

Abstract
The most common mutation in cystic fibrosis (CF) patients is deletion of F508 (ΔF508) in the first nucleotide binding domain (NBD1) of the CF transmembrane conductance regulator (CFTR). ΔF508 causes a decrease in the trafficking of CFTR to the cell surface and reduces the thermal stability of isolated NBD1; it is well established that both of these effects can be rescued by additional revertant mutations in NBD1. The current paradigm in CF small molecule drug discovery is that, like revertant mutations, a path may exist to ΔF508 CFTR correction through a small molecule chaperone binding to NBD1. We, therefore, set out to find small molecule binders of NBD1 and test whether it is possible to develop these molecules into potent binders that increase CFTR trafficking in CF‐patient‐derived human bronchial epithelial cells. Several fragments were identified that bind NBD1 at either the CFFT‐001 site or the BIA site. However, repeated attempts to improve the affinity of these fragments resulted in only modest gains. Although these results cannot prove that there is no possibility of finding a high‐affinity small molecule binder of NBD1, they are discouraging and lead us to hypothesize that the nature of these two binding sites, and isolated NBD1 itself, may not contain the features needed to build high‐affinity interactions. Future work in this area may, therefore, require constructs including other domains of CFTR in addition to NBD1, if high‐affinity small molecule binding is to be achieved.
Keywords: CF, cystic fibrosis, CFTR, cystic fibrosis transmembrane conductance regulator, NBD1, nucleotide binding domain 1
Abbreviations
- 3S mutants,
mutations at F429S, F494N, and Q637R
- ΔF508
deletion of F508
- ΔRI
deletion of residues 405–436 of CFTR (human numbering)
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- NBD1
nucleotide binding domain 1.
Introduction
Cystic fibrosis (CF) is caused by mutations in the CF transmembrane conductance regulator (CFTR), causing inadequate chloride efflux in epithelial cells; loss of chloride efflux leads to chronic lung disease, exocrine pancreatic insufficiency, and male infertility.1 A small minority of CF patients (ca 2500 patients worldwide or 5% of total CF patients) have CFTR, which is appropriately folded/trafficked, but remains inactive at the cell surface; the most common of these is the G551D mutation, which accounts for 4% of CF patients and can be effectively treated by the small molecule potentiator VX‐770 (Kalydeco). However, the vast majority of CF patients (ca. 66,000 patients worldwide or 95% of total CF patients) have mutations decreasing CFTR folding efficiency and trafficking to the cell surface, for which there is currently no curative treatment.2 The most common CF folding mutation is deletion of F508 (ΔF508), occurring in 90% of patients (50%–70% of CF patients have two copies of the ΔF508 allele).3, 4, 5, 6
ΔF508 is in the first nucleotide binding domain (NBD1) of CFTR. In addition to decreasing the trafficking of CFTR, it also destabilizes isolated NBD1.7, 8 Both CFTR trafficking and stabilization of NBD1 can be rescued by revertant mutations within NBD1, such as deletion of residues 405–436 (ΔRI, human numbering), or the mutations F429S, F494N, and Q637R (3S mutants).9, 10, 11, 12, 13, 14 CFTR (both wild type and ΔF508) trafficking in human bronchial epithelial (hBE) cells can also be increased by treatment with the small molecule VX‐809 (Orkambi).15, 16
VX‐809 appears to interact with CFTR directly, as suggested by two distinct lines of evidence. The first is its ability to correct CFTR trafficking in a variety of different cell types—each with its own proteome, but expressing human CFTR as a common feature17, 18; the second is a VX‐809‐dependent increase in ion flux observed in purified CFTR activity assays.19 It, therefore, seems that VX‐809 is a small molecule chaperone of CFTR; this inference is supported by data suggesting an interaction in the first membrane spanning domain of CFTR,20 though the exact site on CFTR and other details of its molecular action are still obscure.
VX‐809 in combination with VX‐770 causes an improvement in ΔF508 patient lung function (+3% FEV) that is sub‐curative (+11%–13% FEV is theorized to be the curative minimum) but encouraging for a first‐generation small molecule chaperone.21 We, among others, have hypothesized that the identification of a new corrector class that binds to NBD1 and stabilizes the thermodynamic defect caused by ΔF508 could form the basis of a therapy complimentary to the VX‐809 mechanism of action and help to cross the curative threshold for ΔF508 CF patients. We, therefore, undertook subset screening of the Pfizer chemical library aimed at the discovery and optimization of NBD1 binders using a combination of nuclear magnetic resonance (NMR) and surface plasmon resonance (SPR) assays against isolated NBD1. In the course of our work, we have discovered two classes of molecules with distinct binding sites on NBD1. Significant additional work to improve the affinities of our initial hits has resulted in a better understanding of the sites of association, but only minor improvements in affinity. These data and their implications for the paradigm of CF correction through NBD1 stabilization are discussed.
Results
NBD1 constructs used have characteristics consistent with previously reported results
Two constructs were used for these studies, a minimal domain of NBD1 (residues 387–646), which does not include the regulatory insertion (RI, residues 405–436, containing helix α1b) or regulatory extension (RE, residues 647–678, containing helix α9b) (ΔRIΔRE construct) and was primarily used for NMR compound mapping experiments; and a larger construct (residues 389–678) containing the RI, the RE, and three solubilizing mutations (F429S, F494N, and Q637R) (3S construct), which was primarily used for NMR fragment screening and SPR experiments.
Our purified NBD1 constructs had TM values and SPR‐derived ATP affinities consistent with previous publications (Fig. 1).18, 22 We also tested the solution‐based affinity of 3S NBD1 for ATP, which we found to be 1.4 ± 0.08 μM, which is in good agreement with SPR values. Because NBD1 is prone to rapid nonreversible thermal aggregation, which is marked by a loss of ATP binding, the ability to bind ATP was used to verify the integrity of our samples prior to crystallization and at the start and stop of compound screening experiments.7, 8
Figure 1.
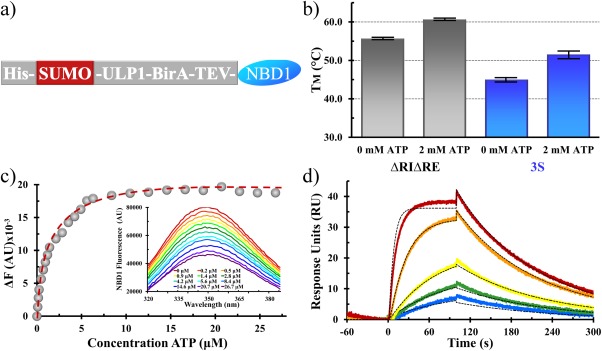
NBD1 constructs and characterization. (a) Expression construct of NBD1, including tags and potential protease cleavage or biotin labeling positions. (b) Thermostability of the ΔRIΔRE (gray) and 3S (blue) NBD1 constructs used in these studies. Values for ΔRIΔRE were 55.3°C (±0.1°C) and 60.3°C (±0.1°C) with no ATP or 2 mM ATP present; values for 3S were 45.1°C (±0.6°C) and 51.1°C (±1.1°C) with no ATP or 2 mM ATP present. (c) Determination of ATP binding affinity to 3S NBD1 from ATP‐dependent changes in tryptophan fluorescence. The fitting function is shown as a dashed red line, and raw fluorescence emission are inset; data were fit with a Kd of 1.4 ± 0.08 µM. (d) Representative SPR data showing the ATP‐dependent changes in 3S NBD1 resonance (colored lines) and fit to a binary association model (black dashed lines); data were fit with a Kd of 2.5 ± 0.2 µM, using k(on) = 2145 M −1 s−1 and k(off) = 0.005 s−1 (t 1/2 of 130 s).
