

Abstract
Contacts with neighboring molecules in protein crystals inevitably restrict the internal motions of intrinsically flexible proteins. The resultant clear electron densities permit model building, as crystallographic snapshot structures. Although these still images are informative, they could provide biased pictures of the protein motions. If the mobile parts are located at a site lacking direct contacts in rationally designed crystals, then the amplitude of the movements can be experimentally analyzed. We propose a fusion protein method, to create crystal contact‐free space (CCFS) in protein crystals and to place the mobile parts in the CCFS. Conventional model building fails when large amplitude motions exist. In this study, the mobile parts appear as smeared electron densities in the CCFS, by suitable processing of the X‐ray diffraction data. We applied the CCFS method to a highly mobile presequence peptide bound to the mitochondrial import receptor, Tom20, and a catalytically relevant flexible segment in the oligosaccharyltransferase, AglB. These two examples demonstrated the general applicability of the CCFS method to the analysis of the spatial distribution of motions within protein molecules.
Keywords: crystal contact effects, crystallographic method, fusion protein, mitochondrial presequence receptor Tom20, oligosaccharyltransferase AglB, protein motional analysis
Short abstract
Introduction
The dynamical view of protein molecules is essential for understanding their biological functions.1, 2, 3 Although NMR spectroscopy is generally considered to be the method of choice to analyze protein dynamics,4 the strong nonlinear distance dependency of NMR data hinders the proper analysis of internal protein motions when large amplitude motions exist.5 An average structure inevitably skews when a single structure is assumed. In contrast, an unbiased spatial view of motions is expected by crystallography. From a practical standpoint, however, the molecular contacts in crystals restrict the internal protein motions, unless the mobile parts are fortuitously located at a site lacking direct contacts with neighboring molecules in the crystal. Consequently, the structures determined by protein crystallography should be regarded as snapshots of fluctuating proteins. A mobile part of a protein or a moving ligand in a bound state is trapped in one possible state/conformation in the protein crystal lattice, and generates a clear electron density that allows model building. These crystallographic snapshot structures provide valuable information about the plasticity, if multiple snapshots are available, but could be misleading by overlooking the dynamic aspects due to the limited sampling of states/conformations.6, 7 X‐ray crystallography at room temperature has recently emerged as a new method to access protein conformational ensembles.8, 9 The contact network analysis of the room‐temperature structures focuses on the analysis of the rotamer distributions of the amino‐acid side chains. To investigate the large amplitude motions within protein molecules, another experimental methodology is required.
An interesting situation arises when the mobile protein segment or ligand is located at a site lacking direct contacts with neighboring molecules in the crystal (Supporting Information Fig. S1). Such crystal contact‐free space (CCFS) occasionally forms in protein crystals, but CCFS has not been used proactively as an analytical tool to study protein motions so far. Here we propose a fusion protein method, to create CCFS in protein crystals and to place the fluctuating segment/ligand in the created CCFS. The idea of using fusion proteins in crystallography is not new; for example, they are used in protein crystallization as crystallization chaperones,10, 11 but we applied the fusion protein‐based approach for a distinct purpose. When few contacts with neighboring molecules exist, the electron densities of the mobile elements become weak or even invisible. In traditional practice, such electron densities have simply been ignored, because an enforced model building for the faint electron density will not be supported by a simulated annealing‐omit map12 or an averaged kick‐omit map construction.13 Here, we showed that the dynamic aspects could be recovered from the abandoned information as smeared electron densities in CCFS, when combined with suitable data processing.
We applied the CCFS method to a presequence peptide in the binding site of the Tom20 protein. Tom20 resides in the mitochondrial outer membrane, where it functions as a receptor for presequences (mitochondrial signal sequences) for efficient import of mitochondrial matrix and inner membrane proteins into mitochondria.14, 15, 16 The analysis of the binding mode of a presequence to Tom20 is particularly suitable as a test case, because the large amplitude motions of a presequence in the bound state are considered to be the structural basis for the promiscuous recognition of mitochondrial presequences.17 We also applied the CCFS method to a catalytically relevant flexible segment in the AglB protein. AglB is an archaeal oligosaccharyltransferase (OST) that catalyzes oligosaccharide chain transfer to asparagine residues in glycoproteins.18 These two examples demonstrated the general applicability of the CCFS method, as an analytical tool for investigating the spatial aspects of protein internal motions.
Results
Rational design of the CCFS in the Tom20 crystals
We developed a special protein design to create CCFS intentionally in protein crystals. The basic idea is the fusion of a target protein with a tag protein, via a rigid linker [Fig. 1(A)]. Rigid linkers are usually used to effectively reduce unfavorable interdomain interactions,19 but in this study, they were used to create isolated space within the framework formed by the rigid fusion protein. We refer to the fusion protein (except for the flexible segment/ligand) as the “CCFS scaffold” [Fig. 1(B)]. Since the crystal packing mode of protein molecules is uncontrollable, the CCFS scaffold must be designed to ensure the formation of a space within a protein molecule that is inaccessible to other molecules.
Figure 1.
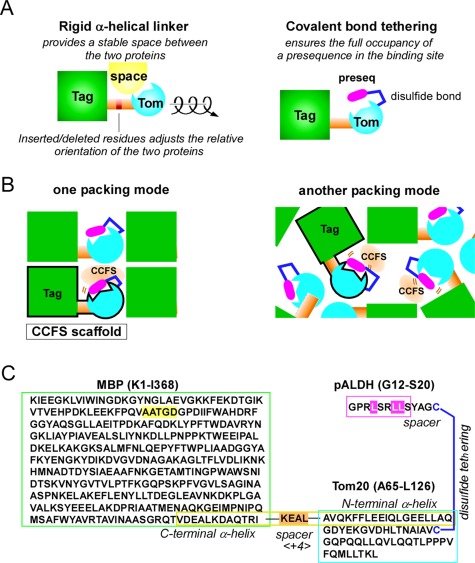
Key techniques to create CCFS in protein crystals. (A) Design of a fusion protein with a rigid α‐helical connection. The relative orientation of the tag and target proteins can be adjusted as a function of the number of residues inserted/deleted at the middle of the connector helix. Covalent‐bond tethering ensures the full occupancy of a weak‐affinity presequence in the binding site. (B) Concept of the CCFS scaffold. The CCFS can be intentionally created in the rigid framework formed by the “CCFS scaffold,” independent of the packing modes of the protein molecules. The CCFS scaffold (surrounded by black borders) is a fusion protein connected by a rigid linker. (C) Amino acid sequence of MBP<+4>Tom20‐SS‐pALDH. The connector helix is enclosed by the yellow box, and the 4‐residue insertion as a spacer is highlighted in orange. A disulfide bond was formed between the cysteine residue attached to the C‐terminus of the pALDH peptide and the single cysteine residue in MBP‐Tom20. The 5‐residue deletion, Δ5, in MBP and the three essential hydrophobic leucine residues in pALDH are highlighted in yellow and magenta, respectively.
