

Abstract
Retroviral DNA integration is mediated by nucleoprotein complexes (intasomes) comprising a pair of viral DNA ends synapsed by a tetramer of integrase. Current integrase inhibitors act on intasomes rather than free integrase protein. Structural and functional studies of intasomes are essential to understand their mechanism of action and how the virus can escape by mutation. To date, prototype foamy virus (PFV) is the only retrovirus for which high‐resolution structures of intasomes have been determined. In the PFV intasome structure, only the core domains of the outer subunits are ordered; the N‐terminal domain, C‐terminal domain, and N‐terminal extension domain are disordered. Are these “missing domains” required for function or are they dispensable? We have devised a strategy to assemble “hetero‐intasomes” in which the outer domains are not present as a tool to assess the functional role of the missing domains for catalysis of integration. We find that the disordered domains of outer subunits are not required for intasome assembly or catalytic activity as catalytic core domains can substitute for the outer subunits in the case of both PFV and HIV‐1 intasomes.
Keywords: HIV‐1, integration, integrase, intasomes
Introduction
Integration of viral DNA into chromosomal DNA is an essential step in the replication of HIV‐1 and other retroviruses (reviewed in Ref. 1). The viral integrase protein is the enzyme that carries out the key DNA cutting and joining steps. The viral DNA made by reverse transcription is initially blunt ended. The first step in the integration process is the removal of two nucleotides from each 3' end of the viral DNA; a reaction termed 3' end processing. In the next step, DNA strand transfer, a pair of transesterification reactions2 inserts the viral DNA ends into a target DNA. In the case of HIV‐1, the sites of insertion on each strand of target DNA are staggered by five nucleotides. Both 3' end processing and DNA strand transfer are carried out by viral integrase protein. The result is an integration intermediate3, 4 in which the 3' ends of the viral DNA are covalently joined to the 5' ends of target DNA at the site of insertion. Repair of the integration intermediate by cellular enzymes completes the integration process. The 5' protruding stagger of the sites of joining on the two target DNA strands results in a short direct repeat of target DNA sequence flanking the integrated provirus.
DNA integration proceeds through a series of stable nucleoprotein complexes that are collectively termed intasomes.5 After reverse transcription, a tetramer of integrase bridges the two viral DNA ends in a nucleoprotein complex termed the Stable Synaptic Complex (SSC). The 3' end processing reaction occurs within the SSC to generate the Cleaved Donor Complex (CDC). The CDC then captures a target DNA and the DNA strand transfer reaction covalently joins viral DNA to target DNA. The product DNA remains stably associated with the integrase tetramer in the resulting Strand Transfer Complex (STC). In vivo, the STC is disassembled by a pathway that is yet to be determined and cellular enzymes complete the integration process.
HIV‐1 integrase is comprised of three domains; the catalytic core domain (CCD) is linked to the N‐terminal and C‐terminal domains by flexible linkers. PFV integrase has an additional N‐terminal extension domain that is not present in HIV‐1 integrase. High‐resolution structures of individual integrase domains have been determined for several retroviral integrases, as have two‐domain structures (reviewed in Ref. 6). However, these structures lack DNA and prototype foamy virus (PFV) is the only retrovirus for which high‐resolution structures of intasomes have been determined to date.7, 8, 9 The structure of PFV intasomes could not have been predicted from the earlier partial structures of integrase protein alone because DNA‐protein interactions play a crucial role in their organization. The overall arrangement of the PFV integrase subunits is similar in the SSC, CDC, and STC [Fig. 1(A)]. Of the four integrase subunits in the complexes, only the inner two subunits are involved in interactions with DNA. The catalytic domain of the outer subunits does not contact DNA and the N‐terminal extension domain, N‐terminal domain (NTD) and C‐terminal domain of the outer subunits are not seen due to their mobility in the crystal lattice. This raises the question of whether the domains that are disordered in the PFV structures are required for catalysis of DNA integration. To address this question, we have devised strategies to assemble heterointasomes in which the domains that are disordered in the PFV structures are not present and test their activity. We find that for both PFV and HIV‐1 intasomes, the domains that are missing in the PFV intasome structures do not contribute to catalytic activity in vitro.