A novel crystal form of human 3S ΔF508 demonstrates the plasticity of RI and RE interactions with the NBD1 core
Using published conditions, we were able to produce crystals of ΔRIΔRE ΔF508,22 but were unsuccessful in producing crystals of 3S ΔF508. We performed our own 3S ΔF508 crystal screen and discovered a novel crystal form of 3S ΔF508, which diffracted to the same resolution (2.05 Ǻ) as the literature 3S ΔF508 crystal form we had sought (Table 1).
Table 1.
Data Collection and Refinement Statistics
| Human CFTR NBD1 (aa389‐678; F429S, F494N, ΔF508, Q637R, ATP•Mg2+) | |
|---|---|
| Data collection | |
| X‐ray source | APS (IMCA) |
| Wavelength (Å) | 1.00 |
| Space group | P43212 |
| Unit cell | |
| a, b, c (Å) | 101.456, 101.456, 58.335 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å)b | 101–2.05 (2.16–2.05) |
| No. of reflections | |
| Total | 226,627 (18,547) |
| Unique | 19,819 (2,829) |
| Completeness (%) | 100.0 (99.7) |
| I/σ (I) | 20.4 (2.2) |
| R meas (%) | 8.8 (87.1) |
| Refinement | |
| Resolution(Å)b | 101–2.05 (2.16–2.05) |
| R work/R free (%) | 19.6/24.3 |
| RMSDs | |
| Bond length (Å) | 0.017 |
| Bond angles (°) | 1.908 |
| No. of atoms | 2,109 |
| Protein | 2,029 |
| Ligands | 32 |
| Water | 48 |
| Ave. B‐factors (Å2) | |
| Protein | 37.3 |
| Water | 29.0 |
| Ramachandran (%) | |
| Favored | 98.4 |
| Allowed | 1.6 |
| Outliers | 0 |
Values in parenthesis are for the highest resolution shells.
Values in parenthesis are for the highest resolution bin.
There have been many structures of NBD1 solved from both human and nonhuman sources in the presence or absence of mutations and in a wide range of precipitant, salt, and pH conditions; all these structures show good overall agreement with our structure, with only local changes apparent upon comparison. We, therefore, compare in detail our structure only with the other published human 3S ΔF508 (pdb 2BBS), which was crystallized in conditions similar to that of ours; differences between our structure and 2BBS should, therefore, provide insight onto the plasticity of human NBD1.
The previous crystal form (2BBS) has two copies of NBD1 in its asymmetric unit our crystal form (4WZ6) has one. Alignment of our structure with the two chains of 2BBS shows good general agreement (root mean square deviation (RMSD) of 0.86 Ǻ or 0.65 Ǻ for our structure to chain A or B of 2BBS; chain A and B of 2BBS align with each other with an RMSD of 0.38 Ǻ). The largest differences occur for the placement of the RI and the RE between these structures [Fig. 2(b)].
Figure 2.
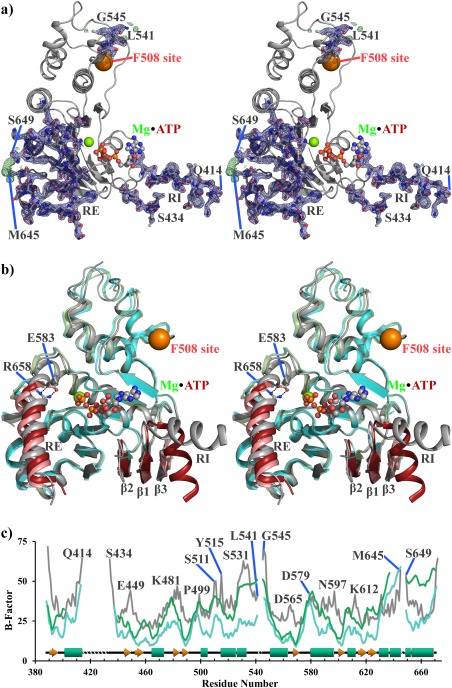
A novel 3S ΔF508 NBD1 structure demonstrates plasticity of RI and RE compared with the rest of NBD1. Stereo view of the 3S ΔF508 NBD1 structure showing either (a) 2Fo‐Fc (blue, 3σ) and Fo‐Fc (green, 1.5σ) density or (b) overlay of our structure (gray) with chains A (cyan) and B (green) of 2BBS. NBD1 is oriented such that the membrane spanning domain interface would be above it, and the NBD2 binding interface is opposite into the plane of the page. The F508 site is indicated as an orange sphere. ATP•Mg2+ is labeled and shown as sticks. The current structure and 2BBS are identical constructs, which crystallized in different space groups but attained the same resolution. Modeled regions of RI and RE are well supported by density and show a significant difference in their interactions with NBD1 than previously seen for human 3S ΔF508 NBD1. (c) B‐factor analysis of the current structure (gray) and 2BBS chain A (cyan) and B (green) with model breaks labeled.
The RI occurs only in NBD1 of CFTR and no other NBDs from other ABC family members and is destabilizing.23 Although it crystallizes as a partial helix, it is partially disordered in solution24 and does not have continuous density in any NBD1 structure. Our new crystal form has continuous density of four more residues of the RI than in 2BBS; three of these new residues are in the disordered loop connecting RI back to the core fold of NBD1 and are stabilized because of crystal packing with P499 from an adjacent unit cell. In our structure, the RI does not interact with the core of NBD1 but instead seems to float off at a significant distance (ca. 25 Ǻ) from the rest of NBD1, emphasizing the flexible nature of this insertion into the canonical NBD1 fold (Fig. 2).
In chain A of 2BBS, the RI folds back toward NBD1 to form a chaste (ca. 45 Ǻ2) interaction with β‐strands β1 and β4, which in turn promote a longer β‐strand interaction among β1, β2, and β4 than seen in our structure. When the RI packs against β4, it places several negatively charged side chains (E403, E407, and E410 of the RI and E476 of β4) in proximity; thus, the net energetic result of this interaction between RI packed against NBD1 in this orientation is likely unfavorable.