We selected Escherichia coli maltose binding protein (MBP) as the fusion partner of the Tom20 protein [Fig. 1(C)]. We focused on the long C‐terminal α‐helix of MBP, which was seamlessly fused to the N‐terminal α‐helix in the cytosolic domain of Tom20 to form a rigid helical connector structure. To place the presequence binding site in the CCFS, the number of amino acid residues inserted or deleted at the junction site in the connector helix provides one degree of freedom, which determines the relative orientation between MBP and Tom20 within the same molecule [Fig. 1(A)]. Prior to the protein expression experiment, we performed a preliminary computer modeling study to estimate the optimal spacer length in the connector helix, by assuming an ideal α‐helical conformation. A sufficient amount of space was predicted to form between MBP and Tom20 with the spacer lengths of −4, 0, +4, and +8 (Supporting Information Fig. S2).
We constructed a series of MBP‐Tom20 fusion proteins with spacers containing different numbers of inserted/deleted residues (−4 to +8) at the junction site (Supporting Information Table S1). The basic spacer sequence, KEALQELA, was designed according to the solvent exposed‐buried pattern, eeebeebe, based on the helical propensity and hydrophobicity of the amino acid residues. All of the constructs were successfully expressed in the recombinant E. coli cells (Supporting Information Fig. S3). We purified the proteins and performed crystallization screening. Protein crystals were obtained for the constructs with spacer lengths of 0, +2, +4, +7, and +8. Among them, the crystal structures were determined for the constructs with spacer lengths of +2 and +4 to resolutions of 2.6 and 2.0 Å, respectively [Fig. 2(A)] (Supporting Information Table S2). We examined the formation of CCFS in the crystals of MBP<+2>Tom20 and MBP<+4>Tom20, where <+2> and <+4> denote the 2‐residue and 4‐residue insertions in the connector helix, respectively. In the MBP<+2>Tom20 crystal, an adjacent molecule blocked the Tom20 binding site, whereas the volume of space around the Tom20 binding site appeared to be sufficient to accommodate a presequence peptide in the MBP<+4>Tom20 crystal [Fig. 2(B)]. Thus, we selected the +4 spacer length for further analysis. This result also confirmed the benefit of performing the computer model building study before the actual experiments.
Figure 2.
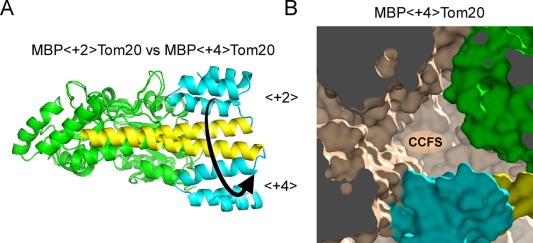
Effects of the different spacer lengths in the connector helix on the structure and the formation of CCFS. (A) Structural comparison of two fusion proteins with spacer lengths of +2 and +4. A long, continuous α‐helical structure (yellow, consisting of the C‐terminal α‐helix of MBP, the spacer, and the N‐terminal α‐helix of Tom20) was formed in the two crystal structures. The Tom20 proteins are located on nearly opposite sides around the helical axis of the connector helix in the two structures, as intended. MBP and Tom20 are colored green and cyan, respectively. (B) Formation of CCFS in the crystal of MBP<+4>Tom20. One molecule in the crystal is colored, and symmetrically related molecules are monochrome.
Tethering for the full occupancy of ligands
In the cases of ligands with weak affinity, the problem of partial ligand occupancy must be considered. The dissociation constant of the presequence peptide derived from rat aldehyde dehydrogenase (pALDH) for Tom20 is about 250 μM.20 The pALDH peptide was tethered onto Tom20, to ensure the full occupancy of the presequence in the binding site [Fig. 1(C)]. We added a cysteine residue at the C‐terminus of pALDH, to form an intermolecular disulfide bond with the single cysteine residue in the fusion protein. The spacer length (3 residues) between pALDH and the C‐terminal cysteine was optimized in the previous peptide library experiment.21 The tethered complex is referred to as MBP<+4>Tom20‐SS‐pALDH. The crystals diffracted to a resolution of 1.7 Å (Supporting Information Table S2).
Deletion in the MBP sequence to expand the CCFS
The structure of the CCFS scaffold, MBP<+4>Tom20, was determined by the molecular replacement method. The extra electron density corresponding to the presequence was clearly observed near the binding site of Tom20, and so a model of the presequence peptide was built (Supporting Information Fig. S4). Close examination of the structure suggested the presence of undesired intramolecular interactions between the presequence peptide and MBP. To enlarge the CCFS, we generated a new construct, Δ5MBP<+4>Tom20, to delete the 5‐residue segment, A51ATGD55, in the MBP sequence [Fig. 1(C)]. The crystal of Δ5MBP<+4>Tom20‐SS‐pALDH diffracted to a resolution of 1.8 Å (Supporting Information Table S3). During the purification, we noticed that the Δ5 deletion reduced the maltose binding to MBP. The MBP in the crystal lacked a bound maltose molecule after the structure refinement of the CCFS scaffold. The apo form of MBP adopts a more open conformation than the maltose‐bound form, which enlarged the CCFS, in addition to the simple expansion effect of the Δ5 deletion.
Defining the location of the mobile presequence in the conventional electron density maps
We did not build a model for the presequence, to avoid model bias. To locate the presequence peptide in the binding site, we used the F o−F c difference Fourier electron density map. However, we did not see any significant electron densities in the binding site of Tom20 in the difference map [Fig. 3(A)]. For confirmation, we attempted to place a helical model in the binding site of Tom20, with subsequent refinement. As expected, the B factors of the presequence atoms exceeded 100 Å2, and were much larger than those in the crystallographic snapshots (15 – 50 Å2).17, 20 After simulated annealing composite omit map construction to remove model bias, no meaningful electron densities remained in the binding site of Tom20. Taken together, the invisibility suggested the large mobility of the presequence peptide in the binding site, beyond the limits of conventional treatment.
Figure 3.
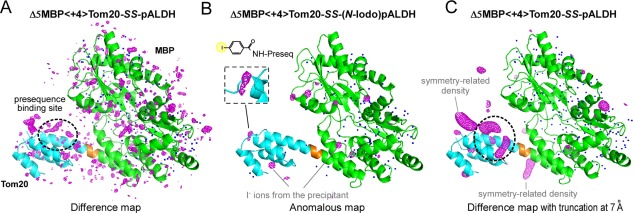
Electron density maps of the disulfide‐bond tethered MBP‐Tom20 complexes. (A) F o−F c difference electron density map of Δ5MBP<+4>Tom20‐SS‐pALDH, contoured at +3σ. (B) Anomalous difference map of Δ5MBP<+4>Tom20‐SS‐(N‐iodo)pALDH, contoured at +5σ to locate iodine atoms/ions. The inset shows an enlarged view of the electron density of the iodine atom in the 4‐iodobenzoyl group attached to the N‐terminus of pALDH. The electron densities of the bound iodine ions from the precipitant solution are also clearly visible, but their spherical shapes are easily discriminated from the iodine atom at the N‐terminus. (C) Difference map with truncation of high‐resolution reflections at 7 Å prior to Fourier transformation, contoured at +3σ. Note that the same X‐ray diffraction data set was used for map generation in (A) and (C). MBP and Tom20 are colored green and cyan, respectively. The 4‐residue spacer is colored orange. Water molecules included in the molecular replacement are depicted as blue dots.
