time course of dimer formation was closely similar to that of enzymatic activity, it was supported that the recovery of enzymatic activity was due to the formation of a heterodimer. This slow dissociation rate also allowed us to analyze its dimer‐monomer ratio by size exclusion HPLC, in which samples are in general diluted several folds, and thus the equilibrium could be shifted to the monomeric state.
From these results, it was concluded that H228 of CN2 is essential in enzymatic reaction. It is to be noted that the present data were mainly obtained from mutant CN2 proteins, which may have some properties different from wild‐type CN2. Since amino acid sequences of dimer interfaces were unchanged both in D132A and H228 mutants, and the overall structure of the mutants were maintained during dimer exchange reaction, it was supposed that the experiments using the mutant proteins reflect the properties of wild‐type CN2. However, further attempts may be desirable to determine the properties of CN2. From this point of view, preparing 15N‐labeled wild type protein may allow us to examine dimer exchange in more native‐like conditions. It would also be valuable to determine the crystal structures of H228A/H228A, H228A/D132A, and D132A/D132A dimers to confirm that the mutants retain the structure of wild‐type CN2.
This mechanism described here is possibly shared by several other M20 family enzymes (Supporting Information Fig. S4). There are some differences, however, in reaction mechanisms among enzymes. In PepD, a histidine residue is also present at the corresponding position of the lid domain, but its replacement to alanine does not affect its enzymatic activity, instead, an arginine residue located in the adjacent loop is essential to the enzymatic activity.7 Furthermore, M28 family enzymes, which share the feature of catalytic domain and active site structure with M20 family, do not have dimerization/lid domain and active as a monomeric form.33 Therefore, the mechanism may have been acquired and modified during the evolution to adapt for environments and substrates to be digested.
The results in this study also demonstrated that ESI‐TOF MS in combination with recombinant DNA technology allows us to monitor dimer exchange reaction of a homodimer. Sample preparation is relatively simple, thus the procedure would be applicable to various studies concerning non‐covalent protein complexes.
Materials and Methods
Preparation of wild‐type and mutant CN2 proteins
A cDNA encoding wild‐type mouse CN2 was cloned into the EcoRI and XhoI sites of pGEX‐6P3 vector (GE Healthcare, Little Chalfont, UK).2 Using this plasmid, cDNAs encoding I319K, H228A, and D132A mutant proteins were prepared by the method of QuickChange (Stratagene, La Jolla). The cDNA encoding D132A mutant protein was further subcloned into pGEX‐4T3 vector (GE Healthcare) using EcoRI and XhoI. Each recombinant protein was expressed in E. coli and purified from the soluble extracts as described previously34 with a few modifications. Briefly, bacterial pellets were sonicated in a buffer containing 25 mM Tris‐HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, and 10 μL/mL aprotinin on ice. After centrifugation, GST‐CN2 fusion protein was trapped on Glutathione‐Sepharose resin (GE Healthcare), washed with the same buffer, and incubated with PreScission protease (GE Healthcare) for 12 to 16 h on ice with constant shaking. Proteins liberated from the resin were collected and further purified on a HiTrap Q column (1 mL bed volume, GE Healthcare) with a linear gradient of 50 to 500 mM NaCl in a buffer containing 25 mM Tris‐HCl, pH 7.4, and 1 mM dithiothreitol (DTT). The samples were then dialyzed against 100 mM ammonium acetate, pH 7.5, and 1 mM DTT at 4°C, concentrated to 2 mg/mL by ultrafiltration (Amicon Ultra, Mw. 30 K cut off, Merck Millipore, Billerica) and stored at −20°C.
Determination of enzymatic activity
An assay mixture containing 20 μg/mL CN2, 100 mM ammonium acetate, 10 mM DTT, 200 μM MnCl2, and 25 mM Tris‐HCl, pH 7.5 was incubated for 30 min at 37°C unless otherwise indicated. After the reaction, free histidine was detected by the reaction with o‐phthalaldehyde in alkaline solution,35 and determined by the absorbance at 405 nm. When pH‐dependency of the enzyme was examined, the same assay conditions were used except that the buffer component was replaced with Tris‐HCl, pH 8.0, 8.8, or 9.5, or sodium phosphate, pH 7.5, 6.5, or 5.5 for respective pH points.
