

Abstract
This work explores the heterogeneity of aggregation of polyglutamine fusion constructs in crude extracts of transgenic Caenorhabditis elegans animals. The work takes advantage of the recent technical advances in fluorescence detection for the analytical ultracentrifuge. Further, new sedimentation velocity methods, such as the multi‐speed method for data capture and wide distribution analysis for data analysis, are applied to improve the resolution of the measures of heterogeneity over a wide range of sizes. The focus here is to test the ability to measure sedimentation of polyglutamine aggregates in complex mixtures as a prelude to future studies that will explore the effects of genetic manipulation and environment on aggregation and toxicity. Using sedimentation velocity methods, we can detect a wide range of aggregates, ranging from robust analysis of the monomer species through an intermediate and quite heterogeneous population of oligomeric species, and all the way up to detecting species that likely represent intact inclusion bodies based on comparison to an analysis of fluorescent puncta in living worms by confocal microscopy. Our results support the hypothesis that misfolding of expanded polyglutamine tracts into insoluble aggregates involves transitions through a number of stable intermediate structures, a model that accounts for how an aggregation pathway can lead to intermediates that can have varying toxic or protective attributes. An understanding of the details of intermediate and large‐scale aggregation for polyglutamine sequences, as found in neurodegenerative diseases such as Huntington's Disease, will help to more precisely identify which aggregated species may be involved in toxicity and disease.
Keywords: analytical ultracentrifugation, Caenorhabditis elegans, multi‐speed method, polyglutamine aggregation, sedimentation velocity, wide distribution analysis
Abbreviations
- BME
β‐mercaptoethanol
- DCVJ
4‐(dicyanovinyl) julolidine
- DIC
differential interference contrast
- DTT
dithiothreitol
- EGTA
ethylene glycol tetraacetic acid
- FCS
fluorescence correlation spectroscopy
- FRAP
fluorescence recovery after photobleaching
- FRET
Förster resonance energy transfer
- GFP
green fluorescent protein
- HEPES
4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid
- MSM
multi‐speed method
- PMSF
phenylmethylsulfonyl fluoride
- polyQ
polyglutamine
- SDD‐AGE
semi‐denaturating detergent agarose gel electrophoresis
- SDS‐PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TAE
Tris‐Acetate‐EDTA
- TBS
Tris‐buffered saline
- WDA
wide distribution analysis
- YFP
yellow fluorescent protein
Introduction
Recent technical developments in fluorescence detection for the analytical ultracentrifuge have made possible the detection of fluorescent protein fusion constructs in complex mixtures for relevant biochemical and biomedical problems.1, 2 One such problem is understanding the degree of aggregation and heterogeneity in polyglutamine sequences, which are found in at least nine human neurodegenerative diseases, with Huntington's Disease and Machado‐Joseph Disease being well‐characterized paradigmatic model systems.3, 4, 5 Proteins in this family that contain long glutamine repeats (typically >40) can lead to disease, and the repeats are caused by unstable repeat expansion at the nucleotide level, resulting in insertions of CAG triplets.
Biochemical work from several other laboratories has provided us with a fairly deep understanding of the polyQ assembly pathway, which arises from intrinsic aggregative tendencies due to glutamine‐glutamine side chain and backbone interactions, and this has been recently reviewed.6, 7 Such biochemical work has led to a detailed understanding of the importance of stable intermediates (such as oligomeric species) in the path to fibril assembly and inclusion body formation.8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 In the case of the huntingtin protein, the causal agent in Huntington's Disease, this assembly process is regulated by a 17‐residue N‐terminal domain, which can transiently adopt a helical conformation, leading to formation of nonspecific coiled‐coil bundles, or oligomers, which substantially impact the assembly process.7 Such oligomers may then form stable higher order aggregates that can be detected by a variety of approaches taking advantage of fluorescent tags, such as Number & Brightness microscopy image analysis,20 FRET,21 FRAP,22 FCS,23 and most recently, sedimentation velocity (SV) with fluorescence detection.24 The major biomedical goal is to identify which of these species are primarily responsible for the disease state.
One value of fluorescence detection is that proteins of interest may be tagged and studied in complex mixtures, and even in samples derived from in vivo systems, using a variety of genetically tractable animal models. There is a significant body of literature identifying the presence of species with intermediate aggregation (or oligomeric) states for polyQ‐containing proteins in such in vivo model systems,20, 25, 26, 27, 28, 29, 30, 31, 32, 33 and this literature has been reviewed recently.5 Similar intermediate aggregation states from a wide variety of proteins involved in other neurodegenerative diseases are also thought to be involved in the disease state.34, 35, 36, 37, 38, 39 In cells and tissues, the fibrils typically become highly entangled, often sequestering other molecular components, to form inclusion bodies. While intermediate aggregate states are now widely thought to be toxic, it has not precluded the possibility that inclusion bodies are also involved as cytotoxic components in disease, and such toxicity has been attributed to either sequestration of various critical molecular factors, or to inhibition of the normal protein degradation processes through the ubiquitin‐proteasome system (UPS) or through autophagy.5 In the case of the huntingtin protein, both the intact protein and presumed proteolytic fragments of this protein have been shown to form heterogeneous aggregate pools either within the cytoplasm or the nucleus of the cell.7
We aim to extend the proof of concept for analysis of crude extracts using SV techniques, coupled with biochemical (SDD‐AGE) and microscopy (imaging of puncta and FRAP) methods to validate this new approach. The advantages of SV methods for the study of smaller aggregative species are their superior precision in establishing polydispersity and defining discrete populations. The focus of the proposed work here is to provide more detailed information about the size of the aggregates for polyQ‐containing sequences as formed in transgenic animal model systems, and to establish the distribution of aggregated species amongst more diffuse soluble oligomers, larger aggregates, and inclusion bodies.
Previous work has shown that the fluorescence detection system in the analytical ultracentrifuge1, 2 could be used to study polyQ protein aggregation in cultured murine neuroblastoma cells.24 In this work, detailed information was provided about the size and heterogeneity of the huntingtin (Htt) exon 1 fragment fused to emerald green fluorescent protein, as expressed in this cell line and extracted as a crude lysate with minimal fractionation. The authors showed that they could identify Htt monomers (2.3 S), soluble oligomers (various species centered around 140 S), and inclusion bodies (320,000 S) for an Htt variant containing a Q46 insertion. A control, using Q25, did not show measurable aggregation. This new SV methodology inspired us to apply a similar approach to test hypotheses of polyQ assembly in C. elegans. C. elegans has been used extensively in the last decade to understand polyQ aggregation and its implications for pathogenicity, and several reviews have been written on this subject.40, 41, 42, 43 Comparison of molecular and cellular impacts on protein aggregation across various model systems will certainly offer the prospect of a general understanding of the mechanism of disease, and provide the ability to study other important factors, such as aging, tissue specificity, and mitigation through over‐expression of chaperones, for example. We believe that the work presented here is the first application of SV methodology to the study of protein aggregation in multicellular organisms. In addition, we take advantage of a relatively new approach in the study of proteins, the multi‐speed method (MSM),44, 45 that allows a broader range of sedimentation coefficients to be assayed in one experiment.
The range of the observable sedimentation coefficients from an experiment carried out at a single rotor speed can be severely limited, and use of MSM can remove such limitations, by starting an ultracentrifuge run at low speed so that the largest particles can be captured, and then increasing the speed in steps during the run so that the smallest species have had an opportunity to clear the meniscus. Such an approach can accommodate in a single experiment a wide range of sedimentation coefficients from about 1.0 S to about 250,000 S.45 Data from all speeds can be combined into a single continuous distribution function using a wide distribution analysis (WDA) method developed originally by the Yphantis group and implemented in Sedanal (v. 6.01).44, 45 The combined MSM‐WDA approach is warranted in resolving the wide distribution of aggregates seen by the use of other methods (e.g., SDD‐AGE, FRAP).