To detect the presequence, we attached an iodine atom to the N‐terminus of the pALDH peptide. To do so, N‐succinimidyl‐4‐iodobenzoate was synthesized and used to modify the N‐terminal α‐amino group of the presequence (Supporting Information Fig. S5). An anomalous difference map clearly showed the blurred electron density of the iodine atom close to the expected N‐terminal position [Fig. 3(B)]. This suggests that the presequence peptide was actually present, at least partly, in the binding site of Tom20. Our interpretation is that an extra, weak interaction of the 4‐iodobenzoate group with the Tom20 protein increased the probability of the iodine atom existing at a particular position close to Tom20 [inset of Fig. 3(B)]. Owing to the enhanced detection ability of the anomalous map, the iodine atom was located at the partially occupied site.
Low‐pass filter effectively located the mobile presequence in the difference electron density maps
We then examined various data‐processing techniques, to determine whether they would be effective to find the location of mobile ligands/segments. We tested the map sharpening to locate the unmodeled presequence (Fig. 4). To our disappointment, only the noise pattern changed due to the over‐sharpening of the residual amplitudes at high resolution, and ripples resulted from the Fourier series termination [Fig. 4(B)]. This is because the blurred local electron density corresponding to a mobile region, such as the presequence, cannot be enhanced effectively using the current overall sharpening method, which assumes one rigid body in the asymmetric unit.22 Thus, for the selective enhancement of the mobile part, map sharpening was applied for the low‐resolution reflections, and map blurring was used for the high‐resolution reflections for noise suppression. We found that the simultaneous combination of sharpening and blurring improved the quality of the electron density in the binding site of Tom20 [Fig. 4(C)]. Simple blurring was also effective [Fig. 4(D)]. Here, we adopted a simpler form of map blurring, using the truncation (to be precise, zero padding) of the high‐resolution reflections, to avoid unnecessary amplitude modulation [Fig. 4(E)], because such amplitude modulation would distort the shape of the smeared electron density in the CCFS. The improvement in the signal‐to‐noise ratio depends on the choice of the resolution limit (r min) of the truncation for map generation. We found that r min = 7 Å was optimal to locate the α‐helical structure of the presequence peptide (Supporting Information Fig. S6). In contrast, the truncation had no obviously favorable effects on the quality of the electron density of the immobile helix in MBP (Supporting Information Fig. S6).
Figure 4.
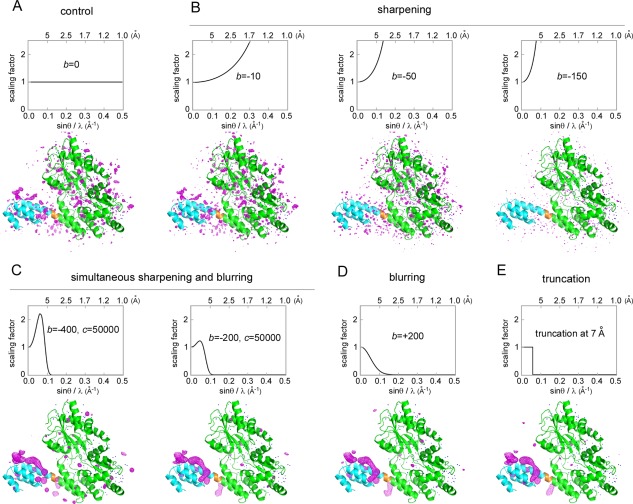
Effects of map sharpening/blurring and truncation on the difference electron density map, contoured at +3σ. (A) Control: no sharpening or blurring was applied. (B) The observed diffraction data (F o) were scaled up using the equation, F sharpened = F o exp[‐b(sinθ/λ)2], where b is a sharpening factor, θ is the scattering angle, and λ is the X‐ray wavelength. (C) Simultaneous application of map sharpening for the low‐resolution reflections and map blurring for the high‐resolution reflections. The bell‐shaped function for scaling is F sharpened = F o exp[‐b(sinθ/λ)2‐c(sinθ/λ)4]. (D) Simple blurring was also effective. (E) Truncation of the high‐resolution reflections, as a simplified version of the simultaneous sharpening and blurring. MBP, Tom20, and the 4‐residue spacer are colored green, cyan, and orange, respectively.
The use of a “free” test data set for cross‐validation caused the quality of the electron densities of the mobile atoms to deteriorate. The severity of the deterioration depended on the choice of the free test set (Supporting Information Fig. S7). As expected, the choice of the free test set had negligible effects on the immobile atoms. Various methods are available to reduce the undesirable effects of the use of the free test set in the electron density maps.23 We adopted the averaging of maps generated with different free test sets, in a procedure referred to as “FreeR averaging” in this study. For a free test set assigned to 5% of the structure factors, 20 maps were calculated and averaged. The specially processed difference map, with the truncation and FreeR averaging, clearly revealed an elongated electron density in the binding site of Tom20 [Fig. 5(A), in stereo], even though the same diffraction data set shown in Figure 3(A) was used.
Figure 5.
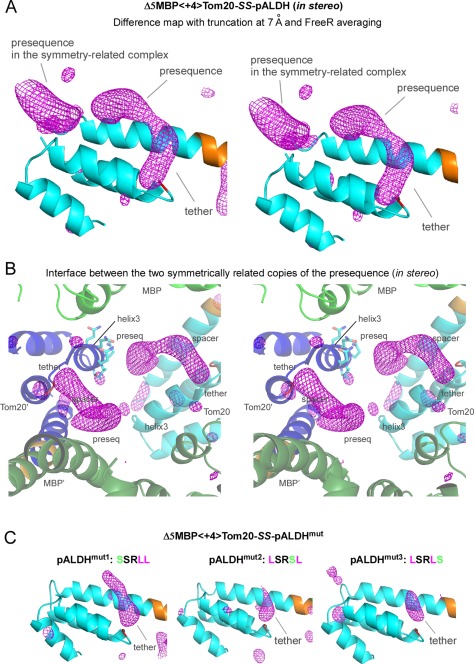
Electron densities in the CCFS corresponding to the presequence peptide and disulfide tether in the binding site of Tom20. (A) Stereo view of the truncated and FreeR‐averaged difference map of Δ5MBP<+4>Tom20‐SS‐pALDH, contoured at +3σ. The electron densities in the binding site of Tom20 were improved by the averaging of 20 maps calculated with different free test sets, as compared with the map without FreeR averaging in Figure 3(C). Tom20 and the 4‐residue spacer are colored cyan and orange, respectively. (B) Stereo close‐up view of the interface between two symmetrically related copies of the elongated electron density. MBP and Tom20 are colored green and cyan in one molecule, and dark green and blue in another molecule, respectively. The side chains of the helix3 of one Tom20 structure are shown in a stick model. (C) Effects of the essential leucine substitutions in pALDH on the electron density in the CCFS. These difference maps were generated and drawn in exactly the same way as in (A). Only the tether parts remained visible in the CCFS.