The positioning and packing of the RE with NBD1 is the other significant difference between our structure and 2BBS. Like the RI, the RE is dynamic and may be partially unfolded or dissociated from the core of isolated NBD1 in solution.25 There are four helical turns of RE; each of these commits at least one hydrophobic side chain to pack against NBD1, with the important exception of the first turn, where instead of a hydrophobic residue the positively charged guanidinium side chain of R658 is positioned toward NBD1. The side chain of R658 is similarly placed in both chains of 2BBS, where it is slightly solvent exposed; in contrast, R658 is buried in our structure and forms a salt bridge with the side chain of E583 in α6 (E583 interacts with the side chain of a partially solvent exposed K606 in 2BBS). E583 and R658 seem to force a binary structural paradigm for RE packing: they can either interact, in which case they are buried and anchor RE closer to the ABCα subdomain of NBD1 (ca. 2.4 or 4.2 Ǻ higher on our structure relative to chain A or chain B of 2BBS), or if not interacting, then NBD1 must adjust to allow solvent access to these otherwise buried side chains by opening up a loop connecting β6 to α6, and the RE is held low on NBD1. These distinct poses suggest that there are likely to be many energetically similar but structurally distinct interactions between NBD1 and RI or RE (Fig. 2). These results are consistent with previous NMR studies on murine NBD1 containing the RI and RE, including the structural perturbations observed upon phosphorylation of these flexible regulatory segments.26
Two compound classes found from a binding‐based NBD1 screen
An NMR‐based binding screen was performed against a fragment library of approximately 2600 compounds. Design parameters of this library have been described.27 Briefly, the average mass of the screening set is 225 Da, and it is expected that binders found from this set will have affinities in the micromolar range at best. Hits from this screen are improved by methods of fragment‐based lead development, which commonly involves additional screening of analogs with high similarity and/or bigger in size to improve affinity while keeping high ligand efficiency (ratio of affinity to compound mass) and desirable absorption, distribution, metabolism, and excretion properties.28, 29, 30, 31
The fragment library was screened by saturation transfer difference (STD) spectra against the 3S form of F508‐containing NBD1 (hereafter called “3S WT”) as the ΔF508 form aggregated too quickly to be used in these experiments. Briefly, STD is performed by saturating the methyl region of the protein; any compound that binds with fast on/off kinetics will be detected in the 1H spectra of the compounds. This has utility in screening because the protein does not need to be 15N or 13C labeled and a mixture of compounds can be screened, but only the bound compounds will be detected. Compounds identified by STD were subsequently tested for binding in an SPR‐based screen and for TM effects using ATP‐bound 3S WT. The majority of the initial STD hits were validated as binders in the SPR platform, though none significantly affected TM; however, this was not considered to be a critical validation parameter because of the weakness of these binders (Kd > 100 μM) and the chance that SYPRO Orange could outcompete weak binders (Fig. 3).
Figure 3.
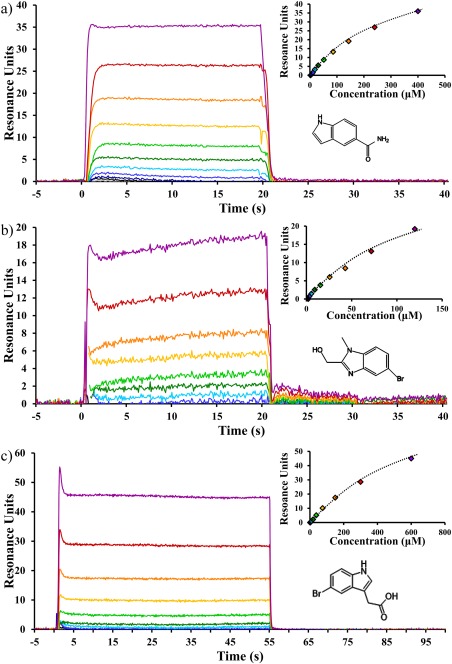
Novel NBD1 binding compounds. Representative SPR sensorgrams showing concentration‐dependent changes in resonance (colored lines in main panel or the markers in the inset panel) and fit to a binary association model (inset) with best fit line (black dashed lines) for (a) the representative α‐site binder Cp‐α1, (b) the representative β‐site binder Cp‐β1, and (c) the BIA compound. The average Kd and standard deviation between experiments were 350 ± 115 μM (n = 24), 230 ± 185 μM (n = 9), and 645 ± 56 μM (n = 27) for Cp‐α1, Cp‐β1, and BIA, respectively. Complete saturation was not achieved because of the limits of compound solubility compared with affinity. Instead, R max, representing complete compound saturation, was defined as the maximum response from ATP controls run prior to compound screening.
Based on both STD and SPR data, compounds were clustered by similarity of chemical structure, and 15N‐1H HSQC chemical shift perturbations were determined against NBD1 ΔRIΔRE (for which resonance assignments were available). Based on chemical shift perturbations, these fragments were classified as belonging to two classes. Class A compounds typically contain a 6‐5 or 6‐6 bicyclic aromatic system with a hydrogen bond acceptor moiety close to the aromatic ring. Fragments in this class cause chemical shift perturbations in the ABCα subdomain of NBD1.23 We refer to this binding site as the “α‐site.”
Class B compounds bear some similarity to Class A because they also typically possess an aromatic bicycle, but they generally lack the carbonyl moiety or other acceptor functionalities adjacent to the ring system. Fragments in this class cause chemical shift perturbations in ABCβ subdomain of NBD1. We refer to this binding site as the “β‐site” (Fig. 4).
Figure 4.
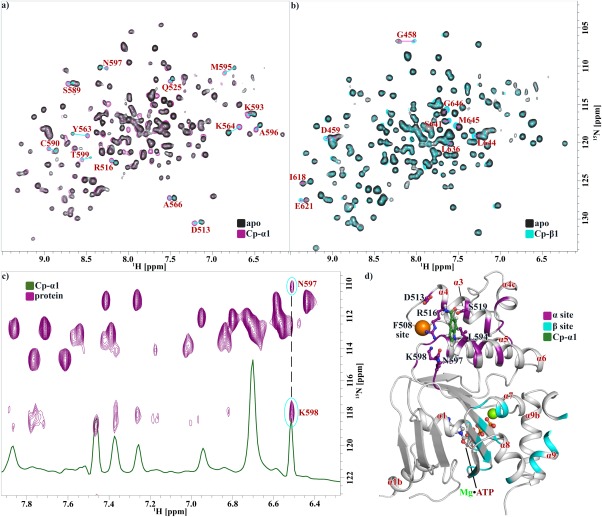
Compounds affect NBD1 residues at two distinct sites. 1H‐15N HSQC spectra of ΔRIΔRE NBD1 as apo (black) or after addition of (a) Cp‐α1 (purple) causing shifts at the α‐site or (b) Cp‐β1 (cyan) causing shifts at the β‐site; residues affected are labeled with a line connecting shifted peaks. (c) 1H‐15N HSQC spectra (purple) or 1H spectra (green) showing NOE between NBD1 and Cp‐α1; correlated shifts are connected by a black dashed line, with residue assignments indicated. (d) 3S ΔF508 NBD1 structure showing residues affected by α‐site (purple) and β‐site (cyan) binders. NBD1 is oriented such that the membrane spanning domain interface would be above it, and the NBD2 binding interface is opposite into the plane of the page. The F508 site is indicated as an orange sphere. ATP•Mg2+ is labeled and shown as sticks and spheres. Docked compound Cp‐α1 is shown in green as sticks, with residues within 5 Å identified and shown as sticks. Alpha helices are numbered in red according to convention established by Lewis et al.32
Class A compounds elicit large chemical shift perturbations in helices α4, α5, and α6; the majority of these perturbations occur at the apex of a helical bundle formed by the N‐terminus of α4 and the C‐termini of α5 and α6. The parsimonious interpretation of these data is that the apex of this bundle is the site of binding, and additional chemical shift perturbations distal from this site (e.g., S549) are perpetrated by compound‐induced rigid rod‐like adjustments in helical packing or other allosteric effects.