Results and Discussion
The goal of this work was to ascertain whether the entire range of polyQ aggregation states, as expressed as fusions with fluorescent proteins in C. elegans, could be observed using fluorescence detection in the analytical ultracentrifuge. Technical validation was performed by an analysis of sedimentation behavior of the monomer species. The upper range of aggregation would involve the particles that appear as puncta in confocal microscopy images of transgenic worms, and methodology was developed to detect these structures using the analytical ultracentrifuge. Finally, a careful analysis of intermediate aggregation states was performed using MSM‐WDA; such aggregates are thought by many to be the toxic species, as described above.
First, expression levels of various polyQn‐YFP constructs in C. elegans crude extracts (using one‐day‐old worms) were estimated with Western blots using a rabbit anti‐GFP antibody [Fig. 1(B) and Supporting Information S1A], which cross‐reacts with YFP, in order to determine appropriate cell‐loading concentrations for the analytical ultracentrifuge. The typical detection limit in Westerns is in the low‐ to high‐femtomole levels, which puts this in the same range as the detection limit of the fluorescence monochromater of ∼300 femtomoles.46 The approximate sizes of the proteins were assessed by comparing them to a molecular weight ladder containing proteins of known size. We detected the presence of the polyglutamine region using a polyQ antibody, in C. elegans strains Q24, Q35, Q40, and Q82 [Supporting Information Fig. S1(B)]. The detection of the polyQ epitope was coincident with detection using a YFP antibody for Q24, Q35, Q46, and Q82 [Fig. 1B and Supporting Information S1A]. Significant differences in expression were noted when comparing loading of relatively equal amounts of total protein (Supporting Information Fig. S1A) to variable loading volumes with the aim of equalizing the amounts of detected fusion protein (Fig. 1B). The molecular weights of the bands were close (within a few kDa) to the predicted molecular weight calculated based on each protein sequence. Extracts from Q0 and Q24 migrated at about 32 kDa, while Q35 migrated around 38 kDa. Extracts of Q35 were occasionally observed as a doublet with a predominant band at 38 kDa and a lesser band at 34 kDa. Q40 samples consistently displayed a doublet at 40 kDa and 38 kDa. Q82 samples migrated at around 48 kDa, as a single band. The presumed degradation products were also observed in earlier work on these strains.47
Figure 1.
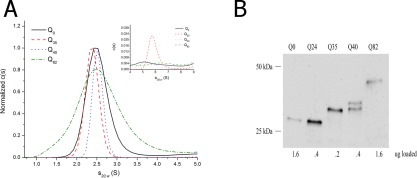
A: c(s) distribution for Qn‐YFP monomers in c. elegans assessed by sedimentation velocity analysis at high speeds (50,000 rpm) in 1x lysis buffer (50 mM HEPES, pH 7.3, 100 mM KCl, 2 mM PMSF, 10 mM DTT, 1 mM EGTA, and supplemented with protease inhibitor 1x PICS) at 20°C. The data were fitted using a c(s) continuous size distribution model (Sedfit v. 14.4d), commonly used to describe the behavior of small diffusing particles. Inset: focus on the 4‐9 S range shows a small amount of slightly aggregated sedimenting material. B: Western blot of Qn‐YFP in C. elegans using one‐day‐old adult worms. Loading concentrations have been adjusted to detect YFP in each of the samples. (Note: Supporting Information Figure S1A shows a Western blot of Qn‐YFP when the same amount of total protein is loaded per well.)
Monomers
The first question addressed was whether the fusion construct could be detected in a complex mixture derived from a worm crude extract, and whether the size could be established with reasonable fidelity. Since the monomer size is well defined, this was used to establish technical feasibility. Detection of monomeric polyQn‐YFP species was attempted using polyQ lengths of Q 0, Q 35, Q 40, and Q 82.
After preparation of crude extracts (with minimal fractionation), total protein loading concentrations were set between 0.5 mg mL−1 and 0.25 mg mL−1 in order to minimize sedimentation artifacts due to crowding effects. The monomer distribution in these extracts was analyzed in the analytical ultracentrifuge using a rotor speed of 50,000 rpm. A well‐populated monomer species was detected for all protein constructs tested, including Q 0‐YFP, Q 35‐YFP, Q 40‐YFP, and Q 82‐YFP. These species all sedimented as monodisperse populations with a sedimentation coefficient centered at s20,w = 2.5 S [Fig. 1(A); the full set of sedimentation profiles are shown in Supporting Information Fig. S2]. This s‐value compares well with that seen in earlier work for sedimentation of a related fluorescent protein either as a purified component24, 46 or in cell extracts.24 Although the experimentally determined frictional coefficient ratio, f/f 0, fell in the range of 1.3 − 1.4, consistent with that expected for a compact β‐barrel as found in YFP,24, 48 it is inappropriate to extract out molecular weight information. This message is driven home by the variations in the breadths of the normalized c(s) distributions for the monomer seen in Figure 1(A). Such differences might be ascribed to changes in the compactness of the polyQ domain, as seen in pure polyQ model systems,49 and in huntingtin exon 1 model systems,50 but could also be due to sedimentation artifacts, and thus should not be over‐interpreted due to the complexity of the mixtures being analyzed. All crude extracts showed minor components with sedimentation coefficients in the range 4‐9 S, which could represent limited oligomerization, or possibly, interactions with other components in the extracts, since sedimentation in this range was also seen for Q 0‐YFP [Fig. 1A, inset].24
Inclusion bodies
Having established technical feasibility for sedimentation analysis of proteins in worm crude extracts, through characterization of the monomer species for the protein constructs tested, the next goal was to establish the range of sizes that could be evaluated. Work from Hatters’ group showed that sizes in the range of 320,000 S could be detected, using samples containing 2 M sucrose, coupled with using low rotor speeds.24 The sizes, as determined by their SV method, were consistent with the sizes of the inclusion bodies that they detected in their mammalian cell line. In an effort to capture such inclusion bodies in our worm model system, a similar approach was taken, using 2 M sucrose and a low speed of 3000 rpm (Fig. 2). As described previously,47, 51 both Q40‐YFP and Q82‐YFP show evidence of inclusion bodies by the presence of puncta as imaged using confocal microscopy (Supporting Information Fig. S4A), and data analysis of the puncta sizes showed a high degree of heterogeneity in size distribution of 4.3 ± 2.7 μm. The smallest punctum observed was about 0.8 μm in diameter, while the largest being almost 10 μm. Further evidence that the puncta contain inclusion bodies was obtained by photobleaching those puncta whose boundaries were clearly distinguishable from the surrounding fluorescence from all sides. After photobleaching, no recovery was seen for Q40‐YFP and Q82‐YFP when the laser was focused on the center of such puncta (Supporting Information Fig. S4B), consistent with results from previous published work in the Morimoto lab.47, 51
Figure 2.