Figure 5(B) shows the close‐up view of the interface between two symmetrically related copies of the elongated electron density. The volume of the CCFS formed at the interface was not as large as we expected, but it provided sufficient room for the presequence peptide to “move” in the binding site. This is evident from the fact that the side chains on the helix3 of the nearest neighboring Tom20 were clearly visible in the nontruncated electron density map, and hence no stable close contacts were present between the presequence peptide and the helix3 in the crystal.
Interpretation of the electron density in the CCFS
Tom20 recognizes a 5‐residue consensus motif, φχχφφ, where φ represents a hydrophobic residue and χ represents any amino acid residue, embedded at various positions within the mitochondrial presequences.21, 24 We performed a mutagenesis study to validate the electron density in the CCFS. For each mutation, one of the three hydrophobic leucine residues at the φ positions was substituted with a hydrophilic serine residue. These substitutions resulted in affinity reductions ranging from 40‐ to 200‐fold.20 The crystals of the three mutant complexes diffracted to resolutions of 2.1, 1.8, and 1.6 Å (Supporting Information Table S3). Although the space group and the number of molecules in the asymmetric unit changed, the CCFS invariably formed. As expected, the electron density corresponding to the presequence part almost disappeared, but that of the tether part remained visible in the binding site [Fig. 5(C)] (Supporting Information Fig. S8). Even though the mutated peptides were still located in the CCFS, without the interactions with Tom20, their large motions were beyond the detection limit of the truncated difference map.
We then compared the electron density in the CCFS with the three crystallographic snapshots of the presequence previously obtained by conventional crystallography.17, 20 The electron density in the CCFS, excluding the tether region, superimposed well on the three snapshots and appeared to correspond to the volume formed by the intersection of overlapping electron densities of the presequence peptide in the binding site of Tom20 [Fig. 6(A)].
Figure 6.
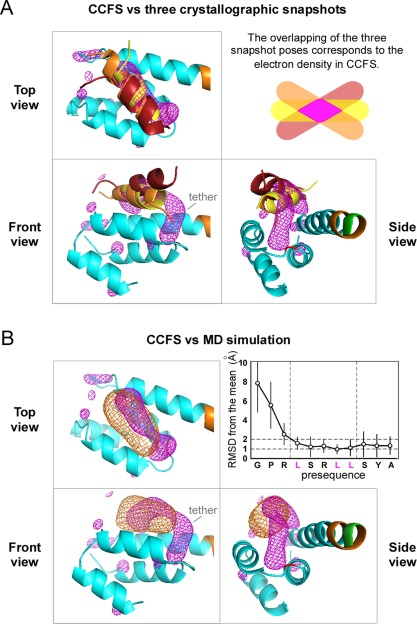
Validation of the electron density in the CCFS of the Δ5MBP<+4>Tom20‐SS‐pALDH crystal. (A) Superimposition of the electron density in the CCFS (magenta mesh) with the three crystallographic snapshots of the pALDH presequence, depicted as brown (PDB codes 2V1S), orange (2V1T), and yellow (3AX3) ribbon models. The 44 Cα atoms (A65‐E79+Y86‐L114) of the first three α‐helices of Tom20 were superimposed. (B) Superimposition of the electron density in the CCFS (magenta mesh) with the electron density calculated from the MD simulation (orange mesh). Tom20 and the 4‐residue spacer are colored cyan and orange, respectively. The graph shows the rmsds of the Cα atoms from the mean positions during the MD simulation.
The molecular dynamics (MD) simulations of the Tom20‐pALDH complex were previously performed in water without tethering.25 The electron density of pALDH calculated from the MD simulation overlapped well with the electron density in the CCFS, with the exclusion of the tether region [Fig. 6(B)]. From the MD simulation, we calculated the amplitude of motions of the presequence in the binding site, with respect to each Cα atom. The average displacement from the mean position was about 1.3 Å (isotropic B‐factor value of 130 Å2) for the Tom20‐binding consensus and tether residues [inset of Fig. 6(B)]. The fewer direct crystal lattice contacts may increase the rigid body disorder of the target protein. In fact, the average B‐factor value of the Tom20 part (70 Å2 for Cα atoms) is larger than that of the MBP part (30 Å2). However, considering the significantly higher value of the estimated average B‐factor of the presequence (130 Å2), the CCFS method is useful to extract the aspects of the presequence dynamics in the binding site of Tom20.
Design of CCFS in the AglB crystal
To demonstrate the general applicability of our approach, we applied the CCFS method to another protein. OST is a membrane‐bound enzyme that catalyzes oligosaccharide transfer to asparagine residues in glycoproteins.18 The C‐terminal globular domain of OST possesses a binding pocket that recognizes Ser and Thr residues in the N‐glycosylation consensus sequence, Asn‐X‐Ser or Asn‐X‐Thr, where X is a nonproline residue.26, 27 The Ser/Thr pocket is part of a segment called the “Turn‐Helix‐Loop (THL)” in the globular domain.28 A large variety of snapshot conformations of the THL segment were observed in the crystals of different OST proteins,28, 29, 30 suggesting its intrinsic flexibility in solution. In fact, an NMR relaxation study revealed that the THL segment of an OST was mobile in solution.28 The restriction of the segment's flexibility by an engineered disulfide bond in another OST reversibly suppressed the enzymatic activity, suggesting that the flexibility of the THL segment is essential for the N‐glycosylation sequon recognition.28
We selected an archaeal OST protein, AglB, from Pyrococcus furiosus. The C‐terminal globular domain of AglB (soluble AglB or sAglB) contains a long N‐terminal α‐helix, which was used as part of the rigid connector helix [Fig. 7(A)]. We first performed a model building study to deduce the appropriate length of the spacer between MBP and sAglB (Supporting Information Fig. S9). The spacer lengths of +1, +16, and +20 were chosen for the proper placement of the THL segment in the CCFS. The amino acid sequences of the spacer were poly‐alanine‐based sequences containing glutamate‐lysine pairs.31 Crystals were obtained for MBP<+16>sAglB and MBP<+20>sAglB, which diffracted to resolutions of 2.6 and 2.1 Å, respectively (Supporting Information Table S4). Close inspection of the MBP<+16>sAglB structure revealed that the relative orientation of MBP and sAglB largely deviated from the expected arrangement (Supporting Information Fig. S10), due to the disruption of the helical structure in the long spacer. In contrast, the MBP<+20>sAglB structure had similar geometry to the model, and was used for further analysis.
Figure 7.