In support of this hypothesis, we observed NOEs between the H3 proton of Cp‐α1, a prototypical Class A compound, and the amide protons of N597 and K598 using 1H‐15N HSQC‐NOESY experiments [Fig. 4(c)]. Based on the NOE between Cp‐α1 and NBD1, we have modeled a binding mode consistent with these data by docking simulations, which places Cp‐α1 in a shallow pocket at the apex of the α4/α5/α6 helical bundle. In this pose, the carbonyl oxygen of Cp‐α1 makes a hydrogen bond with the hydroxyl side chain of S519, which could explain the presence of a hydrogen bond acceptor in Class A compounds. The pose is consistent with the strength of STD signals for H2, H4, H5, and H6 protons, which are pointed toward NBD1 and NOEs between these protons and unassigned methyl peaks from NBD1 (Fig. 5). The structure of NBD1 bound to BIA, a compound similar to Cp‐α1, was reported recently; the pose of BIA is very similar to our NMR‐based docking model.33
Figure 5.
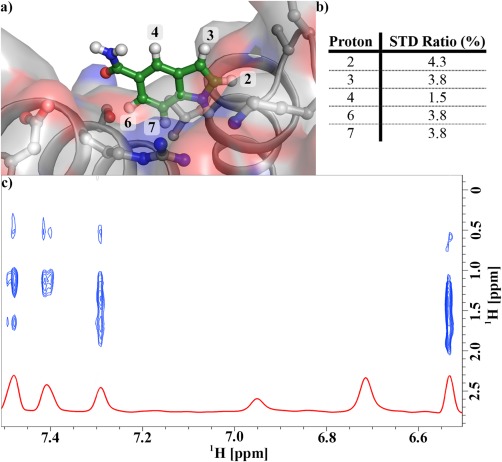
Dock of Cp‐α1 into the 3S ΔF508 NBD1 structure is consistent with chemical shift perturbation and STD data. (a) The dock of Cp‐α1 into the α‐site composed of the apex of helices α4, α5, and α6. (b) STD ratios for the compound protons. STD signal strength is a function of distance from the protein; the protons closest to the protein have the highest STD ratios. (c) NOEs between Cp‐α1 protons and unassigned NBD1 methyl protons.
Class B fragments cause small chemical shift perturbations in the ABCβ subdomain in the C‐terminus of α‐helices α7 and α9 and β‐strands β3, β8, and β9; together, these shifts describe a region formed by a β‐sheet (β3/β8/β9) capped by α‐helices α7 and α9 [Fig. 4(b)]. This is the same region previously seen for the small molecule corrector CFFT‐001, which was docked near β3, β8, and β9.34
Chemical expansion finds several new compounds in both the α‐site and β‐site but no strong binders
In a typical fragment screen outcome, the compounds from the primary screen had modest affinities for NBD1. To increase affinity of the hit scaffolds and our understanding of the chemical structure–activity relationship (SAR), we undertook a hit expansion exercise evaluating affinity of approximately 1000 analogs chemically similar to our top initial hits using an SPR‐based screen. The choice of 1000 compounds is somewhat arbitrary, but, based on past experience, this number was judged to be sufficient to for an unbiased survey of SAR for the best of our initial hits (see below) while also allowing for hypothesis‐driven compound testing.
We chose to expand our top four fragments because these had the best ligand efficiency (the ratio of affinity to compound mass) and representation of both α‐site and β‐site binding modes. The compound selection for hit expansion was based on either a similarity‐based approach where we made matched pair comparisons for SAR development, or a hypothesis‐driven approach based on our compound docked structure. For the former, most relevant analogs were selected on the basis of their similarity scores using ECFP4 for atom pairing and molecular match pair fingerprints. For the latter, file analogs were picked based on medicinal chemistry tactics targeting (a) fragment growth using the existing vectors, (b) isosteric replacement of the existing functional groups, (c) scaffold hopping, and (d) proposed binding mode in the α‐site. The analog selection was done individually for each of the initial parent fragments, and it was noted that selections had noticeable overlap among analogs with decreased similarity scores, which was consistent with the generally similar topology of the initial hits. The diversity of the Pfizer file (ca. 3 million compounds) allowed for selection of analogs to test key hit modification/growth hypotheses without resorting to synthetic chemistry. Overall, 88 unique Murcko scaffolds were present in the hit expansion set, which provided a good balance for evaluation of the effects of scaffold modification and growth directions.35
Through this process, we have identified compounds in both classes that were tighter than 100 μM without adding significantly to the mass of the parent fragment (Fig. 3). However, no compound could be found with an affinity tighter than 10 μM, and the SAR suggests that there are no growth vectors in Class A or Class B compounds that are likely to increase affinity beyond what we have seen. We found that none of the compounds were singularly able to elicit a clear effect on the TM of ATP‐bound 3S NBD1 in a SYPRO assay or on CFTR trafficking levels in patient‐derived hBE cells (data not shown). However, it is worth emphasizing that all tests were done with a single compound at a time; thus, we cannot comment on any effects from combining Class A and Class B compounds.
Discussion
Binding at the α‐site
NBD1 has an F1‐like ATP‐binding core flanked by an alpha helical region called the ABCα subdomain, and a beta strand region called the ABCβ subdomain. For a comprehensive review of the structure of NBD1, see Hunt et al.23
The ABCα subdomain of NBD1 is the region most proximal to the membrane; it contains the F508 site and the residues affected by α‐site binders. The α‐site binders elicit chemical shifts in helices α4, α5, and α6; the majority of these shifts occur at the apex of a helical bundle formed by the N‐terminus of α4 and the C‐termini of α5 and α6. We were not able to determine structures of ΔRIΔRE or 3S NBD1 with any of our fragments, but based on both chemical shift perturbations and direct NOEs between Cp‐α1 and NBD1, we have a high confidence docking model suggesting that the α‐site is a shallow pocket formed at the apex of the α4/α5/α6 helical bundle (Fig. 4). This pocket correlates well with the binding site recently reported for structures of NBD1, with two small molecule binders.33
If the α‐site pocket is indeed as we hypothesize, this model has three important practical considerations. The first is that the docked interaction highlights S519 as a hydrogen bond donor to the keto oxygen of Cp‐α1; this may explain the presence of the keto or ester oxygen moiety seen in Class A compounds. Consistent with this observation, we noted that compounds lacking a hydrogen bond acceptor at this site but otherwise identical to competent α‐site binders lost α‐site binding during our chemical expansion searches (data not shown). Compounds targeting this site may, therefore, need to incorporate a hydrogen bond acceptor for S519.