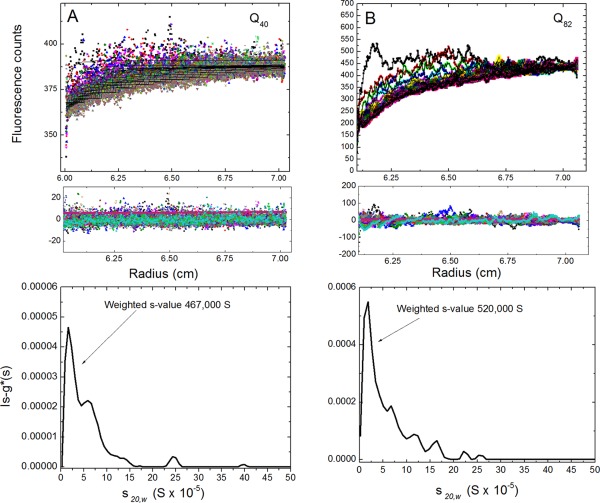
Sedimentation velocity experiments containing 2 M sucrose. A: Aggregates of Q40‐YFP captured in 1x lysis buffer supplemented with 2 M sucrose and run at 3000 rpm (top graph represents raw data; middle graph shows residuals from the fits; and the bottom graph shows the ls‐g*(s) distribution). The data were fit using a ls‐g(s*) model, which is equivalent to the distribution g(s*) from dc/dt as a distribution of nondiffusing particles. B: Aggregates of Q82‐YFP captured under similar conditions as in Q40‐YFP (SV data for Q0‐YFP and Q35‐YFP samples in 2 M sucrose are shown in Supporting Information Fig. S4).
SV data were collected for crude extracts containing the various fusion constructs, and were fitted using an ls‐g*(s) size distribution, which gave a distribution centered at s 20,w = 162,000 S for Q40‐YFP [Fig. 2(A), bottom panel], and at s 20,w = 183,000 S for Q82‐YFP [Fig. 2(B), bottom panel]. The weight‐average s‐values were 467,000 S and 520,000 S respectively for Q40‐YFP and Q82‐YFP [Fig. 2(A,B), bottom panels]. Assuming a partial specific volume of 0.73 mL g−1, sedimentation coefficients of such values correspond to particles of radius ranging from 0.91 μm to 6 μm, thus overlapping with that observed by microscopy. Differences between the size estimates calculated from SV experiments and confocal microscope measurements may be reconciled by recognizing that inclusion bodies are likely to be more asymmetric and less dense, as confocal imaging suggests (Supporting Information Fig. S4A). In contrast, samples from the Q0‐YFP and Q35‐YFP strains showed no boundary formation or boundary movement in the SV experiment (Supporting Information Fig. S5), suggesting that inclusion bodies are only seen in the strains with the polyQ length above the threshold, and their formation being consistent with puncta formation (Supporting Information Fig. S4A). SDD‐AGE supports this result since there is no insoluble material stuck in the wells for Q0‐YFP and Q35‐YFP [Fig. 3(A)]. Furthermore, observation of no sedimentation for Q0‐YFP and Q35‐YFP in 2 M sucrose indicates that the sedimentation boundaries observed for Q40‐YFP and Q82‐YFP were a reflection of the inclusion bodies and not an artifact of general protein aggregation induced by high sucrose concentrations.
Figure 3.
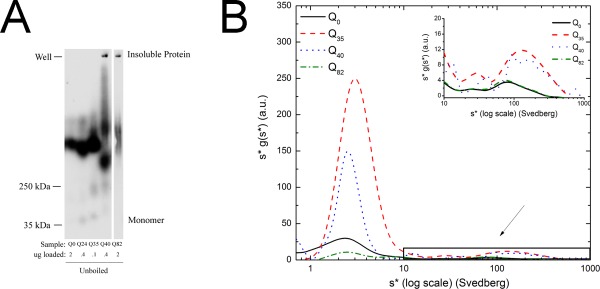
Analysis of aggregates by SDD‐AGE and SV. A: SDD‐AGE experiments. Analysis of Qn‐YFP aggregates by electrophoresis in 1.5% agarose gel. Samples were prepared in 1x sample buffer containing ∼1% SDS. Lanes contain protein extracts from Q0‐YFP, Q24‐YFP, Q35‐YFP, Q45‐YFP, and Q82‐YFP worms. All samples were run on the same gel, and the separation in images indicates that a redundant lane was cropped out. B: Aggregate distribution from multi‐speed data for Q0‐YFP, Q35‐YFP, Q40‐YFP, and Q82‐YFP. Log plot of s*g (s*) vs. (s*) for 0.5 mg/mL Qn‐YFP samples in 1x lysis buffer centrifuged for about 3 h at 10,000 rpm, 30,000 rpm, and continued at 50,000 rpm until the last sedimentation boundary cleared the meniscus. The graph shows the complete distribution of the intermediate aggregates with s‐values ranging from 0.8 to 1,000 S. The inset focuses on the range 10‐1,000 S, to highlight the intermediate aggregates. The MSM data were analyzed using Sedanal v. 6.01 with WDA.
Intermediate aggregation states
Evidence for in vivo intermediate aggregate states for transgenic worms expressing polyQ fusion constructs comes from microscopy experiments, applying FRAP47, 51 and FCS,23 methods. FRAP analysis on the series of polyQ strains suggests such intermediates based on some fluorescence recovery, as shown in Supporting Information Figure S4B, and is consistent with the published work on these strains.47, 51 Intermediates have also been detected in worm crude extracts using SDD‐AGE experiments, and such experiments are described below. Though SDS‐PAGE and SDD‐AGE experiments can measure the abundance of the SDS‐soluble and insoluble protein species, they are not able to resolve the potential heterogeneity within the largest species of the insoluble population as a result of aggregates being trapped within the well. Nevertheless, SDD‐AGE may allow intermediate‐size aggregates to enter the gel and as such, a limited comparison can be made between this method and the SV method.52, 53 Insoluble aggregates were detected for all strains tested, including Q0‐YFP, indicating that the YFP itself is prone to limited aggregation [Fig. 3(A), and Supporting Information Fig. S3, the latter figure showing the monomer species]. The aggregation patterns for Q0‐YFP and Q24‐YFP are about the same, with species present that are somewhat greater than 250 kDa. Q35‐YFP and Q40‐YFP show some evidence of yet higher order aggregation within the resolving power of the gel (∼100 kDa–5 mDa).54 The faster migrating band seen for Q40‐YFP likely represents proteolysis, as such proteolysis is seen in the Western blots (Supporting Information Fig. S1), although we cannot rule out that this faster migrating band may represent a more compact or smaller aggregate state. Both Q40‐YFP, and Q82‐YFP also show large scale aggregation, as detected by material stuck in the wells, and may represent the same materials observed in the 2 M sucrose SV experiments and the FRAP experiments, as described above.
Given the complexity and breadth of the distribution of aggregates as seen by SDD‐AGE [Fig. 3(A)], we chose to use MSM‐WDA to improve the capacity to detect the wide range of the size heterogeneity of the aggregated protein in the analytical ultracentrifuge. In general, MSM‐WDA provides the ability to analyze broad aggregate distributions since data collected at several speeds can be analyzed simultaneously. For comparison, data for the same construct, Q40‐YFP, were studied using either the c(s) method for data collected at a single speed, or MSM‐WDA [Fig. 4(A)]. The MSM‐WDA reveals a broader distribution of aggregates than that seen for the single‐speed method, thus presumably providing a more complete view of the size distributions since it has access to data covering a wider range of gravitational fields.
Figure 4.