Figure 1.
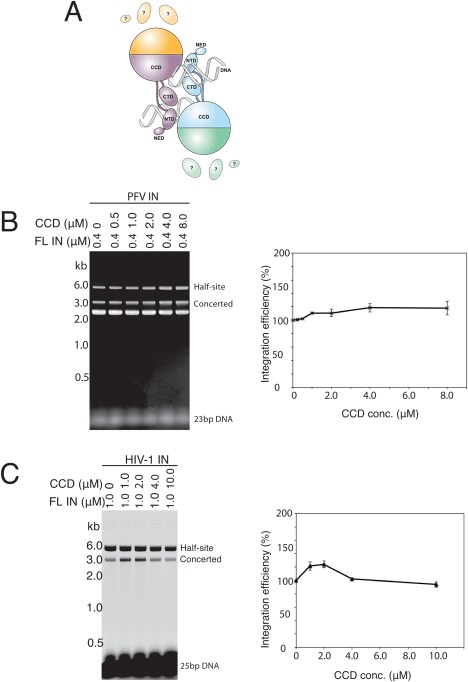
CCD does not inhibit concerted DNA integration. A: Schematic of the PFV intasome structure. The integrase monomers in the intasome are distinguished by their colors. The CCD, NTD, C‐terminal domain (CTD), and N‐terminal extension domain (NED) are distinguished by shape. The pair of viral DNA ends is represented as helices. All the contacts between integrase and viral DNA are with the inner subunits. The CTD, NTD and NED of the outer subunits are disordered. Reproduced from Ref. 16. B: The PFV catalytic domain does not inhibit concerted DNA integration. Reactions were carried out with a constant concentration of full‐length PFV integrase and increasing concentrations of the PFV integrase CCD. The gel was visualized by ethidium bromide staining. B: The HIV‐1 catalytic domain does not inhibit concerted DNA integration. Reactions were carried out with a constant concentration of full‐length HIV‐1 integrase and increasing concentrations of the HIV‐1 integrase CCD. The gel was visualized by fluorescence scanning. Integration products are quantitated in the panels on the right (2 replicates).
Results
CCD does not inhibit concerted integration mediated by PFV integrase and minimally perturbs the HIV‐1 integrase reaction
The crystal structures of PFV intasomes reveal a tetramer of integrase in which only the inner two subunits engage the pair of viral DNA ends.7, 8, 9 The N‐terminal, N‐terminal extension, and C‐terminal domains of the outer two subunits are disordered [Fig. 1(A)]. Are the disordered domains required for catalytic activity or are they dispensable? We first titrated CCD into integration reactions with both PFV [Fig. 1(B)] and HIV‐1 [Fig. 1(C)] integrases. If the CCD can participate in intasome assembly by substituting for the outer full‐length subunits (as we show later), increasing concentrations of core domain would be expected to inhibit the reaction if the outer disordered domains are essential. No inhibition of concerted integration was observed with PFV integrase [Fig. 1(B)]; in fact, addition of the core domain mildly stimulated the reaction. In the case of HIV‐1, the integrase protein contained a fusion of Sulfolobus solfataricus chromosomal protein Sso7d to the N‐terminus. This extra domain greatly enhances the solubility and concerted integration activity of the HIV‐1 integrase.10 Mild stimulation by the core domain was also observed with lower concentrations of HIV‐1 integrase core domain, but the reaction efficiency returned to the basal level at the highest concentration of core domain tested. Although reproducible, the degree of stimulation by the core domain is marginal and we did not further explore this phenomenon. The assay we used can be confounded by the presence of contaminating nuclease that would result in linearized target DNA that co‐migrates with concerted integration products. All protein preparations tested negative for nuclease and product was only observed in the presence of viral DNA substrate (data not shown). The results are consistent with a dispensable role for the outer disordered domains in catalysis of integration.