The second consideration is that this site is, perhaps, not justifiably called a “pocket” but instead is more accurately described as a “dimple” in the surface of NBD1; about 150 Ǻ2 of SASA is expected for high‐affinity binding for a molecule this size, but nearly half that value, 80 Ǻ2, is observed here.36 Therefore, unless additional opening occurs at the α‐site to allow more compound burial, a high‐affinity interaction is not expected at this site.37 Consistent with this observation, Cp‐α1 and the previously reported BIA compound are weak binders (350 ± 115 μM and 645 ± 56 μM) and we were not able to find any compound tighter than 10 μM at this site after an extensive exploration of chemical space around our most potent α‐site binders. Although it is possible, we simply did not find the right key to open this site; further, it seems increasingly unlikely that this site will yield a high‐affinity interaction (Fig. 3).
The final consideration is that the α‐site is distant from the presumed interface of NBD1 with the membrane spanning domain of CFTR and opposite the presumed NBD1:NBD2 interface (for a CFTR model see Rosenberg et al.38); interactions at this site are therefore expected to be well modeled by isolated NBD1. Consistent with this observation, weak binders we found at this site were not able to evoke any increase in trafficking of CFTR in hBE cells, suggesting they did not pick up additional affinity against full length CFTR (data not shown).
Binding at the β‐site
The ABCβ subdomain is the region most distal from the membrane spanning domain; it contains the RI and the RE as well as the residues affected by β‐site binders. The β‐site binders cause chemical shifts in a region formed by a β‐sheet (β3/β8/β9) capped by α‐helices α7 and α9 (Fig. 4). Chemical shifts in the β‐site have been previously seen for the small molecule corrector CFFT‐001,34 where it was hypothesized that the compound bound against the β‐sheet and was displacing α‐helices above it (e.g., α9 and presumably RE in full length CFTR). This model is consistent with the residues affected, which include many of the residues of the helices pointing toward the β‐sheets. This is also an appealing model because it might explain the correction of CFFT‐001 as arising from displacement of RE, which must occur for NBD1 to bind NBD2, an interaction thought to be important for CFTR activation and commonly seen for activation of related ABC transporters.23, 39, 40
In light of this model, the β‐site has three practical considerations for drug development. The first is the potential benefit of displacing the RE from NBD1; because the RE is precluding NBD2 interaction, molecules of this class could promote the association of NBD2 and thereby increase CFTR activity (potentiation). Therefore, if a binder at this site could be developed into a corrector, there is the possibility it would be a dual potentiator/corrector. Interestingly, we have found that compounds we screened in Class B bind both 3S and ΔRIΔRE forms of NBD1 with similar affinities despite the expectation that 3S binding could result in the unfavorable energetic cost of displacing RE. The reason for similar affinities may have its roots in the ability of the RE to adopt multiple packing conformations against NBD1 as see in crystal structures (Fig. 2) and chemical shifts in the disordered region of the spectra [Fig. 4(b)].34 It would seem that the RE does not provide a significant energetic penalty for compounds infiltrating into β‐site of isolated NBD1.
The second consideration is that a clear pocket for binding in the β‐site is not apparent in crystal structures of NBD1, which either have helices α7 and α9 tightly packing against the β‐sheet or in a less tightly packed coil‐like conformation. The structural details of this site are, for the moment, somewhat cryptic, which limits rational drug design for this site.
The third consideration is that this site is largely composed of hydrophobic side chains and hydrogen bond satisfied main‐chain, which is an environment common to the packing core of all soluble proteins; designing molecules specific to this site is, therefore, likely to be a significant challenge. Furthermore, we have observed that several compounds with NBD1 affinities too weak for SPR detection are still capable of causing chemical shifts in the β‐site (data not shown). This leads us to speculate that the β‐site is a promiscuous site capable of absorbing many different compounds and, for that reason, may be hard to optimize through binding‐based screens.
Conclusion
Although we have discovered several compounds that bind at separate sites on NBD1, our SAR studies suggest that, for isolated NBD1, neither of these sites are suitable for the development of high‐affinity ligands needed for small molecule therapeutic agents.
Based on these data, we suspect that screening for correctors against an isolated NBD1 is not the optimal strategy for the discovery of novel direct‐binding correctors of CFTR. In support of this hypothesis, NBD1 binders have been found based on either virtual screening17 or crystallography against isolated NBD1 (BIA and BIAE have been reported to bind NBD1 but no coordinate files are available),33 yet these compounds elicit only modest cell effects at concentrations so high (ca. 1 mM) that it is questionable whether their mode of action is specific to NBD1 binding. Furthermore, the lack of follow‐up on these compounds in the years since their finding suggests that affinities or efficacies may not be readily improved. In contrast, based on its effects on purified CFTR, and the variety of cell types for which it is efficacious, VX‐809 is likely a direct binder of CFTR. Thus, the paradigm of developing a direct‐binder into a CFTR corrector still seems plausible.17, 18, 19 Perhaps the lesson here is that future screens for direct binders need to use more than just isolated NBD1, that they should instead place NBD1 closer to its native context in CFTR, such as with constructs that include the intraprotein domain interfaces of NBD2 or the membrane spanning domains.
Materials and Methods
Protein preparation
The genes for Homo sapiens NBD1 constructs were ordered from GeneWiz (South Plainfield, NJ). Constructs have either residues 387–646 with deletion of 405–436 (ΔRIΔRE construct), or residues 389–678 with the F429S, F494N, and Q637R mutation (3S construct); both constructs were made with or without a deletion of F508, and all constructs are the M470 allelic isoform. Genes were cloned into pET28, which contained an N‐terminal SUMO‐HIS tag, a ULP1 cleavage site, a BIRA recognition sequence, and lastly a tobacco etch virus cleavage site (Fig. 1).
Recombinant proteins were expressed in Escherichia coli BL21(DE3) host cell lines. Cells were grown in liquid broth medium (Invitrogen, Carlsbad, CA) at 37°C to an OD600nm of 1.5–2.0 before protein expression was induced with the addition of 0.5 mM isopropyl‐1‐thio‐β‐d‐galactopyranoside at 15°C for 16–20 h. Cells were collected and immediately transferred to 4°C for the remainder of the purification. The cell pellet was suspended into 50 mM Tris (pH 7.6), 100 mM arginine, 50 mM NaCl, 12.5% (v/v) glycerol, 5 mM MgCl2, 2 mM ATP, and 2 mM DTT and then lysed using a Branson Ultrasonic Disintegrator (VWR Scientific Products, Chicago, IL) with seven rounds of 10 s 10% duty cycle sonication separated by 50 s rest periods. The soluble fraction was separated using centrifugation at 30,000 relative centrifugal force for 1 h, applied to a HisTrap FF column (GE Healthcare), washed with 10 column volumes of buffer containing 20 mM Tris (pH 7.6), 500 mM NaCl, 60 mM imidazole, 12.5% (v/v) glycerol, 5 mM MgCl2, 2 mM ATP, and 2 mM DTT and then eluted using 20 mM Tris (pH 7.6), 250 mM NaCl, 400 mM Imidazole, 12.5% (v/v) glycerol, 5 mM MgCl2, 2 mM ATP, and 2 mM DTT. The elutant was concentrated using a 30‐kDa MWCO Amicon spin column (Millipore, Billerica, MA), buffer exchanged using a HiTrap column (GE Healthcare) into 50 mM Tris (pH 7.6), 150 mM NaCl, 12.5% (v/v) glycerol, 5 mM MgCl2, 2 mM ATP, and 2 mM DTT, and then incubated for 16–20 h with tobacco etch virus (Sigma‐Aldrich, St. Louis, MO) or ULP1 protease (Life Technologies, Carlsbad, CA) and BirA ligase (Avidity, Aurora, CO) to liberate an untagged NBD1 or generate a biotin‐tagged NBD1 for SPR experiments. Samples were then passed through a HisTrap FF column to remove tags/tagged protein, and a final purification was done using a HiLoad Superdex200 column (GE Healthcare). NBD1 is prone to aggregation; glycerol and DTT were all maintained in buffer to reduce cysteine oxidation and aggregation. Protein purity was verified by sodium dodecyl sulfate polyacrylamide gel electrophoresis and ESI‐TOF mass spectrometry.