Size exclusion chromatography
Size exclusion chromatography was carried out using a SEC‐3 column (4.6 × 300 mm, particle size 3 μm, pore size 300 Å, Agilent Technologies, Santa Clara) equipped with an high‐performance liquid chromatography (HPLC) system (JEOL, Model 616, Akishima, Japan). The column was equilibrated with 100 mM ammonium acetate, pH 7.5, and then 5 µL of 10 µM sample protein was injected using a manual injector. Proteins were separated at a flow rate of 0.35 mL/min, and eluted proteins were monitored by absorbance at 280 nm. Molecular weight standards used were thyroglobulin (670 kDa), immunoglobulin G (180 kDa), bovine serum albumin (66 kDa), ovalbumin (45 kDa), myoglobin (17 kDa), cytochrome c (12 kDa), and aprotinin (6.5 kDa).
Mass spectrometry
Electrospray ionization time‐of‐flight mass spectrometry (ESI‐TOF MS) was carried out using AccuTOF® mass spectrometer (JEOL). To optimize the transmission of high‐m/z protein complex ions, the radio‐frequency (RF) power supply for the quadrupole‐type ion guide, situated between the atmospheric pressure ionization interface and the TOF mass analyzer, was modified to lower the RF frequency from standard 3 MHz to 0.8 MHz, and the pressure of the first differential pumping region was raised from about 200 Pa to 320 Pa. Electrospray ionization was achieved using a nanospray tip (30 μm tip ID and 50 μm tubing ID, SilicaTip^TM, New Objective, Woburn) connected to a microsyringe set on a syringe pump, and positioned using an XYZ‐stage. Sample proteins were prepared at 10 µM in a solution containing 100 mM ammonium acetate, pH 7.5, and 1 mM dithiothreitol unless otherwise indicated. The sample was injected into the electrospray ion source at a flow rate of 600 nL/min using the syringe pump, and a voltage of 1650 volts was applied to the nanospray tip. Mass measurement was carried out on the TOF mass analyzer in the positive ion mode.
Kinetic analysis
Formation of H228A/D132A heterodimer after mixing of the same concentrations of H228A and D132A homodimer solutions was subjected to regression analysis. Given that the association and dissociation rate constants of H228A‐H228A, H228A‐D132A, and D132A‐D132A are indistinguishable, the expected initial concentration of each component could be expressed as follows:
where [A] and [B] are concentrations of H228A and D132A monomers; [AA] and [BB] are those of respective dimers; and k on and k off are association and dissociation rate constants, respectively. When the reaction is reached the equilibrium, the concentrations of the components can be expressed as:
Association of A and B to form AB would be at a constant rate if [A] and [B] can be considered to be constant throughout the reaction. On the other hand, dissociation rate of AB is proportional to [AB]. Thus heterodimer formation rate can be described as:
This could be converted to as follows:
where r is the fraction of [AB] in the total dimer concentration. Actual data of heterodimer concentration obtained from enzymatic activity and ESI‐TOF MS analysis were fitted to the equation by nonlinear regression analysis using the function, nls, of the software, R (The R Foundation for Statistical Computing, Vienna, Austria).
Supporting information
Supporting Information
Acknowledgment
The authors thank Dr. Masami Kusunoki and Hideaki Unno for helpful discussions regarding the crystal structure and reaction mechanism.