The CCD assembles heterointasomes with full‐length integrase
To directly test whether the N‐terminal, N‐terminal extension, and C‐terminal domains of the outer two subunits of PFV integrase are required for catalysis of integration, we assembled hetero‐intasomes by including an excess of CCD in the intasome assembly reaction mixture. The CCD contained a His‐tag to enable purification of intasomes containing the CCD by nickel affinity chromatography [Fig. 2(A)]. After assembly, the intasomes were purified away from free full‐length integrase (FL‐IN) and CCD by size exclusion chromatography [Fig. 2(B)]. Intasomes assembled in the presence of an excess of CCD contained both full‐length integrase and CCD confirming that the CCD can be incorporated into intasomes [Fig. 2(C)]. Quantitation relative to standards revealed the purified PFV heterointasomes contained a molar ratio of 1:0.8 for full‐length integrase and CCD; the corresponding value for HIV‐1 heterointasomes was 1:0.6 (data not shown). Since intasomes assembled from only full‐length integrase are not recovered after Ni affinity chromatography (data not shown), we infer that a fraction of the intasomes have only one of the outer subunits substituted by CCD. Since the N‐terminal, N‐terminal extension, and C‐terminal domains of the inner two subunits are required for interaction with viral DNA,7 we infer that the CCD substitutes for the outer subunits only. Full‐length PFV integrase is monomeric in solution11 while the PFV core domain exists as a reversible monomer‐dimer equilibrium in solution (data not shown). HIV‐1 integrase exists as a monomer‐oligomer equilibrium (data not shown). Before comparing the activity of the hetero‐intasomes with the activity of intasomes assembled with full‐length integrase only, we first confirmed that such exchange of subunits does not occur after intasome assembly. Intasomes assembled with full‐length integrase only were incubated with an excess of CCD and then subjected to size exclusion chromatography. The intasome peak fractions revealed no CCD demonstrating the integrase subunits do not exchange once the intasomes have been assembled [Fig. 2(C)]. Subunit exchange was not tested for HIV‐1 intasomes.
Figure 2.
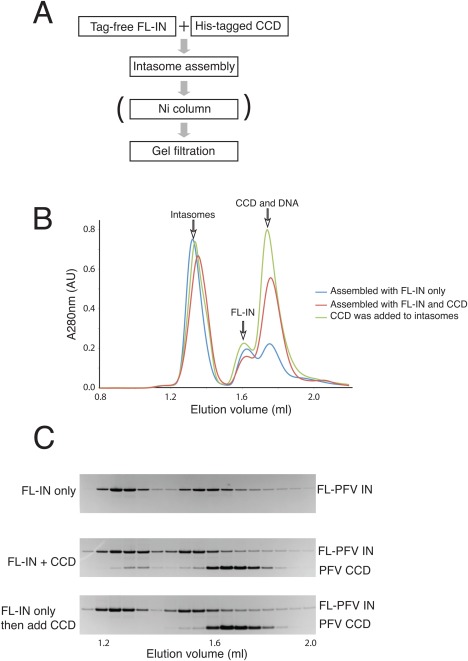
Assembly of PFV heterointasomes (SSC). A: Strategy for assembly and purification of heterointasomes. The Ni column step was included when purifying heterointasomes. FL‐IN, full‐length IN; CCD, catalytic core domain. B: Size‐exclusion chromatography of PFV intasomes. C: SDS‐PAGE of fractions eluted from the gel filtration column.