Thermal denaturation
Thermal denaturation of NBD1 was monitored by changes in SYPRO Orange (Sigma‐Aldrich) fluorescence at 600 nm using an excitation wavelength of 300 nm; data were collected at an interval of 0.25°C using a thermal ramp of 2°C/min on a Cary Eclipse Fluorescence Spectrophotometer (Varian, Palo Alto, CA). Protein concentrations were 0.15 g/L (5.3 or 4.3 μM for ΔRIΔRE or 3S constructs) in buffer containing 50 mM Tris (pH 7.6), 150 mM NaCl, 12.5% glycerol, 5 mM MgCl2, and 2 mM DTT; when present, ATP was at 2 mM and compounds were at 0.75 mM (all compounds tested appeared visibly soluble at this concentration). TM corresponds to the midpoint of the fluorescence profiles, as determined by the maxima of the first derivative of the fluorescence:
| (1) |
where F ( T ) is fluorescence at temperature T. Data were fit using Eq. (1) in GraphPad Prism v5 [Fig. 1(b)].
Fluorescence measurements
Monitoring of ATP‐dependent changes in ΔRIΔRE or 3S NBD1 intrinsic tryptophan fluorescence was done at 15°C with a constant protein concentration (6 μM) and variable ATP (0.08 to 26.75 μM over 25 injections). Data were collected on a FluoroMax‐3 (Horiba Jobin Yvon, Edison, NJ) using excitation at 290 nm with 2 nm slit widths, a 0.5 s integration time, and emission monitoring between 320 and 385 nm. The fluorescence spectra were collected in the ratio mode (signal/reference) to correct for potential wavelength‐dependent changes in intensity, and sample fluorescence was subtracted from ATP injected into buffer. A somewhat broad fluorescence peak occurs between 344 and 354 nm, with the maximum emission at 349 nm; change in fluorescence was, therefore, averaged over 344–354 nm. A small linear change in fluorescence occurs for ATP concentrations above 10 μM; all data were, therefore, corrected through subtraction of the slope determined between 10 and 26.75 μM. Final data are the change in fluorescence between apo protein and protein after an injection, ΔF, versus ATP and were fit to the quadratic equation to obtain the apparent dissociation constant (Kd) for the NBD1•ATP complex:
| (2) |
where F 0 is intrinsic tryptophan fluorescence in the absence of compound, E is the concentration of NBD1, F max is the maximal change in fluorescence upon saturation of ligand, E•L is the NBD1•ATP complex. Data were fit using Eq. (2) in GraphPad Prism v5 [Fig. 1(c)].
Surface plasmon resonance
SPR experiments were conducted using CM5 sensor chips in a BIACORE T100 (GE Healthcare). Surfaces were prepared by immobilizing neutravidin (Pierce, Waltham, MA) through random‐amine coupling (GE Healthcare) at 25°C to a final density of 25,000–30,000 resonance units (RUs; one RU is equivalent to one picogram of protein per square millimeter of the surface), followed by biotinylated NBD1 immobilization at 4°C to a final density of 3000–8000 RUs. A single channel containing only neutravidin was used as a control from which NBD1 data were subtracted to remove non‐NBD1 compound interactions. The compounds were injected over the immobilized protein for a period of 30 s, and dissociation was measured for 30 s with a flow rate of 60 μL/min at 4 °C in buffer containing 10 mM HEPES (pH 7.5), 150 mM NaCl, 10% glycerol, 3 mM MgCl2, 2 mM ATP, 1 mM tris(2‐carboxyethyl)phosphine and 2% dimethylsulfoxide.
All SPR compound data presented were collected on ATP‐bound 3S NBD1. Only ATP curves had association (k(on)) or dissociation (k(off)) rates slow enough to be measured; thus, all compound data were fit to equilibrium association, Kd, determined by fit to a binary association model assuming one transient:
| (3) |
where, R ( C ) is the response at concentration C, R max is the maximum response from ATP controls run prior to compound screening, and R 0 is a global data offset from zero. Data were fit using Eq. (3) in the BIA T100 Evaluation program.
NBD1 activity (R %) was approximated by the percent response of the surface to an analyte relative to the amount of protein immobilized:
| (4) |
where, R imm is the amount of NBD1 immobilized in RUs, and M analyte and M protein are the mass of the analyte and the protein in daltons. Values were determined using Eq. (4) fit in Microsoft Excel.
The R % for ATP was typically 50% at the start of an experiment and was sampled again after compound screening experiments; experiments were discounted if the ATP R % was less than half of the initial ATP R % or the Kd was greater than 10‐fold divergent from the average values for the construct, which typically occurred no sooner than 48 h after the experimental start.
Crystallization
Crystals of ΔRIΔRE were grown as previously reported.22 A novel crystal form of 3S NBD1 was found from reagent 6 of the Hampton Crystal Screen matrix and eventually optimized to 0.2M MgCl2, 0.1M Tris (pH 8.5), and 33% PEG 4K at 10°C. Crystals were observed between 0.2 and 0.4 mM (7–15 g/L) NBD1 in buffer containing 20 mM Tris (pH 8), 150 mM NaCl, 10% glycerol, 10% ethylene glycol, 5 mM MgCl2, 3 mM ATP, and 2 mM TCEP. Crystals were flash frozen in liquid nitrogen without cryoprotectant, and data collected at the Argonne National Lab (IMCA) beamline. Refinement statistics are presented in Table 1.
Nuclear magnetic resonance
All NMR data were on a Bruker Avance III series 600 MHz spectrometer (Bruker, Billerica, MA) using a 1.7 mm cryoprobe. The Pfizer fragment library was screened against 3 μM 3S NBD1 in 50 mM deuterated‐Tris (pH 7.6), 12.5% deuterated‐glycerol, 150 mM NaCl, 5 mM MgCl2, 2 mM ATP, and 2 mM dithiothreitol fragments were at 300 μM in mixtures of either five or 10 and were screened using STD experiments.41 For each STD, 512 scans were collected over 0.5 h. The temperature was 285 K for fragment screening and 293 K for all other NMR experiments.
1H‐15N NBD1 chemical shift perturbation experiments were performed using ΔRIΔRE NBD1 from assignments determined using standard NMR experiments.42 1H‐15N TROSY experiments were performed with 100 μM ΔRIΔRE NBD1 in 50 mM phosphate (pH 7.5), 2% deuterated‐glycerol, 5 mM MgCl2, and 5 mM ATP. Spectra were recorded using Echo‐Anti‐Echo phase discrimination of 128 increments defined by 64 scans and 2048 points over a total experiment time of 2.5 h.43 Water flip‐back pulses were used in the sequence to minimize water signal.