References
- 1. Teufel M, Saudek V, Ledig JP, Bernhardt A, Boularand S, Carreau A, Cairns NJ, Carter C, Cowley DJ, Duverger D, Ganzhorn AJ, Guenet C, Heintzelmann B, Laucher V, Sauvage C, Smirnova T (2003) Sequence identification and characterization of human carnosinase and a closely related non‐specific dipeptidase. J Biol Chem 278:6521–6531. [DOI] [PubMed] [Google Scholar]
- 2. Otani H, Okumura N, Hashida‐Okumura A, Nagai K (2005) Identification and characterization of a mouse dipeptidase that hydrolyzes L‐carnosine. J Biochem 137:167–175. [DOI] [PubMed] [Google Scholar]
- 3. Otani H, Okumura A, Nagai K, Okumura N (2008) Colocalization of a carnosine‐splitting enzyme, tissue carnosinase (CN2)/cytosolic non‐specific dipeptidase 2 (CNDP2), with histidine decarboxylase in the tuberomammillary nucleus of the hypothalamus. Neurosci Lett 445:166–169. [DOI] [PubMed] [Google Scholar]
- 4. Okumura N. Carnosine dipeptidase II In: Rawlings ND, Salvesen GS, Eds. (2013) Handbook of proteolytic enzymes. Oxford: Academic Press, pp 1596–1600. [Google Scholar]
- 5. Unno H, Yamashita T, Ujita S, Okumura N, Otani H, Okumura A, Nagai K, Kusunoki M (2008) Structural basis for substrate recognition and hydrolysis by mouse carnosinase CN2. J Biol Chem 283:27289–27299. [DOI] [PubMed] [Google Scholar]
- 6. Jozic D, Bourenkow G, Bartunik H, Scholze H, Dive V, Henrich B, Huber R, Bode W, Maskos K (2002) Crystal structure of the dinuclear zinc aminopeptidase PepV from Lactobacillus delbrueckii unravels its preference for dipeptides. Structure 10:1097–1106. [DOI] [PubMed] [Google Scholar]
- 7. Chang CY, Hsieh YC, Wang TY, Chen YC, Wang YK, Chiang TW, Chen YJ, Chang CH, Chen CJ, Wu TK (2010) Crystal structure and mutational analysis of aminoacylhistidine dipeptidase from Vibrio alginolyticus reveal a new architecture of M20 metallopeptidases. J Biol Chem 285:39500–39510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Girish TS, Gopal B (2010) Crystal structure of Staphylococcus aureus metallopeptidase (Sapep) reveals large domain motions between the manganese‐bound and apo‐states. J Biol Chem 285:29406–29415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hakansson K, Miller CG (2002) Structure of peptidase T from Salmonella typhimurium . Eur J Biochem 269:443–450. [DOI] [PubMed] [Google Scholar]
- 10. Singh AK, Singh M, Pandya VK, G LB, Singh V, Ekka MK, Mittal M, Kumaran S (2014) Molecular basis of peptide recognition in metallopeptidase Dug1p from Saccharomyces cerevisiae . Biochemistry 53:7870–7883. [DOI] [PubMed] [Google Scholar]
- 11. Lindner HA, Lunin VV, Alary A, Hecker R, Cygler M, Menard R (2003) Essential roles of zinc ligation and enzyme dimerization for catalysis in the aminoacylase‐1/M20 family. J Biol Chem 278:44496–44504. [DOI] [PubMed] [Google Scholar]
- 12. Lundgren S, Gojkovic Z, Piskur J, Dobritzsch D (2003) Yeast beta‐alanine synthase shares a structural scaffold and origin with dizinc‐dependent exopeptidases. J Biol Chem 278:51851–51862. [DOI] [PubMed] [Google Scholar]
- 13. Meinnel T, Schmitt E, Mechulam Y, Blanquet S (1992) Structural and biochemical characterization of the Escherichia coli argE gene product. J Bacteriol 174:2323–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nocek BP, Gillner DM, Fan Y, Holz RC, Joachimiak A (2010) Structural basis for catalysis by the mono‐ and dimetalated forms of the dapE‐encoded N‐succinyl‐L,L‐diaminopimelic acid desuccinylase. J Mol Biol 397:617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lindner HA, Alary A, Boju LI, Sulea T, Menard R (2005) Roles of dimerization domain residues in binding and catalysis by aminoacylase‐1. Biochemistry 44:15645–15651. [DOI] [PubMed] [Google Scholar]
- 16. Ganem B, Li YT, Henion JD (1991) Detection of noncovalent receptor ligand complexes by mass‐spectrometry. J Am Chem Soc 113:6294–6296. [Google Scholar]