Heterointasomes (SSC) and homointasomes (SSC) exhibit indistinguishable catalytic activity
We next compared the activity of both SSC PFV and HIV‐1 heterointasomes and homointasomes in a concerted in vitro DNA integration assay. The expected concerted integration product is a linearized target DNA flanked by viral DNA ends [Fig. 3(A)]. Preassembled intasomes, purified by size exclusion chromatography [Fig. 3(B,C)], were incubated with a circular target DNA in the presence of Mg2+. The activities of heterointasomes and homointasomes were indistinguishable for both PFV and HIV‐1 intasomes [Fig. 3(D)]. As previously noted,5 although HIV‐1 integrase tends to promote half‐site integration in vitro, purified HIV‐1 intasomes catalyze exclusively concerted DNA integration. Sequencing the integration products of reactions with PFV and HIV‐1 heterointasomes revealed predominantly 4 and 5 bp target site duplications, respectively (Supporting Information Table 1), as expected for integration carried out by PFV and HIV‐1 integrases. The PFV reintegration products (see below) may have a lower fidelity of target site duplication than products formed from the conventional reaction pathway, but the number of sequenced products is too small to definitively draw this conclusion. A lower fidelity would not be surprising considering Mn2+ is required as the divalent metal ion cofactor. Thus, integration mediated by heterointasomes or homointasomes exhibits similar fidelity.
Figure 3.
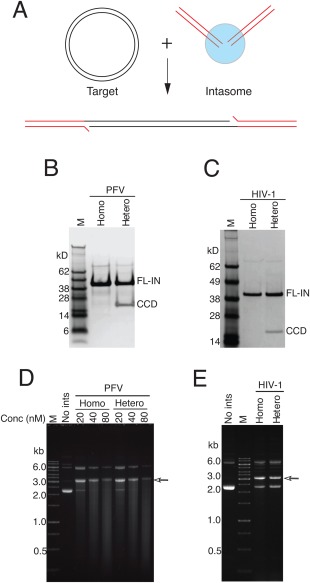
Activity of heterointasomes. A: Assembled intasomes were mixed with a circular target DNA in the presence of Mg2+. The integration product is linearized target DNA flanked by a pair of viral DNA ends. B: SDS‐PAGE of PFV heterointasomes. C: SDS‐PAGE of HIV heterointasomes. D: Activity of PFV heterointasomes. E: Activity of HIV heterointasomes. The final concentration of intasomes in the reaction mixture was 20 nM. The arrow indicates the migration position of the concerted integration product in agarose gel electrophoresis.
STC heterointasomes and homointasomes exhibit similar properties
PFV STCs can be assembled directly on DNA that mimics the integration product,12 bypassing the normal forward reaction pathway [Fig. 4(A)]. STCs assembled in this manner are identical to STCs made by the normal integration pathway.12 We assembled PFV STCs with full‐length integrase in the presence and absence of an excess of CCD and purified heterointasomes (STC) as described for heterointasomes (SSC). Assembly was carried out in the presence of Ca2+ to prevent catalysis. As expected, no reaction was observed when Mg2+ was added to either homointasomes (STC) or heterointasomes (STC) as the reaction is essentially irreversible with integrase remaining tightly associated with the product DNA. In the presence of Mn2+ both homointasomes (STC) and heterointasomes (STC) catalyzed “disintegration reactions” with comparable efficiency (data not shown). In the presence of a target DNA, some of the disintegration products reintegrate [Fig. 4(B)]. Like SSC intasomes, substituting the CCD for the outer subunits does not affect catalytic activity.
Figure 4.
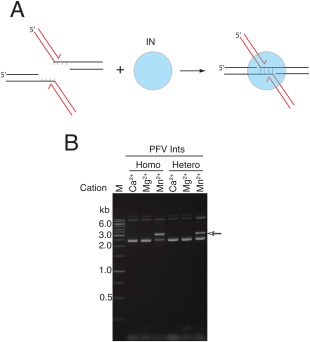
Heterointasomes (STC) and homointasomes (STC) exhibit similar properties. A: PFV STCs were assembled on branched DNA that mimics the integration products using full‐length PFV integrase or a mixture of full‐length and CCD and purified as in Figure 2A. B: The STCs were incubated in the presence of a circular target DNA and the indicated divalent metal ion. The STCs are stable in the presence of Mg2+, but disintegrate in the presence of Mn2+. The arrow indicates the migration position of the products reintegration of disintegration products in agarose gel electrophoresis.