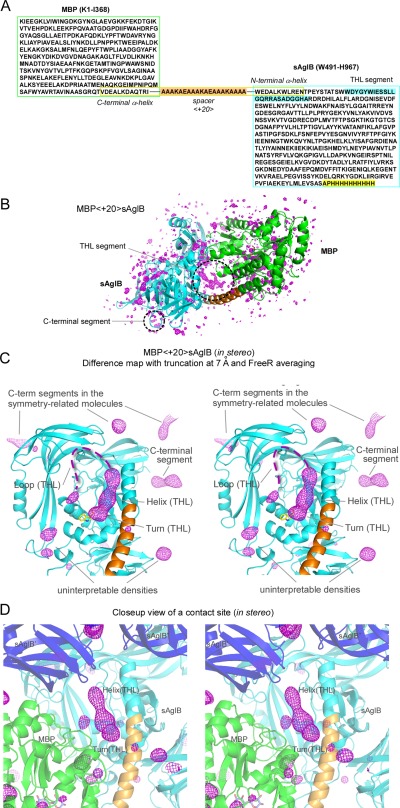
Electron density maps of the MBP‐sAglB fusion protein. (A) Amino acid sequence of MBP<+20>sAglB. The connector helix is enclosed by the yellow box, and the +20 spacer is highlighted in orange. The THL segment and the C‐terminal segment, which were not included in the CCFS scaffold, are highlighted in cyan and yellow, respectively. (B) F o−F c difference map, (C) truncated and FreeR‐averaged difference map (in stereo), and (D) close‐up view of a crystal contact site (in stereo) of MBP<+20>sAglB, all contoured at +3σ. Note that the same X‐ray diffraction data set was used for map generation in (B–D). The 3‐residue turn structure of the THL segment consists of the underlined residues in the well‐conserved WWDYG motif. The first Trp residue of the WWDYG motif is part of the CCFS scaffold, and its side chain is depicted as a yellow stick model in (C). The invisible, highly mobile loop of the THL segment is shown as dashed lines. MBP, sAglB, and the 20‐residue spacer are colored green, cyan, and orange in one molecule, respectively. sAglBs in other fusion protein molecules are colored blue, and labeled with sAglB′ and sAglB″.
In the conventional difference Fourier map of MBP<+20>sAglB, there were no continuous electron densities corresponding to the THL segment in the CCFS [Fig. 7(B)]. In the truncated and FreeR‐averaged difference map, clear electron densities corresponding to the turn (3 res) and subsequent helix (9 res) were visible, but the loop (12 res) remained invisible except for 1 or 2 C‐terminal residues [Fig. 7(C)]. The CCFS formed at the interface between symmetrically related MBP‐sAglB molecules appeared to have sufficient volume, as planned [Fig. 7(D)]. For reference, the r min dependency and the effect of the choice of the free data set are shown in Supporting Information Figures S11 and S12. These results showed that the turn and helix parts have narrower spatial distributions than that of the loop.
Discussion
We have proposed a fusion protein method for the creation of CCFS in protein crystals [Figs. 5(B) and 7(D)], independent of the protein molecule packing mode in crystals (Fig. 1). We selected MBP as a fusion partner among many tag proteins, to construct a rigid CCFS scaffold. We successfully used α‐helical spacers up to 20‐residues in length, to fuse the C‐terminal α‐helix of MBP and the N‐terminal α‐helix of the target proteins firmly. The present study has confirmed that the CCFS in protein crystals can be rationally created with reasonable efforts. In addition, a preliminary model building study prior to protein expression is quite useful to reduce the number of experimental trials (Supporting Information Figs. 2 and 9).
The presequence peptide in an α‐helical conformation with relatively large amplitude motions in the binding site of Tom20 was hardly visible in the created CCFS in the conventional difference electron density map [Fig. 3(A)], but was detected as a continuous, elongated electron density in the difference map with the truncation of high‐resolution reflections [Fig. 5(A)]. The dynamical segment in the C‐terminal globular domain of the AglB protein in the created CCFS was also visible in the truncated difference map [Fig. 7(C)]. Map averaging was dispensable in both cases, but it effectively solved the problem of the use of the free test set for cross‐validation (Supporting Information Figs. S7 and S12). We obtained an averaged rmsd value of 1.3 Å from the mean positions during the MD simulation, as the amplitude of the presequence peptide movement [Fig. 6(B)]. This estimated amplitude value suggested that the difference map with truncation has the potential to analyze motions up to 1.5 Å rmsd from the mean positions. Obviously, without any proactive CCFS designs, the truncation of high‐resolution reflections prior to map generation and FreeR averaging of the generated maps should be effective to find the location of loosely‐bound small ligands and short segments, in the context of larger proteins that provide a rigid framework, if such mobile parts are fortuitously located in a spontaneously formed CCFS. In fact, the truncated difference map revealed the mobile C‐terminal tail segment of sAglB, in addition to the THL segment [Fig. 7(C)]. The size of the electron density suggested that it corresponds to the first 4 or 5 residues of the invisible 12‐residue segment (4 sAglB residues and 8 His‐tag residues). Finally, although we used truncation at r min = 7 Å in this study, the resolution limit of the diffraction measurement (d min) should be as high as possible, for the accurate molecular replacement and refinement of the CCFS scaffold. In our case, d min was 1.6–2.6 Å (Supporting Information Tables S3 and S4).
The bulk solvent is the disordered water and ions filling the intermolecular space between protein molecules, and it attenuates low‐resolution reflections. Bulk solvent correction is necessary to account for the attenuation effect.32, 33 The bulk solvent correction may have significant impacts on the quality and accuracy of the electron density in the CCFS, since the truncated difference maps are generated only from low‐resolution data. We compared the map generated by the program REFMAC5 [Fig. 5(A)] with those generated by the programs CNS and PHENIX, using the same CCFS scaffold (MBP‐Tom20) structure refined by the program REFMAC5 (Supporting Information Fig. S13). The three programs produced similar shapes and volumes of the electron density in the CCFS. This comparison indicated that different implementations of the bulk solvent correction did not cause significant differences in the electron density in the CCFS. Improvements of bulk solvent correction methods will benefit CCFS crystallography in the future.
Our current working hypothesis is that “a rapid equilibrium of multiple states with partial recognitions” is the molecular basis for the promiscuous binding of the Tom20 receptor to diverse mitochondrial presequences with nearly equal affinities.17, 20 The smeared electron density in the difference electron density map corresponded to the partially‐overlapped volume among the multiple poses of the presequence helix [Fig. 6(A)], but did not allow us to estimate the amplitude of the presequence helix motions in the binding site. Thus, the results presented in this work are mostly confirmatory of our previous structural studies. In the future, better diffraction measuring and data processing will improve the signal‐to‐noise ratio of the difference electron density maps, and reveal the spatial distribution of the moving α‐helical presequence peptide experimentally. Obviously, the influences of the non‐physiological solution conditions required for crystallization must be considered when interpreting the electron density in the CCFS.
In protein crystallography, protein crystals are routinely frozen prior to X‐ray diffraction measurements, to avoid radiation damage. Thus, the presequences in the CCFS do not literally “move,” but thermal motions in solution are observed as static disorder in a glassy state in frozen crystals. Consequently, the electron density in the CCFS obtained at a cryogenic temperature does not fully represent the kinetically trapped ensemble in a room‐temperature equilibrium, because a substate population shift is unavoidable during the flash‐cooling of crystals, due to unit cell contraction and water remodeling.34, 35 In fact, a small displacement was found between the experimental electron density in the CCFS and the simulated electron density [Fig. 6(B)]. A promising solution is room‐temperature X‐ray diffraction measurement. A detailed comparison of the high‐resolution electron density maps of dihydrofolate reductase (DHFR), measured at room and cryogenic temperatures, revealed that cryocooling induced artificial biases in the rotamer distributions of the side chains.35 At the main‐chain level, a narrower distribution of a loop structure was also detected after cryocooling, but the movement of the averaged position was not evident. Room‐temperature X‐ray diffraction measurements of protein crystals with CCFS will reveal the unbiased movements of protein segments in the future.