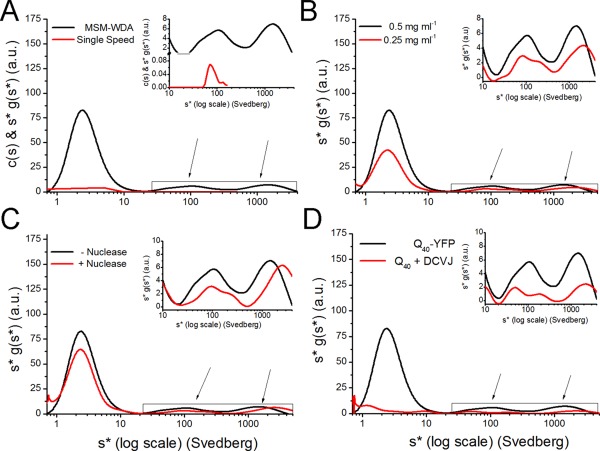
Analysis of intermediate aggregation of Q40‐YFP. A: Aggregate distribution of 0.5 mg/mL Q40‐YFP prepared in 1x lysis buffer, comparing a single speed to MSM. MSM involved centrifugation at 3000 rpm, 10,000 rpm, 30,000 rpm, and continued at 50,000 rpm until the smallest species cleared the meniscus. The single speed data were analyzed using Sedfit v. 14.4d while the MSM data were analyzed using Sedanal v. 6.01 using wide distribution analysis (WDA). The graph represents the complete distribution with s‐values ranging from 0.8‐4,000 S. B: Protein concentration dependence for the aggregate distribution of 0.5 mg/mL Q40‐YFP. C: Aggregate distribution for Q40‐YFP in 1x lysis buffer treated with and without universal nuclease centrifuged using MSM‐WDA. D: Comparison of aggregate distribution of Q40‐YFP and Q40 stained with 10 μM DCVJ, in 1x lysis buffer. The insets show the distribution profiles of intermediate aggregates, with s‐values ranging from 10‐4,000 S.
MSM data were analyzed using a “wide distribution analysis” (WDA) method incorporated as part of the dc/dt module in Sedanal v. 6.01.45 The dc/dt algorithm, based on the time derivative method developed by Stafford,55 eliminates both the time independent and radially independent noise, and results in improved precision. MSM data for Q0‐YFP, Q35‐YFP, Q40‐YFP, and Q82‐YFP strains indicated polydisperse distributions [Fig. 3(B)] once again confirming the SDD‐AGE results [Fig. 3(A)], with the majority of the protein forming aggregates [note that the X‐axis in Fig. 3(B) is a log scale, thus compressing the profiles of the larger scale materials]. Since both Q0‐YFP and Q82‐YFP lysates contained peaks with s‐values at around 80 S, we ascribe such species as arising independently of the polyQ aggregation, corresponding either to small oligomers driven by the fluorescent protein itself, or due to interactions with other cellular components as mentioned above [Fig. 3(B)]. The 80 S distribution is not due to problems with autofluorescence since wild‐type worms showed no sedimentation boundaries in this range. In addition to these species, SV analysis of Q35‐YFP and Q40‐YFP lysates identified peaks with s‐values in the range of 100‐1,000 S [Fig. 3(B)]. These peaks disappear in looking at the s‐value distribution for Q82‐YFP, whereas those that we ascribe due to YFP aggregation do not. Therefore, we suggest that small nonspecific oligomers in the 80 S range are not sequestered in the inclusion bodies, while the ones in the 100 − 1000 S range are. All strains showed a single dominant peak centered at an s‐value of 2.5 S in the MSM runs, which corresponds to the monomer species as described above [inset Fig. 3(B); see also Fig. 1(A)].
To validate the accuracy of the distribution, a variety of conditions was tested. The classic approach to testing for dynamic equilibrium effects on protein distribution is to test the concentration dependence of the distribution of s‐values. Due to instrument detection limits and problems with nonideality at high protein concentrations, we only tested a two‐fold difference in total protein concentration. The sedimentation profiles collected at 0.5 mg mL−1 and 0.25 mg mL−1 total protein showed no significant impact on the sedimentation rate of species as a function of concentration [Fig. 4(B)], including the monomer at 2.5 S and both populations of oligomers centered around 100 S and 1000 S. The presence of natural molecular crowding agents such as nucleic acids can have a significant impact on sedimentation behavior due to the excluded volume effect and an increase in viscosity.56 In addition to the effects on crowding and viscosity caused by nucleic acids, this macromolecule can also have an impact: (1) if the ionic strength is very low (<1 mM), such that nucleic acid becomes a significant co‐ion or counterion; (2) if the nucleic acids sediment at a rate that fall in the range of s we are looking at; or (3) if the nucleic acid binds to aggregates significantly. We can rule out point 1 simply because the buffer contains a significant salt concentration. Points 2 and 3 can be addressed by testing the effect of breaking down the nucleic acids into much smaller pieces. We observed no significant changes in the oligomer distributions of Q40‐YFP lysates after treatment with a universal nuclease. Both distributions look similar, containing prominent peaks with s‐values centered around 110 − 120 S and above 1000 S [Fig. 4(C)]. To test for the general effects of crowding on sedimentation behavior in the crude extracts, glycerol (a known molecular crowding agent) was added to Q40‐YFP lysates, using the same buffer as that used for the monomer analysis. The monomer species appears largely to be gone (Supporting Information Fig. S6), suggesting that the glycerol induced further aggregation in the sample. Sedimentation profiles looking at the intermediate range of aggregation behavior indicated polydisperse populations (Supporting Information Fig. S6 inset) with the distribution skewed to smaller s‐values, indicative of large‐scale changes in sedimentation behavior. These results suggest that reagents such as glycerol should be used with caution in such experiments, probably owing to non‐ideality associated with glycerol concentration boundaries.
A long‐standing concern in the field has been the potential effect of fusion partners on the rate, degree, and heterogeneity of aggregation. Our SDD‐AGE and MSM results of Q0‐YFP suggest that some aggregation due to the fluorescence protein partner itself does occur. To assess the effect of the YFP on polyQ aggregation as observed using SV, we looked at the sedimentation profiles of Q40‐YFP and a Q40 construct with no protein fluorescence partner, fluorescently labeled instead with the dye, DCVJ. This dye, like ThioT,57 binds with high specificity to cross‐β‐sheet fibrils,58, 59, 60 and has been used in sedimentation velocity experiments as an effective detection method.60 We show here that worms will take up this dye simply by soaking them in a solution of DCVJ for a short period of time prior to sample preparation.
In order to construct the Q40‐expressing transgenic worms without YFP, a separate marker for following the expression was required, and mCherry was chosen because its excitation wavelength is significantly different from that of DCVJ. Worms soaked with DCVJ show puncta that are essentially indistinguishable from those imaged with the YFP partner present [Fig. 5(A)]. Q40 and mCherry do not co‐localize in their expression, as evidenced in an image highlighting the fluorescence of both proteins, with mCherry being diffusely expressed [Fig. 5(B)]. As a control, worms only expressing mCherry showed no DCVJ staining [Fig. 5(C,D)]. Interestingly, Q40 expression seems to actually exclude co‐localization of mCherry, since a zoom on a single Q40 punctum shows an absence of mCherry fluorescence in that location [Fig. 5(E,F)].
Figure 5.