Discussion
Determination of the crystal structures of PFV intasomes was a pivotal advance in understanding the molecular mechanism of retroviral DNA integration. A striking feature of the intasome is the very different contacts of the inner and outer subunits. Whereas the inner subunits engage DNA and reach across to hold the tetramer together, the N‐terminal, N‐terminal extension, and C‐terminal domains of the outer two subunits are disordered and the catalytic domain of the outer subunits appears to play only a scaffold role to stabilize the structure. Nonequivalent roles for subunits containing active sites, and redundancy of active sites, is not unusual in nucleoprotein complexes that mediate DNA recombination reactions. For example, the Tn3 resolvase complex contains six active sites, only two of which are directly involved in catalysis of recombination.13 The disorder of the noncatalytic domains in the outer subunits of retroviral intasomes immediately raises the question of whether these domains are functionally involved in catalysis of integration, play some secondary role or are functionally irrelevant.
Our strategy to answer this question was to assemble intasomes in which the disordered domains in the PFV intasome structure are not present. Heterointasomes exhibit catalytic activity that is identical to that of intasomes assembled with full‐length integrase. We conclude that the outer domains that are disordered in the PFV intasomes structures are not involved in catalysis of DNA integration. We assembled heterointasomes by including both full‐length integrase and an excess CCD with a His tag in the assembly reaction. We cannot exclude the possibility that some intasomes have only one of the outer subunits substitutes by the CCD but in the presence of an excess of CCD these are likely to be a minor fraction. We see no diminution of the efficiency of catalytic activity even over a wide range of ratios of catalytic core excess (data not shown).
Attempts to crystallize HIV‐1 intasomes have been frustrated by their propensity to aggregate. Based on the assumption that HIV‐1 intasomes have a similar overall organization as PFV intasomes, we speculated that the outer domains corresponding to the disordered domains in the PFV structure might be responsible for this aggregation. We, therefore, compared the aggregation properties of HIV‐1 homointasomes and heterointasomes. The gel filtration profiles of these two types of intasomes were identical (data not shown), demonstrating that the “outer” domains are not responsible for aggregation. Nevertheless, since the “disordered outer domains” are dispensable for key steps of integration, heterointasomes provide a further tool that may aid high‐resolution structural studies of HIV‐1 intasomes. It remains to be determined whether these domains play a functional role for DNA integration in vivo but, consistent with a role in chromatin interaction, deletion of the NTD and CTD domains of the outer subunits of PFV intasomes reduces their ability to pull‐down nucleosomes.14
Materials and Methods
Oligonucleotides and expression vectors
Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, Iowa). DNA substrate for assembly of PFV SSCs was prepared by annealing PFV‐U5 AATATACAAAATTCCATGACA and PFV‐U5R ATTGTCATGGAATTTTGTATATT. PFV DNA substrates for assembly of STCs on product DNA were assembled by annealing PFV‐dis, AATATACAAAATTCCATGACAGTACGATCCGTCTTGGCTCCTA, PFV‐U5R, and PFV‐disR, TAGGAGCCAAGACGGATC. The corresponding oligonucleotides for assembling the HIV substrates are HIV‐U5, AGCGTGGGCGGGAAAATCTCTAGCA; HIV‐U5R, ACTGCTAGAGATTTTCCCGCCCACGCT; HIV‐dis AGCGTGGGCGGGAAAATCTCTAGCAGTTACAGTCAGCGTACGTCGGCA; HIV‐ disR TGCCGACGTACGCTGACT; HIV‐U5R. Fluorescent DNA substrates were prepared by attaching 6‐FAM fluorophor at 5′ end of oligonucleotides.