Finally, the concept of CCFS is not limited to the analysis of protein dynamics. Other potential applications include correcting the distorted protein conformations induced by the crystallographic contact effects; allowing ligands to soak into otherwise occluded sites in protein crystals, and studying the time‐resolved large motions that are normally constrained in conventional crystal lattices.
Materials and Methods
Protein expression and purification
The DNA sequence encoding MBP (residues K1‐T366) was amplified by PCR, using the plasmid pMAL‐c5x (New England Biolabs) as the template. The reverse primer contained codons encoding Arg‐Ile, to restore the original MBP sequence (K1‐T366R367I368), and a 20‐bp overlap with the 5′ region of the Tom20 sequence. The DNA sequence of the cytosolic domain (A65‐L126) of Tom20 from Rattus norvegicus (UniProt: TOM20_RAT, AC: Q62760) was amplified by PCR from the expression plasmid encoding GST‐Tom20, generated in the previous study.36 The two DNA fragments were combined by the SOEing PCR method.37 The final PCR product was cloned into the NdeI‐SalI digested pET‐41b(+) vector (Novagen), using an In‐Fusion Advantage PCR Cloning Kit (Clontech). The resultant expression plasmid was used for the production of MBP<0>Tom20. Then, one to eight amino acid residues were inserted or one to four residues were deleted at the junction of the two proteins, by the inverse PCR method.38 The amino acid sequences at the junction site are summarized in Supporting Information Table S1. The deletion of Δ5 (A51ATGD55) in MBP was also generated by the inverse PCR method. The DNA sequence (codon‐optimized for E. coli expression) of the C‐terminal globular domain (W491‐H967 plus a C‐terminal 8×His) of AglB from Pyrococcus furiosus was synthesized by Life Technologies. The amplified PCR product was cloned into the SmaI‐SalI digested pET‐47b(+) vector (Novagen). The expression plasmid thus obtained was used for the expression of MBP<0>sAglB. One (Alanine), 16, and 20 spacer residues were inserted at the junction of the two proteins by the inverse PCR method. The amino acid sequences of the long spacers are listed in Supporting Information Table S4.
The fusion proteins were expressed in the E. coli host strain BL21(DE3) (Stratagene). The E. coli cells were grown at 310 K in LB medium, supplemented with 30 mg L−1 kanamycin. When the A600 reached 0.3‐0.5, isopropyl‐1‐thio‐β‐d‐thiogalactopyranoside was added at a final concentration of 0.5 mM. After 3–12 h induction at 289 K, the cells were harvested by centrifugation and disrupted by sonication. The MBP‐Tom20 fusion proteins containing the wild‐type MBP were purified by affinity chromatography on amylose resin (New England Biolabs) in TS buffer (50 mM Tris‐HCl, pH 8.0, 0.1M NaCl, with 10 mM maltose added for elution), gel filtration chromatography on a Superdex 200 10/300 column (GE Healthcare) in TS buffer, and anion‐exchange chromatography on a MonoQ 5/50 column (GE Healthcare) in 20 mM Tris‐HCl, pH 8.0, with a salt gradient from 0 to 1M NaCl. In spite of the absence of maltose in the crystal structures, the MBP‐Tom20 fusion proteins with the Δ5 deletion weakly adsorbed to the amylose resin. Thus, the affinity chromatography on amylose resin was also used for the Δ5 deletion‐containing MBP‐Tom20 fusion proteins. The eluted proteins were desalted and concentrated with an Amicon Ultra‐15 centrifugal filter unit (Millipore, 30 kDa NMWL) to 10 mg mL−1 in 20 mM MES buffer, pH 6.5, for crystallization. To purify MBP‐sAglB, the fusion proteins eluted from the amylose resin were denatured by 8M urea, refolded by gel filtration chromatography on a Superdex 200 HiLoad 26/60 column in TS buffer, and finally purified by anion‐exchange chromatography on a MonoQ 5/50 column in 20 mM Tris‐HCl, pH 8.0, with a salt gradient from 0 to 1M NaCl. The eluted proteins were desalted and concentrated with an Amicon Ultra‐15 unit (50 kDa NMWL) to 10 mg/mL in 20 mM MES buffer, pH 6.5, for crystallization. In spite of the urea treatment, a maltose molecule was bound to the MBP in the fusion proteins.
ALDH presequence peptides and tethering to Tom20
The presequence (G12PRLSRLLS20) is derived from rat mitochondrial aldehyde dehydrogenase (ALDH, AC: P11884). The residue numbering starts at 12, since the sequence corresponds to the C‐terminal half of the ALDH presequence. A cysteine residue, Cys24, was attached to the C‐terminus via a three‐residue spacer, Y21A22G23.21 The peptide and its sequence variants were synthesized with an N‐terminal acetyl group and a C‐terminal amide group, by Hokkaido System Science (Sapporo, Japan). An intermolecular disulfide bond was formed between the Cys24 residue of the presequence and a single cysteine residue (Cys100) of Tom20 in high pH buffer (0.1M Tris‐HCl, pH 9.0). Note that MBP contains no cysteine residues. The formation of the disulfide bond was monitored by native‐PAGE and reverse‐phase HPLC analyses in the absence and presence of 1 mM dithiothreitol.
For the preparation of an N‐terminally iodine‐labeled pALDH peptide, N‐succinimidyl‐4‐iodobenzoate was synthesized by carbodiimide coupling.39 Briefly, to a solution of 4‐iodobenzoic acid and N‐hydroxysuccinimide in dry dichloromethane, 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide hydrochloride was added. The mixture was refluxed for two hours. After extraction with a saturated bicarbonate solution, the organic layer was concentrated and the resulting residue was washed with a small volume of chloroform. The product was obtained as a white powder. The purity was monitored by 1H and 13C‐NMR. The pALDH peptide for iodine labeling was synthesized without an acetyl group at the N‐terminus. The α‐amino group of the peptide was reacted with 0.1M N‐succinimidyl‐4‐iodobenzoate in 40% dimethylsulfoxide. The modified peptide was purified by reverse‐phase HPLC. The modification was confirmed by monitoring the increase of the molecular mass corresponding to the 4‐iodobenzoyl group, IC6H4CO, using MALDI‐TOF‐MS. The iodinated pALDH peptide was tethered to Δ5MBP<+4>Tom20 through a disulfide bond in the high pH buffer. The resultant Δ5MBP<+4>Tom20‐SS‐(N‐iodo)pALDH was purified by anion exchange chromatography.
Crystallization, data collection, and structure determination
Initial crystallization screening was performed by the sitting drop vapor diffusion method. To search for optimized crystallization conditions, we performed grid screening and additive screening (Hampton Research). X‐ray diffraction data were collected at beamlines BL32XU, BL44XU, and BL26B1 of SPring‐8 (Harima, Japan), and beamlines PF‐BL1A, PF‐BL5A, PF‐BL17A, and AR‐NW12A of the Photon Factory (Tsukuba, Japan). Diffraction data were collected with a wavelength of 0.9000 or 1.000 Å, and with a 1.6000 Å wavelength for the detection of the anomalous iodine signal. The crystals were cooled to 95–100 K. The final crystallization conditions and the cryo‐conditions are summarized in Supporting Information Tables S2–S4.