- 17. Loo RRO, Goodlett DR, Smith RD, Loo JA (1993) Observation of a noncovalent ribonuclease S‐protein S‐peptide complex by electrospray ionization mass‐spectrometry. J Am Chem Soc 115:4391–4392. [Google Scholar]
- 18. Benesch JL, Ruotolo BT, Simmons DA, Robinson CV (2007) Protein complexes in the gas phase: technology for structural genomics and proteomics. Chem Rev 107:3544–3567. [DOI] [PubMed] [Google Scholar]
- 19. Yin S, Loo JA (2009) Mass spectrometry detection and characterization of noncovalent protein complexes. Methods Mol Biol 492:273–282. [DOI] [PubMed] [Google Scholar]
- 20. Heck AJ (2008) Native mass spectrometry: a bridge between interactomics and structural biology. Nat Methods 5:927–933. [DOI] [PubMed] [Google Scholar]
- 21. Kaltashov IA, Bobst CE, Abzalimov RR (2013) Mass spectrometry‐based methods to study protein architecture and dynamics. Protein Sci 22:530–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sobott F, Benesch JL, Vierling E, Robinson CV (2002) Subunit exchange of multimeric protein complexes. Real‐time monitoring of subunit exchange between small heat shock proteins by using electrospray mass spectrometry. J Biol Chem 277:38921–38929. [DOI] [PubMed] [Google Scholar]
- 23. Yamazaki Y, Takao T (2008) Metalation states versus enzyme activities of Cu, Zn‐superoxide dismutase probed by electrospray ionization mass spectrometry. Anal Chem 80:8246–8252. [DOI] [PubMed] [Google Scholar]
- 24. Fukuda M, Takao T (2012) Quantitative analysis of deamidation and isomerization in beta(2)‐microglobulin by O‐18 labeling. Anal Chem 84:10388–10394. [DOI] [PubMed] [Google Scholar]
- 25. Boeri Erba E, Petosa C (2015) The emerging role of native mass spectrometry in characterizing the structure and dynamics of macromolecular complexes. Protein Sci 24:1176–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schrodinger: LLC; (2010) The PyMOL Molecular Graphics System, Version 1.3r1. [Google Scholar]
- 27. Konermann L, Douglas DJ (1997) Acid‐induced unfolding of cytochrome c at different methanol concentrations: electrospray ionization mass spectrometry specifically monitors changes in the tertiary structure. Biochemistry 36:12296–12302. [DOI] [PubMed] [Google Scholar]
- 28. Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105–132. [DOI] [PubMed] [Google Scholar]
- 29. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lundgren S, Andersen B, Piskur J, Dobritzsch D (2007) Crystal structures of yeast beta‐alanine synthase complexes reveal the mode of substrate binding and large scale domain closure movements. J Biol Chem 282:36037–36047. [DOI] [PubMed] [Google Scholar]
- 31. Vis H, Dobson CM, Robinson CV (1999) Selective association of protein molecules followed by mass spectrometry. Protein Sci 8:1368–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chevreux G, Atmanene C, Lopez P, Ouazzani J, Van Dorsselaer A, Badet B, Badet‐Denisot MA, Sanglier‐Cianferani S (2011) Monitoring the dynamics of monomer exchange using electrospray mass spectrometry: the case of the dimeric glucosamine‐6‐phosphate synthase. J Am Soc Mass Spectrom 22:431–439. [DOI] [PubMed] [Google Scholar]
- 33. Gilboa R, Spungin‐Bialik A, Wohlfahrt G, Schomburg D, Blumberg S, Shoham G (2001) Interactions of Streptomyces griseus aminopeptidase with amino acid reaction products and their implications toward a catalytic mechanism. Proteins 44:490–504. [DOI] [PubMed] [Google Scholar]
- 34. Yamashita T, Unno H, Ujita S, Otani H, Okumura N, Hashida‐Okumura A, Nagai K, Kusunoki M (2006) Crystallization and preliminary crystallographic study of carnosinase CN2 from mice. Acta Crystallogr F62:996–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bando K, Shimotsuji T, Toyoshima H, Hayashi C, Miyai K (1984) Fluorometric assay of human serum carnosinase activity in normal children, adults and patients with myopathy. Ann Clin Biochem 21:510–514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information