The PFV integrase expression vector pSSH6P‐PFV‐IN‐FL was generously provided by Peter Cherepanov. The vector for expression of the PFV core domain was constructed by synthesizing the coding sequence and cloning into pET15B (GenScript). The vector for expression of HIV integrase with the Sso7d domain fused to the N‐terminus (Sso7d‐IN) has been previously described.10
Protein expression and purification
Full‐length PFV integrase was expressed and purified as described.11 The hyperactive HIV‐1 integrase Sso7d‐IN was used for this study. HIV‐1 integrases and the PFV core domain were expressed and purified essentially as described15 with minor modifications. Briefly, integrase was expressed in E. coli BL21(DE3) and the cells were lysed in buffer containing 20 mM HEPES, pH 7.5, 2 mM 2‐mercaptoethanol, 20 mM imidazole,1M NaCl, and 10% glycerol. The protein was then purified by nickel‐affinity chromatography and, for full‐length integrase, the His‐tag was removed with thrombin. Aggregated protein was removed by gel filtration on a Hiload 26/60 Superdex‐200 column (GE Healthcare) for full‐length protein or Hiload 26/60 Superdex‐75 for CCDs; columns were equilibrated with 20 mM Hepes pH 7.5, 5 mM DTT, 1 mM EDTA, 1M NaCl, and 10% glycerol. Proteins were concentrated using an Amicon Centrifugal concentrator (EMD Millipore) if necessary, flash‐frozen in liquid nitrogen and stored at −80°C.
Intasome assembly and purification
PFV homointasomes were assembled by dialysis as described,7 except the protein and DNA concentrations were 25 and 10 µM, respectively. HIV‐1 homointasomes were assembled by mixing 1.0 μM integrase, 0.5 μM DNA substrate, 20 mM HEPES pH 7.5, 5 mM CaCl2, 10 mM DTT, 4 μM ZnCl2, 100 mM NaCl, and 25% glycerol (final), followed by incubation at 37°C for 1 h. NaCl was then added to 500 mM, and after incubation at room temperature for 15 min the mixture was centrifuged at 15,000g for 15 min. The supernatant was concentrated using a Vivaspin centrifugal concentrator (Satorius Stedim Biotech). Intasomes were separated from free protein and DNA by size‐exclusion chromatography on a Superdex 200 PC 3.2/30 column (GE Healthcare) equilibrated with 20 mM HEPES pH 7.5, 5 mM DTT, 500 mM NaCl, and 20% glycerol.
Heterointasomes were prepared as described above (same full‐length final protein concentration), except His‐tagged CCD was included in the assembly mixture at a ratio of 8:1 for PFV and 3:1 for HIV‐1. The His‐tagged heterointasomes were subjected to the additional purification steps of Ni‐affinity chromatography and anion exchange chromatography prior to the size exclusion chromatography. The intasome mixture was first loaded onto a HisTrap HP column (GE Healthcare) previously equilibrated with 20 mM HEPES pH 7.5, 20 mM imidazole, 5 mM 2‐mercaptoethanol, 0.5M NaCl, and 20% glycerol, and then washed extensively with the same buffer including 50 mM imidazole. Heterointasomes were eluted with a linear gradient of 50 to 500 mM imidazole. Intasome‐containing fractions were combined and diluted with 20 mM Tris pH 8.0, 0.5 mM TCEP, and 20% glycerol to lower the NaCl to 200 mM and then quickly loaded onto a Mono Q column (GE Healthcare) equilibrated with 20 mM Tris, pH 8.0, 0.5 mM TCEP, 200 mM NaCl, and 20% glycerol. Heterointasomes were eluted with a linear gradient of 200 mM to 1M NaCl. Fractions were pooled, and further purified by size exclusion chromatography on a Superdex 200 column as described for homointasomes.