The diffraction data were processed with the program HKL2000.40 Initial phases were obtained by the molecular replacement method with the program PHASER in CCP4, 41 using the structures of MBP (PDB code 1ANF for the maltose‐bound form of MBP,42 and PDB code 3PUY for the apo form of MBP43), Tom20 (PDB code 2V1T 17), and sAglB (PDB code 2ZAI, without the THL segment29) as the search models. We did not build a model for the presequence (and the disulfide tether part plus the waters bound to Tom20), to avoid model bias. The coordinates of the fusion proteins plus water molecules bound to MBP were refined with the program REFMAC5 ver. 5.7 in CCP4 44 The final manual modeling was performed using the program COOT.45 To create the anomalous difference Fourier map, the phases were calculated from the model of Δ5MBP<+4>Tom20 in the Δ5MBP<+4>Tom20‐SS‐pALDH crystal, by the program SFALL in CCP4, and the map was generated by the program FFT in CCP4. For the tethered complex crystals with mutated presequence peptides, pALDH(L18S) and pALDH(L19S), pseudomerohedral twinning was detected using the program Xtriage in the PHENIX program package.46 Xtriage indicated that the twin law was a mirror index (h,‐k,‐h‐l) for P21. The estimated twin fractions were 0.18 for L18S and 0.25 for L19S. The data were detwinned with DETWIN in the CCP4 program package,47 and were used for the refinement calculations. Data collection and refinement statistics are summarized in Supporting Information Tables S2–S4.
Electron density map to locate the mobile peptide/segment
The 20 refinement calculations were rerun with 20 different free test sets, using the refined model of the CCFS scaffolds (fusion protein plus water molecules bound to MBP). No model was placed for the smeared electron density corresponding to the presequence peptide or the THL segment, and no additional water molecules were picked. A sigma A‐weighted F o−F c (mF o‐DF c) difference electron density map was calculated, using Fourier amplitudes (DELFWT) and phases (PHDELWT) outputted by the program REFMAC5. The program SFTOOLS in CCP4 was used to perform the truncation and zero padding of the high‐resolution structure factor amplitudes, by using the keywords, “SELECT RESOL < 7” and “CALC COL DELFWT = 0.” Since the truncation of high‐resolution reflections results in a coarse mesh size of the electron density map, zero padding maintains the original mesh spacing size simply for cosmetic reasons. The truncation threshold was empirically selected. This value, r min= 7 Å, seems suitable for finding the location of the α‐helical structures. FFT was used for map generation. The average of the truncated F o−F c difference maps generated with different 20 free test sets (comprised of 5% of the total reflections) was calculated, by using the MAPS ADD function of the program MAPMASK in CCP4. The overall task workflow is provided in Supporting Information Fig. S14.
Figures were generated with the program PyMOL (Schrödinger). Multiple structure fitting and rmsd calculations in batch mode were performed with the program ProFit, version 3.1 (Martin, A.C.R., and Porter, C.T., http://www.bioinf.org.uk/software/profit/, 2009).
Model building for the estimation of the optimal length of the spacer
To estimate the optimal spacer length in the connector helix, computer modeling was performed using PyMOL. An ideal α‐helical conformation was assumed for the connector helix between MBP (PDB code 4MBP 42) and Tom20/sAglB. The structure of Tom20 was adopted from one of the three crystallographic snapshot structures (PDB code 2V1T 17). The structure of sAglB was the C‐terminal globular domain of the Pyrococcus furiosus AglB (PDB code 2ZAI 29).
Calculation of electron density from MD simulation
The 50,000 snapshots of the Tom20‐pALDH complex, obtained from the previous replica‐exchange MD simulation,25 were classified into four major clusters with different probabilities (I: 4.76%, II: 5.13%, III: 80.0%, IV: 4.23%). Based on the probabilities, 100 structures were randomly chosen and superimposed to fit the 40 Cα atoms (F69‐E79+Y86‐L114) of the first three α‐helices of Tom20. The coordinates of the presequence were extracted and combined into one file. The occupancy and isotropic B factor of all atoms were uniformly set to 0.01 and 1.00, respectively. The program SFALL was used to calculate the structure factors from the coordinate file, and the structure factors were used to generate a simulated map with a resolution limit of 7 Å, using the program FFT. The GRID SAMPLE parameter was set to 8 for fine grid spacing.
Accession Numbers
The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 5AZ6, 5AZ8, 3FRX, 5AZ9, and 5AZA).
Supporting information
Supporting Information
Acknowledgments
The authors thank Drs. Hiroshi Nonaka and Shinsuke Sando (Graduate School of Engineering, The University of Tokyo) for the chemical synthesis of N‐succinimidyl‐4‐iodobenzoate, and Dr. Shunsuke Matsumoto for the plasmid constructions and pilot expression experiments. The experiments at the Photon Factory were performed with the approval of the Photon Factory Program Advisory Committee, as proposal 2013G023, and those at SPring‐8 were performed under the Cooperative Research Program of the Institute for Protein Research, Osaka University, as proposals 20126719, 20136820, and 20146922 and under the Budding Researchers Support Proposal of the Japan Synchrotron Radiation Research Institute (JASRI) as proposal 2012B1657.
Conflict of Interest: The authors declare no competing financial interests.