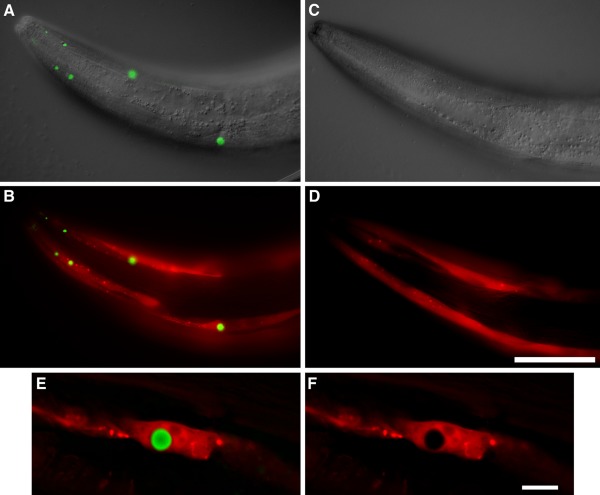
Poly Q inclusions detected with DCVJ. Transgenic strains expressing either Punc‐54::Q40 + Pmyo‐3::mCherry (CL1781) or Pmyo‐3::mCherry only (CL6617) were stained in parallel with DCVJ, and DCVJ fluorescence was imaged using identical exposure settings for the two strains. A: CL1781, overlay of DIC and DCVJ images. B: CL1781, DCVJ and mCherry fluorescence. C: CL6617, overlay of DIC and DCVJ images. D: CL6617 DCVJ and mCherry fluorescence. E: Higher magnification image of single DCVJ‐positive inclusion in strain CL1781, single optical section from digitally deconvolved image. F: Same as (E), mCherry channel only. Note: DCVJ only detects Q40 inclusions. Size bar = 50 μm, panels A−D; = 10 μm, panels E and F. DIC and epifluorescence images were acquired on a Zeiss Axiophot compound microscope equipped with a computer‐controlled Z‐drive and software from Intelligent Imaging Innovations. Photoshop software (Adobe) was used to fuse DIC and epifluorescence images.
Interestingly, as an aside, the specificity of binding extends further to certain types of cross‐β‐sheet fibrils, as exemplified by the fact that DCVJ does not bind cross‐β‐sheet fibrils made up of the Aβ peptide (Supporting Information Fig. S7C and D) whereas, X‐34, a dye known to bind cross‐β‐sheet fibrils made up of the Aβ peptide,61 does not bind to polyQ cross‐β‐sheet fibrils (Supporting Information Fig. S7A and B).
DCVJ‐stained worm extracts of Q40 prepared in the same way as for the Qn‐YFP animals were subjected to sedimentation velocity, and the resultant sedimentation profile in the 100‐1,000 S range looks similar to those seen for Q40‐YFP [Fig. 4(D)]. Such a result suggests that the sedimenting fluorescent materials in the range of 100‐1,000 S are due solely to polyQ‐induced aggregation. It is interesting to note that the monomer species does not appear, consistent with the understanding that DCVJ (and other such dyes) does not bind this state. Finally, while it was initially surprising to note the presence of an aggregate pool in the 100 S region in the absence of the YFP component, this may not be unusual given the presence of such smaller intermediate species noted in the biochemical literature. A careful analysis reveals the presence of a 110 S peak that is specific to polyQ aggregation (this peak is not seen for Q0‐YFP), and a new peak at about 50 S that is occluded by the YFP aggregate peak at around 80 S. Such peaks are consistent with limited polyQ aggregate, or oligomer, sizes as seen in the literature.19, 29, 62, 63 In particular, work from Legleiter's lab62 on both a fragment of the huntingtin protein and a simplified polyQ peptide sequence, has shown spheroidal aggregates that fall within the same range as that which we observed by SDD‐AGE and SV, which over time can then convert to fibrils, thus depleting the oligomer pools (as we see as well) that are then ultimately sequestered into inclusion bodies.
In conclusion, we have shown that SV methods using fluorescence detection can be extended to the study of polyQ aggregation in transgenic animal model systems, building on the important work of the Hatters group in the study of such aggregation in a tissue culture system.24 An analysis of the polyQ monomer pool provides proof of principle that the size of proteins can be extracted with some reliability in complex mixtures. While such a result is perhaps surprising, it may simply be due to the degree of dilution of third‐component materials in such extracts. High‐density solutions run at low speed in the analytical ultracentrifuge allow us to identify species that likely correspond to inclusion bodies, as the predicted size of such particles is coincident with that measured by confocal microscopy. Another major advance that we describe is the ability to use multi‐speed methods coupled with wide distribution analysis to study highly heterogeneous mixtures in crude extracts. We find that a single‐speed experiment is often insufficient for identifying the wide distribution of aggregated species, however the multi‐speed method allows one to improve the ability to detect a wide range of heterogeneous mixtures in the 2 − 1000 S size range (and larger). To our knowledge, this experimental approach has not been applied before to such complex biological mixtures, particularly crude extracts from animal model systems. Future work is being planned to compare polyQ aggregation between C. elegans and D. melanogaster to determine to what degree the work is generalizable, and to begin exploring correlations between toxicity and aggregate heterogeneity.
Materials and Methods
C. elegans strains and maintenance
Worms were maintained according to standard methods, at 20° C on nematode growth media (NGM) with OP50 E. Coli.64 The polyglutamine strains, expressing different lengths of CAG‐repeats fused with YFP, have been described elsewhere: wild‐type (wt) Bristol strain N2; Q0AM134 (rmls126[Punc‐54::Q0::YFP]X; Q24AM138 (rmIs130[Punc‐54::Q24::YFP]II); Q35AM140 (rmIs132[Punc‐54::Q35::YFP]I; Q40AM141 (rmIs133[Punc‐54::Q40::YFP]X).23, 47, 51, 65 The Q82‐YFP strain, harboring an integrated extrachromosomal array, was created in our laboratory using a plasmid donated from the Morimoto laboratory.66 To construct transgenic worms expressing a polyglutamine protein without a fluorescent fusion partner, the unc‐54 promoter/polyQ40::YFP plasmid generated by the Morimoto lab was first subjected to in vitro mutagenesis. Using a Stratagene QuikChange 11 site‐directed mutagenesis kit (cat#200523), a lysine‐to‐stop (AAG to TAG) mutation was introduced at the fourth codon of YFP. After confirmation by sequencing, this construct was co‐injected with Pmyo‐3::mCherry), and a transmitting extrachromosomal line (strain CL1781) was recovered for our experimental work.
Synchronized populations for crude extracts were obtained from gravid adults after treatment with 20% (v/v) alkaline hypochlorite solution (3.0 mL Bleach, 3.75 mL 1M NaOH, 8.25 mL ddH2O) for 5 min and allowing eggs to hatch in M9 buffer overnight. On the next day, the synchronized populations were transferred on fresh NGM plates.
Worms that did not have the YFP fusion partner were stained with the molecular dye, DCVJ, which was carried out as follows. Stock solutions of DCVJ were prepared in a 50 : 50 (v/v) ethanol:water mixture and stored in the dark at −20 °C. Concentrations of DCVJ stock solutions were determined using an extinction coefficient of 65,900 M−1 cm−1 at 453 nm.60 Live 4th larval stage transgenic worms were incubated in a drop of saturated DCVJ solution (∼200 μM in 10 mM Tris‐HCl, pH 7.5) for 2 h at 20 °C, then destained by transfer to a seeded NGM plate and overnight incubation at 16 °C. Worms were mounted in 100 mM sodium azide and imaged using a Zeiss Axiophot using FITC and CY3 filter sets. Digital overlays and brightness adjustments were done using Photoshop.
Preparation of crude lysates
After synchronization, day‐one‐old adult worms were harvested and washed three times with M9 Buffer (22 mM sodium phosphate, 22 mM potassium phosphate, 85 mM NaCl, 1 mM MgSO4). Worms were snap frozen in liquid nitrogen and kept at −80 °C. After 3 cycles of freezing and thawing, protein was extracted using lysis buffer (100 mM HEPES, pH 7.3, 200 mM KCl, 2 mM EGTA, 2 mM PMSF, 2 mM DTT, and 2x Protease Inhibitor Cocktail (Roche)). Lysis was supplemented with 5 cycles of 30 s each of motorized homogenization (Kontes), followed by a 5‐min incubation on ice. Sedimentation via gravity was allowed to occur for 30 min, after which the supernatant was removed. Total protein concentration was calculated using Coomassie Plus Protein Assay Reagent (Thermo Scientific). Samples were snap frozen and stored at −80 °C.