Integration assays
Integration assays were performed as follows; 20 μL reaction mixtures contained 1.0 μM integrase, 0.5 μM viral DNA substrate, 20 mM HEPES pH 7.5, 5 mM MgCl2, 10 mM DTT, 4 μM ZnCl2, 100 mM NaCl, and 25% glycerol and 300 ng target plasmid DNA (pGEM‐9zf). The reaction mixture was incubated at 37°C for 1 h. The reaction was stopped by addition of SDS (0.2% final), EDTA (10 mM final), 5 μg proteinase K and incubation was continued at 37°C for a further 1 h. The DNA was then recovered by ethanol precipitation and subjected to electrophoresis in a 1.5% agarose gel in 1× TBE buffer. Gels were visualized either by ethidium bromide staining or by using a Typhoon 8600 fluorescence scanner (GE Healthcare). Activity assays of homointasomes and heterointasomes were carried out in the same way except intasomes were substituted for integrase and viral DNA substrate.
Sequencing of integration products
Integration products were sequenced as described.11 Briefly, linear DNA corresponding to concerted integration products was separated and isolated from a 1× TBE agarose gel, purified and ligated to a DNA fragment containing the Tn5 kanamycin resistance gene. The plasmids were then transformed into competent E. coli TOP10 cells (Invitrogen). Plasmids were recovered from kanamycin resistant clones and those with the expected size were sequenced and analyzed.
Supporting information
Supporting Information
Acknowledgment
The authors thank Alison Burgess Hickman for comments on the manuscript.
References
- 1. Brown PO, Retroviruses. In: Coffin JM, Hughes SH, Varmus HE, Ed. (1997) Ch. Integration. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, pp 161–203. [Google Scholar]
- 2. Engelman A, Mizuuchi K, Craigie R (1991) HIV‐1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell 67:1211–1221. [DOI] [PubMed] [Google Scholar]
- 3. Brown PO, Bowerman B, Varmus HE, Bishop JM (1989) Retroviral integration: structure of the initial covalent product and its precursor, and a role for the viral IN protein. Proc Natl Acad Sci USA 86:2525–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fujiwara T, Mizuuchi K (1988) Retroviral DNA integration: structure of an integration intermediate. Cell 54:497–504. [DOI] [PubMed] [Google Scholar]
- 5. Li M, Mizuuchi M, Burke TR, Craigie R (2006) Retroviral DNA integration: reaction pathway and critical intermediates. Embo J 25:1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Andrake MD, Skalka AM (1996) Retroviral integrase, putting the pieces together. J Biol Chem 271:19633–19636. [DOI] [PubMed] [Google Scholar]
- 7. Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P (2010) Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maertens GN, Hare S, Cherepanov P (2010) The mechanism of retroviral integration from X‐ray structures of its key intermediates. Nature 468:326–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hare S, Maertens GN, Cherepanov P (2012) 3 '‐Processing and strand transfer catalysed by retroviral integrase in crystallo. Embo J 31:3020–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li M, Jurado KA, Lin S, Engelman A, Craigie R (2014) Engineered hyperactive integrase for concerted HIV‐1 DNA integration. Plos One 9:#S [PAGE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Valkov E, Gupta SS, Hare S, Helander A, Roversi P, McClure M, Cherepanov P (2009) Functional and structural characterization of the integrase from the prototype foamy virus. Nucleic Acids Res 37:243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yin Z, Lapkouski M, Yang W, Craigie R (2012) Assembly of prototype foamy virus strand transfer complexes on product DNA bypassing catalysis of integration. Protein Sci 21:1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sherratt D, Mobile DNA. In: Berg DE, Howe MM, Ed. (1989) Washington, DC: American Society for Microbiology, pp 163–184. [Google Scholar]
- 14. Maskell DP, Renault L, Serrao E, Lesbats P, Matadeen R, Hare S, Lindemann D, Engelman AN, Costa A, Cherepanov P (2015) Structural basis for retroviral integration into nucleosomes. Nature 523:366–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li M, Craigie R (2009) Nucleoprotein complex intermediates in HIV‐1 integration. Methods 47:237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Craigie R (2012) The molecular biology of HIV integrase. Future Virol 7:679–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information