15N‐edited NOESY experiments detecting the NOE between the amide protons of ΔRIΔRE NBD1 and the protons of Cp‐α1 were performed with 170 μM ΔRIΔRE NBD1 and 1 mM Cp‐α1 in 50 mM phosphate (pH 7.5), 2% deuterated‐glycerol, 5 mM MgCl2, and 5 mM ATP; data were collected using an NOE mixing time of 300 ms and an 1H‐15N heteronuclear single quantum correlation nuclear Overhauser effect spectroscopy (HSQC‐NOESY) pulse sequence with relaxation filters to reduce intramolecular NOEs. NOE spectra were collected with and without 15N decoupling in the direct dimension to distinguish amide proton peaks from compound peaks. Protons of Cp‐α1 were assigned using standard proton NOESY and correlation spectroscopy experiments. Spectra were processed and analyzed with TopSpin (Bruker).
Docking of Cp‐α1 to NBD1
Cp‐α1 was docked into our 3S ΔF508 ATP‐bound structure using the Glide docking protocol within the Maestro software from Schrodinger Inc. (New York, NY). Briefly, the compound was placed near the putative pocket based on the observed NOEs, and a search grid of the protein was created using a 10 Ǻ distance cutoff from the ligand. The distances between the residues involved in the observed NOEs were constrained to be less than 6 Ǻ during docking; the docking search was performed 16 times and was found to converge to a similar pose each time, which is the pose presented in this report.
Acknowledgments
The authors acknowledge Chris Bulawa, John Mathias, Marko Pregel, and Ryan Tyler for key scientific insights. They thank advisors from CFFT for thoughtful discussions. Author Contributions: The research was designed by AEA and JDH; protein was produced and purified by SS; fluorescence and TM experiments were developed and performed by JDH; SPR was performed by JDH and KW; NMR assignments of NBD1 were provided by PAC and JDF‐K; NMR experiments and Cp‐α1 docking were designed and executed by HW; SAR analysis and subsequent chemical expansion were performed by IVE; crystallography was performed by LJB and JDH; manuscript and figures were prepared by LJB and JDH, and the final version was read and approved by all authors. The authors declare no competing financial interests.
Grant sponsors: Pfizer and CFFT.
References
- 1. Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63:827–834. [DOI] [PubMed] [Google Scholar]
- 2. Riordan JR (2008) CFTR function and prospects for therapy. Ann Rev Biochem 77:701–726. [DOI] [PubMed] [Google Scholar]
- 3. Bobadilla JL, Macek MJ, Fine JP, Farrell PM (2002) Cystic fibrosis: a worldwide analysis of CFTR mutations–correlation with incidence data and application to screening. Hum Mutat 19:575–606. [DOI] [PubMed] [Google Scholar]
- 4. Carlile GW, Robert R, Zhang D, Teske KA, Luo Y, Hanrahan JW, Thomas DY (2007) Correctors of protein trafficking defects identified by a novel high‐throughput screening assay. Chembiochem 8:1012–1020. [DOI] [PubMed] [Google Scholar]
- 5. Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM, Du K, di Bernardo S, Liu Y, Konermann L, Roldan A, Lukacs GL (2012) Correction of both NBD1 energetics and domain interface is required to restore DeltaF508 CFTR folding and function. Cell 148:150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mendoza JL, Schmidt A, Li Q, Nuvaga E, Barrett T, Bridges RJ, Feranchak AP, Brautigam CA, Thomas PJ (2012) Requirements for efficient correction of DeltaF508 CFTR revealed by analyses of evolved sequences. Cell 148:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Protasevich I, Yang Z, Wang C, Atwell S, Zhao X, Emtage S, Wetmore D, Hunt JF, Brouillette CG (2010) Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide‐binding domain 1. Protein Sci 19:1917–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang C, Protasevich I, Yang Z, Seehausen D, Skalak T, Zhao X, Atwell S, Spencer Emtage J, Wetmore DR, Brouillette CG, Hunt JF (2010) Integrated biophysical studies implicate partial unfolding of NBD1 of CFTR in the molecular pathogenesis of F508del cystic fibrosis. Protein Sci 19:1932–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thibodeau PH, III Richardson JM, , Wang W, Millen L, Watson J, Mendoza JL, Du K, Fischman S, Senderowitz H, Lukacs GL, Kirk K, Thomas PJ (2010) The cystic fibrosis‐causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J Biol Chem 285:35825–35835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aleksandrov AA, Kota P, Aleksandrov LA, He L, Jensen T, Cui L, Gentzsch M, Dokholyan NV, Riordan JR (2010) Regulatory insertion removal restores maturation, stability and function of DeltaF508 CFTR. J Mol Biol 401:194–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Teem JL, Berger HA, Ostedgaard LS, Rich DP, Tsui LC, Welsh MJ (1993) Identification of revertants for the cystic fibrosis delta F508 mutation using STE6‐CFTR chimeras in yeast. Cell 73:335–346. [DOI] [PubMed] [Google Scholar]
- 12. Teem JL, Carson MR, Welsh MJ (1996) Mutation of R555 in CFTR‐delta F508 enhances function and partially corrects defective processing. Recept Channels 4:63–72. [PubMed] [Google Scholar]
- 13. DeCarvalho AC, Gansheroff LJ, Teem JL (2002) Mutations in the nucleotide binding domain 1 signature motif region rescue processing and functional defects of cystic fibrosis transmembrane conductance regulator delta f508. J Biol Chem 277:35896–35905. [DOI] [PubMed] [Google Scholar]
- 14. Pissarra LS, Farinha CM, Xu Z, Schmidt A, Thibodeau PH, Cai Z, Thomas PJ, Sheppard DN, Amaral MD (2008) Solubilizing mutations used to crystallize one CFTR domain attenuate the trafficking and channel defects caused by the major cystic fibrosis mutation. Chem Biol 15:62–69. [DOI] [PubMed] [Google Scholar]
- 15. Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu PA (2011) Correction of the F508del‐CFTR protein processing defect in vitro by the investigational drug VX‐809. Proc Natl Acad Sci USA 108:18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, Ballmann M, Boyle MP, Bronsveld I, Campbell PW, De Boeck K, Donaldson SH, Dorkin HL, Dunitz JM, Durie PR, Jain M, Leonard A, McCoy KS, Moss RB, Pilewski JM, Rosenbluth DB, Rubenstein RC, Schechter MS, Botfield M, Ordonez CL, Spencer‐Green GT, Vernillet L, Wisseh S, Yen K, Konstan MW (2012) Results of a phase IIa study of VX‐809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del‐CFTR mutation. Thorax 67:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Odolczyk N, Fritsch J, Norez C, Servel N, da Cunha MF, Bitam S, Kupniewska A, Wiszniewski L, Colas J, Tarnowski K, Tondelier D, Roldan A, Saussereau EL, Melin‐Heschel P, Wieczorek G, Lukacs GL, Dadlez M, Faure G, Herrmann H, Ollero M, Becq F, Zielenkiewicz P, Edelman A (2013) Discovery of novel potent DeltaF508‐CFTR correctors that target the nucleotide binding domain. EMBO Mol Med 5:1484–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, Roldan A, Verkman AS, Kurth M, Simon A, Hegedus T, Beekman JM, Lukacs GL (2013) Mechanism‐based corrector combination restores DeltaF508‐CFTR folding and function. Nat Chem Biol 9:444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eckford PD, Ramjeesingh M, Molinski S, Pasyk S, Dekkers JF, Li C, Ahmadi S, Ip W, Chung TE, Du K, Yeger H, Beekman J, Gonska T, Bear CE (2014) VX‐809 and related corrector compounds exhibit secondary activity stabilizing active F508del‐CFTR after its partial rescue to the cell surface. Chem Biol 21:666–678. [DOI] [PubMed] [Google Scholar]
- 20. Ren HY, Grove DE, De La Rosa O, Houck SA, Sopha P, Van Goor F, Hoffman BJ, Cyr DM (2013) VX‐809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane‐spanning domain 1. Mol Biol Cell 24:3016–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vertex Pharmaceuticals (2012) Vertex corrects and provides additional data from recent interim analysis of phase 2 combination study of VX‐809 and Kalydeco™ (ivacaftor) in people with cystic fibrosis who have two copies of the F508del mutation. http://investors.vrtx.com/releasedetail.cfm?ReleaseID=677520.