Western blot analysis
Sample loading buffer was added for a final concentration of 125 mM Tris‐HCl pH 6.8, 1% (v/v) SDS, 10% (v/v) glycerol, 5% (v/v) BME, and bromophenol blue. Samples were boiled for 5 min and subsequently loaded on a 12.5% (v/v) acrylamide gel. Samples containing 400 ng of total protein were added to each lane, unless otherwise indicated. After transfer to nitrocellulose membrane (Thermo Scientific 88018) and blocking using blotto (5% (w/v) nonfat dry milk in PBS containing 0.001% (v/v) Tween‐20), protein expression was detected using a primary antibody against GFP (Invitrogen A11122) and goat anti‐rabbit HRP as a secondary antibody (Cell Signaling 7074S). For some experiments, expression was detected using a primary antibody against the polyQ sequence itself (Millipore MAB1574) using anti‐mouse HRP as a secondary antibody (Cell Signaling Technology 7076S). An antibody against gamma tubulin (Abcam ab50721) was used as a loading control. Membranes were prepared using Super Signal West Femto Maximum Sensitivity Substrate (Thermo Scientific 34095), and imaged using a FluorChem HD2 Imager (Alpha Innotech).
SDD‐AGE analysis
Sample loading buffer was added to crude lysate for a final concentration of 125 mM Tris‐HCl pH 6.8, 1% (v/v) SDS, 10% (v/v) glycerol, 5% (v/v) BME, and bromophenol blue and incubated for 5 min at room temperature. A 1.5% horizontal agarose gel containing 0.1% (v/v) SDS (in 1xTAE) (40 mM Tris‐HCl, 20 mM acetic acid, 1 mM EDTA) was loaded with indicated amounts of total protein. Samples were run at 45 V in a running buffer containing 0.1% (v/v) SDS (in 1xTAE).52 The amount of protein loaded per lane was adjusted based on the intensity of bands as seen in the Western experiments, in order to assure detection of aggregates. Protein was transferred overnight to nitrocellulose membrane using capillary action in 1xTBS (50 mM Tris‐HCl and 150 mM NaCl). Blocking and protein detection followed the same procedure as that used for SDS‐PAGE.
Confocal microscopy and fluorescence after photobleaching (FRAP)
Day‐one‐old worms were transferred to a 4% (w/v) agar pad on a glass slide and paralyzed in a drop of M9 + 20 mM NaN3 solution. Worms were imaged manually on a Nikon C1 Confocal Microscope using the 488 nm laser and the 20x objective. Images were collected at 70% transmission with a gain of 4 (6 for Q0‐YFP) and the pinhole set to large. For FRAP analysis, a 10 μm2 area was selected and imaged 6 times every 20 s at 10% transmission and 512x512 step size to assess prebleach fluorescence intensity. Subsequently, a 1 μm2 area at the center of the prebleach area was bleached at 100% transmission for 15 iterations every 0.8 s with 32 × 32 step size. Then, the original 10 μm2 area was imaged with the exact same settings as during prebleach six times every 20 s at 10% transmission. Relative fluorescence intensity (RFI) was determined in the NIS‐Elements based on the formula RFI = T t/T 0, where T 0 represents fluorescence intensity in the bleached area before bleaching and T t fluorescence intensity in the bleached area at time t after bleaching.23 Since our confocal microscope did not support an automated protocol, it was not possible to monitor a control unbleached area in tandem with the bleached area.
Analytical ultracentrifugation
Lysates were prepared in 2x lysis buffer with no addition of SDS (100 mM HEPES, pH 7.3, 200 mM KCl, 2 mM PMSF, 2 mM DTT, 2 mM EGTA, and 2x Protease Inhibitor Cocktail (Roche)) and frozen in −80 °C (see above protocol for more details). Samples were adjusted to 0.5 or 0.25 mg/mL total protein concentration in 1x lysis buffer and supplemented with 20% (v/v) glycerol, or 2 M sucrose. A 3 M sucrose stock solution was prepared as previously described.24 Sucrose stock solution concentrations were verified by density measurement (ρ = 0.1257 × M + 1.00).67
Samples (350 μL) were loaded into two‐channel quartz window charcoal/Epon sedimentation velocity cells with 50 μL FC43 fluorinert heavy oil. Cells were placed in an 8‐hole AnTi rotor (Beckman‐Coulter) and equilibrated to 20 °C in an XL‐A analytical ultracentrifuge (Beckman‐Coulter) fitted with a fluorescence detection system. Radial fluorescence scans were collected continuously at 3000 rpm, 10,000 rpm, 30,000 rpm, and 50,000 rpm using a 488 nm laser for excitation and 520 nm cut off emission filter using a constant photomultiplier voltage (voltage: 2234, Gain: 8, Range 4) in all experiments. The run was continued at 50,000 rpm until it was obvious that the solution was cleared of the protein or until the meniscus was depleted.
Samples containing 1x lysis buffer, and 20% (v/v) glycerol were corrected to s 20,w using Sednterp. v 1.09,68 while samples containing 2 M sucrose s 20,w values were corrected using the reported values for density and viscosity of concentrated sucrose solutions.24, 69 Temperature‐dependent partial specific volumes: Q0‐YFP = 0.7315 mL g−1; Q35‐YFP = 0.7228 mL g−1; Q40‐YFP = 0.7217 mL g−1; and Q82‐YFP = 0.7143 mL g−1. Solution densities: 1x lysis buffer (1.00685 g mL−1); 10% glycerol (1.0355 g mL−1); 20% glycerol (1.06446 g mL−1); and 2 M sucrose solution (1.250 g mL−1). Viscosities: 1x lysis buffer (1.0322 × 10−2 Pa s); 10% glycerol (1.4056 × 10−2 Pa s); 20% glycerol (2.0052 × 10−2 Pa s); and 2 M sucrose (0.65 × 10° Pa s).
Data were analyzed using Sedfit v. 14.470 (Peter Schuck, National Institute of Biomedical Imaging and Bioengineering, NIH), and SedAnal v. 6.0171 (Peter Sherwood and Walter Stafford, Boston Biomedical Research Institute, Watertown, MA).
High angular velocity data were fitted using a c(s) distribution, using the Sedfit program.71 Data were fitted without radial independent and time independent noise, baseline floated and with confidence levels (F‐ratio) set at 0.95. For samples containing 2 M sucrose, a ls‐g*(s) distribution was used with radial independent and time independent noise and with confidence level set at 0.95. The meniscus position was allowed to vary in the fitting procedure while the bottom positions were fixed.
Multiple‐speed data were fitted using dc/dt (concentration profile time‐derivative analysis),56 with the wide distribution analysis option selected, in SedAnal v. 6.01.45 Instead of plotting all of the fluorescence data directly, we chose to plot for radii every 0.01 cm from a 6.40 to 6.59 cm window, which gave us data from 20 radial positions to average thus providing us with a better signal‐to‐noise ratio. An additional 5% smoothing algorithm was applied to the distribution within this window that contains the data from all speeds.
Supporting information
Supporting Information
Acknowledgments
The authors thank Richard Morimoto for the worm strains harboring the Q0, Q24, Q35, and Q40 fusion constructs and for the plasmid containing the Q82 fusion construct. They thank Jessica Tanis for creating the Q82‐containing worm strain, and Naomi Chaqueco, Tim Chaya, and George Neusch for able technical support. They also thank Aviv Biomedical, Inc. for the use of their facilities to collect data early in the development of this work.