References
- 1. Singh P, Abeysinghe T, Kohen A (2015) Linking protein motion to enzyme catalysis. Molecules 20:1192–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ramanathan A, Savol A, Burger V, Chennubhotla CS, Agarwal PK (2014) Protein conformational populations and functionally relevant substates. Acc Chem Res 47:149–156. [DOI] [PubMed] [Google Scholar]
- 3. van den Bedem H, Fraser JS (2015) Integrative, dynamic structural biology at atomic resolution–it's about time. Nat Methods 12:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gobl C, Madl T, Simon B, Sattler M (2014) NMR approaches for structural analysis of multidomain proteins and complexes in solution. Prog Nucl Magn Reson Spectrosc 80:26–63. [DOI] [PubMed] [Google Scholar]
- 5. Vranken WF (2014) NMR structure validation in relation to dynamics and structure determination. Prog Nucl Magn Reson Spectrosc 82:27–38. [DOI] [PubMed] [Google Scholar]
- 6. Zhang XJ, Wozniak JA, Matthews BW (1995) Protein flexibility and adaptability seen in 25 crystal forms of T4 lysozyme. J Mol Biol 250:527–552. [DOI] [PubMed] [Google Scholar]
- 7. Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KF, Becker S, Meiler J, Grubmuller H, Griesinger C, de Groot BL (2008) Recognition dynamics up to microseconds revealed from an RDC‐derived ubiquitin ensemble in solution. Science 320:1471–1475. [DOI] [PubMed] [Google Scholar]
- 8. Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T (2009) Hidden alternative structures of proline isomerase essential for catalysis. Nature 462:669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T (2011) Accessing protein conformational ensembles using room‐temperature X‐ray crystallography. Proc Natl Acad Sci U S A 108:16247–16252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smyth DR, Mrozkiewicz MK, McGrath WJ, Listwan P, Kobe B (2003) Crystal structures of fusion proteins with large‐affinity tags. Protein Sci 12:1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhan Y, Song X, Zhou GW (2001) Structural analysis of regulatory protein domains using GST‐fusion proteins. Gene 281:1–9. [DOI] [PubMed] [Google Scholar]
- 12. Hodel A, Kim S‐H, Brunger AT (1992) Model bias in macromolecular crystal structures. Acta Cryst A48:851–858. [Google Scholar]
- 13. Praznikar J, Afonine PV, Guncar G, Adams PD, Turk D (2009) Averaged kick maps: less noise, more signal and probably less bias. Acta Cryst D65:921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmidt O, Pfanner N, Meisinger C (2010) Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol 11:655–667. [DOI] [PubMed] [Google Scholar]
- 15. Endo T, Yamano K, Kawano S (2011) Structural insight into the mitochondrial protein import system. Biochim Biophys Acta 1808:955–970. [DOI] [PubMed] [Google Scholar]
- 16. Abe Y, Shodai T, Muto T, Mihara K, Torii H, Nishikawa S, Endo T, Kohda D (2000) Structural basis of presequence recognition by the mitochondrial protein import receptor Tom20. Cell 100:551–560. [DOI] [PubMed] [Google Scholar]
- 17. Saitoh T, Igura M, Obita T, Ose T, Kojima R, Maenaka K, Endo T, Kohda D (2007) Tom20 recognizes mitochondrial presequences through dynamic equilibrium among multiple bound states. Embo J 26:4777–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jarrell KF, Ding Y, Meyer BH, Albers SV, Kaminski L, Eichler J (2014) N‐linked glycosylation in Archaea: a structural, functional, and genetic analysis. Microbiol Mol Biol Rev 78:304–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen X, Zaro JL, Shen WC (2013) Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev 65:1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saitoh T, Igura M, Miyazaki Y, Ose T, Maita N, Kohda D (2011) Crystallographic snapshots of Tom20‐mitochondrial presequence interactions with disulfide‐stabilized peptides. Biochemistry 50:5487–5496. [DOI] [PubMed] [Google Scholar]
- 21. Obita T, Muto T, Endo T, Kohda D (2003) Peptide library approach with a disulfide tether to refine the Tom20 recognition motif in mitochondrial presequences. J Mol Biol 328:495–504. [DOI] [PubMed] [Google Scholar]
- 22. Liu C, Xiong Y (2014) Electron density sharpening as a general technique in crystallographic studies. J Mol Biol 426:980–993. [DOI] [PubMed] [Google Scholar]
- 23. Kleywegt GJ, Jones TA (1997) Model building and refinement practice. Methods Enzymol 277:208–230. [DOI] [PubMed] [Google Scholar]
- 24. Muto T, Obita T, Abe Y, Shodai T, Endo T, Kohda D (2001) NMR identification of the Tom20 binding segment in mitochondrial presequences. J Mol Biol 306:137–143. [DOI] [PubMed] [Google Scholar]
- 25. Komuro Y, Miyashita N, Mori T, Muneyuki E, Saitoh T, Kohda D, Sugita Y (2013) Energetics of the presequence‐binding poses in mitochondrial protein import through Tom20. J Phys Chem B 117:2864–2871. [DOI] [PubMed] [Google Scholar]
- 26. Lizak C, Gerber S, Numao S, Aebi M, Locher KP (2011) X‐ray structure of a bacterial oligosaccharyltransferase. Nature 474:350–355. [DOI] [PubMed] [Google Scholar]
- 27. Matsumoto S, Shimada A, Nyirenda J, Igura M, Kawano Y, Kohda D (2013) Crystal structures of an archaeal oligosaccharyltransferase provide insights into the catalytic cycle of N‐linked protein glycosylation. Proc Natl Acad Sci U S A 110:17868–17873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nyirenda J, Matsumoto S, Saitoh T, Maita N, Noda NN, Inagaki F, Kohda D (2013) Crystallographic and NMR evidence for flexibility in oligosaccharyltransferases and its catalytic significance. Structure 21:32–41. [DOI] [PubMed] [Google Scholar]
- 29. Igura M, Maita N, Kamishikiryo J, Yamada M, Obita T, Maenaka K, Kohda D (2008) Structure‐guided identification of a new catalytic motif of oligosaccharyltransferase. EMBO J 27:234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matsumoto S, Igura M, Nyirenda J, Matsumoto M, Yuzawa S, Noda N, Inagaki F, Kohda D (2012) Crystal structure of the C‐terminal globular domain of oligosaccharyltransferase from Archaeoglobus fulgidus at 1.75 Å resolution. Biochemistry 51:4157–4166. [DOI] [PubMed] [Google Scholar]
- 31. Arai R, Ueda H, Kitayama A, Kamiya N, Nagamune T (2001) Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng 14:529–532. [DOI] [PubMed] [Google Scholar]
- 32. Afonine PV, Grosse‐Kunstleve RW, Adams PD, Urzhumtsev A (2013) Bulk‐solvent and overall scaling revisited: faster calculations, improved results. Acta Cryst D69:625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brunger AT (2007) Version 1.2 of the Crystallography and NMR system. Nat Protoc 2:2728–2733. [DOI] [PubMed] [Google Scholar]
- 34. Dunlop KV, Irvin RT, Hazes B (2005) Pros and cons of cryocrystallography: should we also collect a room‐temperature data set? Acta Cryst D61:80–87. [DOI] [PubMed] [Google Scholar]
- 35. Keedy DA, van den Bedem H, Sivak DA, Petsko GA, Ringe D, Wilson MA, Fraser JS (2014) Crystal cryocooling distorts conformational heterogeneity in a model Michaelis complex of DHFR. Structure 22:899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Igura M, Maita N, Obita T, Kamishikiryo J, Maenaka K, Kohda D (2007) Purification, crystallization and preliminary X‐ray diffraction studies of the soluble domain of the oligosaccharyltransferase STT3 subunit from the thermophilic archaeon Pyrococcus furiosus . Acta Cryst F63:798–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Horton RM, Cai ZL, Ho SN, Pease LR (1990) Gene splicing by overlap extension: tailor‐made genes using the polymerase chain reaction. Biotechniques 8:528–535. [PubMed] [Google Scholar]
- 38. Ochman H, Gerber AS, Hartl DL (1988) Genetic applications of an inverse polymerase chain reaction. Genetics 120:621–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shell TA, Mohler DL (2005) Selective targeting of DNA for cleavage within DNA‐histone assemblies by a spermine‐[CpW(CO)3Ph]2 conjugate. Org Biomol Chem 3:3091–3093. [DOI] [PubMed] [Google Scholar]
- 40. Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- 41. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Cryst 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Quiocho FA, Spurlino JC, Rodseth LE (1997) Extensive features of tight oligosaccharide binding revealed in high‐resolution structures of the maltodextrin transport/chemosensory receptor. Structure 5:997–1015. [DOI] [PubMed] [Google Scholar]
- 43. Oldham ML, Chen J (2011) Crystal structure of the maltose transporter in a pretranslocation intermediate state. Science 332:1202–1205. [DOI] [PubMed] [Google Scholar]
- 44. Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst D67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 46. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Cryst D66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Cryst D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information