- 22. Atwell S, Brouillette CG, Conners K, Emtage S, Gheyi T, Guggino WB, Hendle J, Hunt JF, Lewis HA, Lu F, Protasevich II, Rodgers LA, Romero R, Wasserman SR, Weber PC, Wetmore D, Zhang FF, Zhao X (2010) Structures of a minimal human CFTR first nucleotide‐binding domain as a monomer, head‐to‐tail homodimer, and pathogenic mutant. Protein Eng Des Sel 23:375–384. [DOI] [PubMed] [Google Scholar]
- 23. Hunt JF, Wang C, Ford RC (2013) Cystic fibrosis transmembrane conductance regulator (ABCC7) structure. Cold Spring Harb Perspect Med 3:a009514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chong PA, Kota P, Dokholyan NV, Forman‐Kay JD (2013) Dynamics intrinsic to cystic fibrosis transmembrane conductance regulator function and stability. Cold Spring Harb Perspect Med 3:a009522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lewis HA, Wang C, Zhao X, Hamuro Y, Conners K, Kearins MC, Lu F, Sauder JM, Molnar KS, Coales SJ, Maloney PC, Guggino WB, Wetmore DR, Weber PC, Hunt JF (2010) Structure and dynamics of NBD1 from CFTR characterized using crystallography and hydrogen/deuterium exchange mass spectrometry. J Mol Biol 396:406–430. [DOI] [PubMed] [Google Scholar]
- 26. Kanelis V, Hudson RP, Thibodeau PH, Thomas PJ, Forman‐Kay JD (2010) NMR evidence for differential phosphorylation‐dependent interactions in WT and DeltaF508 CFTR. EMBO J 29:263–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lau WF, Withka JM, Hepworth D, Magee TV, Du YJ, Bakken GA, Miller MD, Hendsch ZS, Thanabal V, Kolodziej SA, Xing L, Hu Q, Narasimhan LS, Love R, Charlton ME, Hughes S, van Hoorn WP, Mills JE (2011) Design of a multi‐purpose fragment screening library using molecular complexity and orthogonal diversity metrics. J Comput Aided Mol Des 25:621–636. [DOI] [PubMed] [Google Scholar]
- 28. Scott DE, Coyne AG, Hudson SA, Abell C (2012) Fragment‐based approaches in drug discovery and chemical biology. Biochemistry 51:4990–5003. [DOI] [PubMed] [Google Scholar]
- 29. Jordan JB, Poppe L, Xia X, Cheng AC, Sun Y, Michelsen K, Eastwood H, Schnier PD, Nixey T, Zhong W (2012) Fragment based drug discovery: practical implementation based on (1)(9)F NMR spectroscopy. J Med Chem 55:678–687. [DOI] [PubMed] [Google Scholar]
- 30. Murray CW, Rees DC (2009) The rise of fragment‐based drug discovery. Nat Chem 1:187–192. [DOI] [PubMed] [Google Scholar]
- 31. Kumar A, Voet A, Zhang KY (2012) Fragment based drug design: from experimental to computational approaches. Curr Med Chem 19:5128–5147. [DOI] [PubMed] [Google Scholar]
- 32. Lewis HA, Zhao X, Wang C, Sauder JM, Rooney I, Noland BW, Lorimer D, Kearins MC, Conners K, Condon B, Maloney PC, Guggino WB, Hunt JF, Emtage S (2005) Impact of the deltaF508 mutation in first nucleotide‐binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J Biol Chem 280:1346–1353. [DOI] [PubMed] [Google Scholar]
- 33. He L, Aleksandrov AA, An J, Cui L, Yang Z, Brouillette CG, Riordan JR (2015) Restoration of NBD1 thermal stability is necessary and sufficient to correct F508 CFTR folding and assembly. J Mol Biol 427:106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hudson RP, Chong PA, Protasevich II, Vernon R, Noy E, Bihler H, An JL, Kalid O, Sela‐Culang I, Mense M, Senderowitz H, Brouillette CG, Forman‐Kay JD (2012) Conformational changes relevant to channel activity and folding within the first nucleotide binding domain of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 287:28480–28494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bemis GW, Murcko MA (1996) The properties of known drugs. 1. Molecular frameworks. J Med Chem 39:2887–2893. [DOI] [PubMed] [Google Scholar]
- 36. Cavallo L, Kleinjung J, Fraternali F (2003) POPS: a fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res 31:3364–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng AC, Coleman RG, Smyth KT, Cao Q, Soulard P, Caffrey DR, Salzberg AC, Huang ES (2007) Structure‐based maximal affinity model predicts small‐molecule druggability. Nat Biotech 25:71–75. [DOI] [PubMed] [Google Scholar]
- 38. Rosenberg MF, O'Ryan LP, Hughes G, Zhao Z, Aleksandrov LA, Riordan JR, Ford RC (2011) The cystic fibrosis transmembrane conductance regulator (CFTR): three‐dimensional structure and localization of a channel gate. J Biol Chem 286:42647–42654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eckford PD, Li C, Ramjeesingh M, Bear CE (2012) Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX‐770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation‐dependent but ATP‐independent manner. J Biol Chem 287:36639–36649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jih KY, Hwang TC (2013) Vx‐770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc Natl Acad Sci USA 110:4404–4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mayer B, Peters T (1999) Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew Chem Int Ed 38:1784–1788. [DOI] [PubMed] [Google Scholar]
- 42. Chong PA, Farber PJ, Vernon RM, Hudson RP, Mittermaier AK, Forman‐Kay JD (2015) Deletion of phenylalanine‐508 in the first nucleotide binding domain of the cystic fibrosis transmembrane conductance regulator increases conformational exchange and inhibits dimerization. J Biol Chem 290:22862–22878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pervushin K, Riek R, Wider G, Wuthrich K (1997) Attenuated T2 relaxation by mutual cancellation of dipole‐dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA 94:12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]