References
- 1. Kingsbury JS, Laue TM (2011) Fluorescence‐detected sedimentation in dilute and highly concentrated solutions. Methods Enzymol 492:283–304. [DOI] [PubMed] [Google Scholar]
- 2. Kroe RR, Laue TM (2009) NUTS and BOLTS: applications of fluorescence‐detected sedimentation. Anal Biochem 390:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gatchel JR, Zoghbi HY (2005) Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet 6:743–755. [DOI] [PubMed] [Google Scholar]
- 4. Mitas M (1997) Trinucleotide repeats associated with human disease. Nucleic Acids Res 25:2245–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arrasate M, Finkbeiner S (2012) Protein aggregates in Huntington's disease. Exp Neurol 238:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kodali R, Wetzel R (2007) Polymorphism in the intermediates and products of amyloid assembly. Curr Opin Struct Biol 17:48–57. [DOI] [PubMed] [Google Scholar]
- 7. Wetzel R (2012) Physical chemistry of polyglutamine: intriguing tales of a monotonous sequence. J Mol Biol 421:466–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perutz MF, Pope BJ, Owen D, Wanker EE, Scherzinger E (2002) Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine‐rich and asparagine‐rich domains of Sup35 and of the amyloid beta‐peptide of amyloid plaques. Proc Natl Acad Sci USA 99:5596–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE (2001) Accumulation of mutant huntingtin fragments in aggresome‐like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell 12:1393–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE (1999) Self‐assembly of polyglutamine‐containing huntingtin fragments into amyloid‐like fibrils: implications for Huntington's disease pathology. Proc Natl Acad Sci USA 96:4604–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE (1997) Huntingtin‐encoded polyglutamine expansions form amyloid‐like protein aggregates in vitro and in vivo. Cell 90:549–558. [DOI] [PubMed] [Google Scholar]
- 12. Bhattacharyya A, Thakur AK, Chellgren VM, Thiagarajan G, Williams AD, Chellgren BW, Creamer TP, Wetzel R (2006) Oligoproline effects on polyglutamine conformation and aggregation. J Mol Biol 355:524–535. [DOI] [PubMed] [Google Scholar]
- 13. Chen S, Berthelier V, Hamilton JB, O'Nuallain B, Wetzel R (2002) Amyloid‐like features of polyglutamine aggregates and their assembly kinetics. Biochemistry 41:7391–7399. [DOI] [PubMed] [Google Scholar]
- 14. Jayaraman M, Kodali R, Sahoo B, Thakur AK, Mayasundari A, Mishra R, Peterson CB, Wetzel R (2011) Slow amyloid nucleation via alpha‐helix‐rich oligomeric intermediates in short polyglutamine‐containing Huntingtin fragments. J Mol Biol 415:881–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kar K, Jayaraman M, Sahoo B, Kodali R, Wetzel R (2011) Critical nucleus size for disease‐related polyglutamine aggregation is repeat‐length dependent. Nat Struct Mol Biol 18:328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mishra R, Jayaraman M, Roland BP, Landrum E, Fullam T, Kodali R, Thakur AK, Arduini I, Wetzel R (2011) Inhibiting the nucleation of amyloid structure in a Huntingtin fragment by targeting alpha‐helix‐rich oligomeric intermediates. J Mol Biol 415:900–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, Byeon IJ, Anjum DH, Kodali R, Creamer TP, Conway JF, Gronenborn AM, Wetzel R (2009) Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol 16:380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kelley NW, Huang X, Tam S, Spiess C, Frydman J, Pande VS (2009) The predicted structure of the headpiece of the Huntingtin protein and its implications on Huntingtin aggregation. J Mol Biol 388:919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vitalis A, Pappu RV (2011) Assessing the contribution of heterogeneous distributions of oligomers to aggregation mechanisms of polyglutamine peptides. Biophys Chem 159:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ossato G, Digman MA, Aiken C, Lukacsovich T, Marsh JL, Gratton E (2010) A two‐step path to inclusion formation of huntingtin peptides revealed by number and brightness analysis. Biophys J 98:3078–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Behrends C, Langer CA, Boteva R, Bottcher UM, Stemp MJ, Schaffar G, Rao BV, Giese A, Kretzschmar H, Siegers K, Hartl FU (2006) Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol Cell 23:887–897. [DOI] [PubMed] [Google Scholar]
- 22. Weiss KR, Kimura Y, Lee WC, Littleton JT (2011) Huntingtin aggregation kinetics and their pathological role in a Drosophila Huntington's disease model. Genetics 190:581–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beam M, Silva MC, Morimoto RI (2012) Dynamic imaging by fluorescence correlation spectroscopy identifies diverse populations of polyglutamine oligomers formed in vivo. J Biol Chem 287:26136–26145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Olshina MA, Angley LM, Ramdzan YM, Tang J, Bailey MF, Hill AF, Hatters DM (2010) Tracking mutant huntingtin aggregation kinetics in cells reveals three major populations that include an invariant oligomer pool. J Biol Chem 285:21807–21816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee CC, Walters RH, Murphy RM (2007) Reconsidering the mechanism of polyglutamine peptide aggregation. Biochemistry 46:12810–12820. [DOI] [PubMed] [Google Scholar]
- 26. Miller J, Arrasate M, Brooks E, Libeu CP, Legleiter J, Hatters D, Curtis J, Cheung K, Krishnan P, Mitra S, Widjaja K, Shaby BA, Lotz GP, Newhouse Y, Mitchell EJ, Osmand A, Gray M, Thulasiramin V, Saudou F, Segal M, Yang XW, Masliah E, Thompson LM, Muchowski PJ, Weisgraber KH, Finkbeiner S (2011) Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol 7:925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mukai H, Isagawa T, Goyama E, Tanaka S, Bence NF, Tamura A, Ono Y, Kopito RR (2005) Formation of morphologically similar globular aggregates from diverse aggregation‐prone proteins in mammalian cells. Proc Natl Acad Sci USA 102:10887–10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Poirier MA, Li H, Macosko J, Cai S, Amzel M, Ross CA (2002) Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J Biol Chem 277:41032–41037. [DOI] [PubMed] [Google Scholar]
- 29. Sathasivam K, Lane A, Legleiter J, Warley A, Woodman B, Finkbeiner S, Paganetti P, Muchowski PJ, Wilson S, Bates GP (2009) Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock‐in mouse models of Huntington's disease. Hum Mol Genet 19:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weiss A, Klein C, Woodman B, Sathasivam K, Bibel M, Regulier E, Bates GP, Paganetti P (2008) Sensitive biochemical aggregate detection reveals aggregation onset before symptom development in cellular and murine models of Huntington's disease. J Neurochem 104:846–858. [DOI] [PubMed] [Google Scholar]
- 31. Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810. [DOI] [PubMed] [Google Scholar]
- 32. Tyedmers J, Mogk A, Bukau B (2010) Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol 11:777–788. [DOI] [PubMed] [Google Scholar]
- 33. van Ham TJ, Holmberg MA, van der Goot AT, Teuling E, Garcia‐Arencibia M, Kim HE, Du D, Thijssen KL, Wiersma M, Burggraaff R, van Bergeijk P, van Rheenen J, van Veluw GJ, Hofstra RMW, Rubinsztein DC, Nollen EAA (2010) Identification of MOAG‐4/SERF as a regulator of age‐related proteotoxicity. Cell 142:601–612. [DOI] [PubMed] [Google Scholar]
- 34. Baglioni S, Casamenti F, Bucciantini M, Luheshi LM, Taddei N, Chiti F, Dobson CM, Stefani M (2006) Prefibrillar amyloid aggregates could be generic toxins in higher organisms. J Neurosci 26:8160–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, Sato K (2003) Spherical aggregates of beta‐amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase‐3beta. Proc Natl Acad Sci USA 100:6370–6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ferreira ST, Vieira MN, De Felice FG (2007) Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases. IUBMB Life 59:332–345. [DOI] [PubMed] [Google Scholar]
- 37. Stefani M, Dobson CM (2003) Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med 81:678–699. [DOI] [PubMed] [Google Scholar]
- 38. Goldsbury C, Frey P, Olivieri V, Aebi U, Muller SA (2005) Multiple assembly pathways underlie amyloid‐beta fibril polymorphisms. J Mol Biol 352:282–298. [DOI] [PubMed] [Google Scholar]
- 39. Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL (2000) Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289:1317–1321. [DOI] [PubMed] [Google Scholar]
- 40. Brignull HR, Moore FE, Tang SJ, Morimoto RI (2006) Polyglutamine proteins at the pathogenic threshold display neuron‐specific aggregation in a pan‐neuronal Caenorhabditis elegans model. J Neurosci 26:7597–7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brignull HR, Morley JF, Morimoto RI (2007) The stress of misfolded proteins: C. elegans models for neurodegenerative disease and aging. Adv Exp Med Biol 594:167–189. [DOI] [PubMed] [Google Scholar]
- 42. Marsh JL, Lukacsovich T, Thompson LM (2009) Animal models of polyglutamine diseases and therapeutic approaches. J Biol Chem 284:7431–7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Teschendorf D, Link CD (2009) What have worm models told us about the mechanisms of neuronal dysfunction in human neurodegenerative diseases? Mol Neurodegener 4:38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Runge MS, Laue TM, Yphantis DA, Lifsics MR, Saito A, Altin M, Reinke K, Williams RCJ (1981) ATP‐induced formation of an associated complex between microtubules and neurofilaments. Proc Natl Acad Sci USA 78:1431–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stafford WF, Braswell EH (2004) Sedimentation velocity, multi‐speed method for analyzing polydisperse solutions. Biophys Chem 108:273–279. [DOI] [PubMed] [Google Scholar]
- 46. MacGregor IK, Anderson AL, Laue TM (2004) Fluorescence detection for the XLI analytical ultracentrifuge. Biophys Chem 108:165–185. [DOI] [PubMed] [Google Scholar]
- 47. Nollen EA, Garcia SM, van Haaften G, Kim S, Chavez A, Morimoto RI, Plasterk RH (2004) Genome‐wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci USA 101:6403–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Battistutta R, Negro A, Zanotti G (2000) Crystal structure and refolding properties of the mutant F99S/M153T/V163A of the green fluorescent protein. Proteins 41:429–437. [DOI] [PubMed] [Google Scholar]
- 49. Crick SL, Jayaraman M, Frieden C, Wetzel R, Pappu RV (2006) Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc Natl Acad Sci USA 103:16764–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Peters‐Libeu C, Miller J, Rutenber E, Newhouse Y, Krishnan P, Cheung K, Hatters D, Brooks E, Widjaja K, Tran T, Mitra S, Arrasate M, Mosquera LA, Taylor D, Weisgraber KH, Finkbeiner S (2012) Disease‐associated polyglutamine stretches in monomeric huntingtin adopt a compact structure. J Mol Biol 421:587–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Morley JF, Brignull HR, Weyers JJ, Morimoto RI (2002) The threshold for polyglutamine‐expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci USA 99:10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Halfmann R, Lindquist S (2008) Screening for amyloid aggregation by semi‐denaturing detergent‐agarose gel electrophoresis. J Vis Exp 17:838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brignull HR, Morley JF, Garcia SM, Morimoto RI (2006) Modeling polyglutamine pathogenesis in C. elegans . Methods Enzymol 412:256–282. [DOI] [PubMed] [Google Scholar]
- 54. Bagriantsev SN, Kushnirov VV, Liebman SW (2006) Analysis of amyloid aggregates using agarose gel electrophoresis. Methods Enzymol 412:33–48. [DOI] [PubMed] [Google Scholar]
- 55. Stafford WF3 (1992) Boundary analysis in sedimentation transport experiments: a procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile. Anal Biochem 203:295–301. [DOI] [PubMed] [Google Scholar]
- 56. Laue T, Demeler B (2011) A postreductionist framework for protein biochemistry. Nat Chem Biol 7:331–334. [DOI] [PubMed] [Google Scholar]
- 57. Lindgren M, Sorgjerd K, Hammarstrom P (2005) Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys J 88:4200–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bertoncini CW, Celej MS (2011) Small molecule fluorescent probes for the detection of amyloid self‐assembly in vitro and in vivo. Curr Protein Pept Sci 12:205–220. [DOI] [PubMed] [Google Scholar]
- 59. Kokona B, Rosenthal ZP, Fairman R (2014) Role of the coiled‐coil structural motif in polyglutamine aggregation. Biochemistry 53:6738–6746. [DOI] [PubMed] [Google Scholar]
- 60. Mok YF, Ryan TM, Yang S, Hatters DM, Howlett GJ, Griffin MD (2011) Sedimentation velocity analysis of amyloid oligomers and fibrils using fluorescence detection. Methods 54:67–75. [DOI] [PubMed] [Google Scholar]
- 61. Link CD, Johnson CJ, Fonte V, Paupard M, Hall DH, Styren S, Mathis CA, Klunk WE (2001) Visualization of fibrillar amyloid deposits in living, transgenic Caenorhabditis elegans animals using the sensitive amyloid dye, X‐34. Neurobiol Aging 22:217–226. [DOI] [PubMed] [Google Scholar]
- 62. Legleiter J, Mitchell E, Lotz GP, Sapp E, Ng C, DiFiglia M, Thompson LM, Muchowski PJ (2010) Mutant huntingtin fragments form oligomers in a polyglutamine length‐dependent manner in vitro and in vivo. J Biol Chem 285:14777–14790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jayaraman M, Mishra R, Kodali R, Thakur AK, Koharudin LM, Gronenborn AM, Wetzel R (2012) Kinetically competing huntingtin aggregation pathways control amyloid polymorphism and properties. Biochemistry 51:2706–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Silva MC, Fox S, Beam M, Thakkar H, Amaral MD, Morimoto RI (2011) A genetic screening strategy identifies novel regulators of the proteostasis network. PLoS Genet 7:e1002438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Satyal SH, Schmidt E, Kitagawa K, Sondheimer N, Lindquist S, Kramer JM, Morimoto RI (2000) Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci USA 97:5750–5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lide DR (1996). CRC handbook of chemistry and physics.Boca Raton, FL: CRC Press, Inc. [Google Scholar]
- 68. Schuck P, Zhao H (2011). Editorial for the special issue of methods “Modern Analytical Ultracentrifugation”. Methods. United States, pp. 1–3. [DOI] [PubMed]
- 69. Mok YF, Ryan TM, Yang S, Hatters DM, Howlett GJ, Griffin MD (2011). Sedimentation velocity analysis of amyloid oligomers and fibrils using fluorescence detection. Methods. United States: Elsevier Inc, pp 67–75. [DOI] [PubMed]
- 70. Dam J, Velikovsky CA, Mariuzza RA, Urbanke C, Schuck P (2005) Sedimentation velocity analysis of heterogeneous protein‐protein interactions: Lamm equation modeling and sedimentation coefficient distributions c(s). Biophys J 89:619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stafford WF, Sherwood PJ (2004) Analysis of heterologous interacting systems by sedimentation velocity: Curve fitting algorithms for estimation of sedimentation coefficients, equilibrium and kinetic constants. Biophys Chem 108:231–243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information