

Abstract
The N‐end rule pathway uses an evolutionarily conserved mechanism in bacteria and eukaryotes that marks proteins for degradation by ATP‐dependent chaperones and proteases such as the Clp chaperones and proteases. Specific N‐terminal amino acids (N‐degrons) are sufficient to target substrates for degradation. In bacteria, the ClpS adaptor binds and delivers N‐end rule substrates for their degradation upon association with the ClpA/P chaperone/protease. Here, we report the first crystal structure, solved at 2.7 Å resolution, of a eukaryotic homolog of bacterial ClpS from the malaria apicomplexan parasite Plasmodium falciparum (Pfal). Despite limited sequence identity, Plasmodium ClpS is very similar to bacterial ClpS. Akin to its bacterial orthologs, plasmodial ClpS harbors a preformed hydrophobic pocket whose geometry and chemical properties are compatible with the binding of N‐degrons. However, while the N‐degron binding pocket in bacterial ClpS structures is open and accessible, the corresponding pocket in Plasmodium ClpS is occluded by a conserved surface loop that acts as a latch. Despite the closed conformation observed in the crystal, we show that, in solution, Pfal‐ClpS binds and discriminates peptides mimicking bona fide N‐end rule substrates. The presence of an apicoplast targeting peptide suggests that Pfal‐ClpS localizes to this plastid‐like organelle characteristic of all Apicomplexa and hosting most of its Clp machinery. By analogy with the related ClpS1 from plant chloroplasts and cyanobacteria, Plasmodium ClpS likely functions in association with ClpC in the apicoplast. Our findings open new venues for the design of novel anti‐malarial drugs aimed at disrupting parasite‐specific protein quality control pathways.
Keywords: ClpS, N‐end rule, N‐degron, crystal structure, malaria, plasmodium, apicoplast, plastid, organelle, phylogeny, Clp chaperone, Clp protease, drug target, carrier‐assisted crystallization
Short abstract
PDB Code(s): 4O2X
Introduction
Intracellular proteolysis plays an essential role in the regulation of various biological processes, including cell cycle progression, gene expression, and protein‐quality control.1, 2 ATP‐dependent proteases, such as the 26S proteasome in eukaryotes and the caseinolytic peptidase in bacteria (Clp proteases), selectively degrade various proteins.3 Clp proteases are highly conserved serine proteases found in most bacteria and in eukaryotic organelles (e.g., mitochondria and chloroplasts), but not in the archaea, mollicutes or some fungi species.4 They are composed of a core tetradecameric proteolytic chamber (ClpP) flanked by one of several possible hexameric ATPase rings (e.g., ClpA and ClpX in E. coli) that belong to the AAA+ (ATPase associated with a variety of cellular activities) protein superfamily.3
The most obvious feature of proteolysis is that it is irreversible. Hence, the recognition of substrates for degradation must be tightly regulated in order to protect cells from uncontrolled proteolysis. In bacteria, substrates are generally recognized via small peptide sequences encoded in the primary sequence of the polypeptide that is targeted for degradation. These short peptide sequences are referred to as “degrons” and are often located either at the N‐terminus (e.g., N‐degrons), C‐terminus (e.g., SsrA tag), or the internal region of a substrate.5, 6 AAA+ proteins can directly recognize these signals and physically prepare the tagged substrates for subsequent translocation into the associated protease for proteolysis. Additionally, adaptor proteins that bind and deliver a specific substrate to its cognate protease for degradation7, 8, 9 can also mediate substrate recognition. Adaptor proteins constitute a diverse group of proteins that regulate protein quality control by enhancing or expanding the substrate recognition abilities of their cognate protease.10 Thus, understanding the mechanism behind adaptor‐assisted substrate delivery, as well as how adaptor proteins have co‐evolved with their partner proteases, is an important aspect of solving the complex puzzle of how proteases recognize their appropriate substrates. ClpS is a bacterial adaptor that regulates protein degradation by recognizing and delivering substrates for degradation by the ClpAP machinery. ClpS and ClpAP are the major components of the N‐end rule degradation pathway.5, 11, 12 The N‐end rule pathway is a highly conserved pathway that determines the stability (or half‐life) of proteins according to the identity of their N‐terminal residues.13, 14 In general, amino acids are categorized as either destabilizing or stabilizing, with the destabilizing residues acting as degradation‐targeting signals. N‐degrons can be classified into type 1, composed of basic residues (arginine, lysine, and histidine), and type 2, composed of bulky hydrophobic residues (phenylalanine, tryptophan, tyrosine, and leucine). In E. coli, the presence of a destabilizing residue (phenylalanine, tyrosine tryptophan, leucine) at the N‐terminus of a polypeptide signals its rapid degradation.15 ClpS functions by binding directly to these so‐called type 2 N‐degrons16 and delivering the associated substrate to the ClpAP protease.5, 17 Previous studies have elucidated the molecular basis for N‐end rule substrate recognition by ClpS11, 12, 18, 19 and the modalities of the specific interaction between ClpS and the N‐terminal domain of the ClpA chaperone.20, 21 This complex then mediates the staged delivery of the substrates from the adaptor ClpS to the ClpAP protease.22
The apicomplexan parasite Plasmodium falciparum is the causative agent of malaria, which accounts for almost 1 million fatalities worldwide every year and about 500 million infections, mostly in Sub‐Saharan Africa, Asia, and South America. The spread of drug resistance constitutes a serious threat to the effective control or eradication of the disease. This eukaryotic pathogen has evolved a complex network of Clp chaperones and proteases including the conserved proteolytic subunit ClpP, the inactive proteolytic subunit ClpR, and four Clp ATPases (ClpB1, ClpB2, ClpC, and ClpM).23, 24 Clp chaperones and proteases have been shown to be important for regulating the pathogenicity and survival of many pathogens. In Plasmodium, all Clp proteins, except ClpB2/HSP101, are localized in the apicoplast, a unique organelle that is evolutionarily related to the chloroplasts of plants, and a defining feature of the Apicomplexa group of parasites. ClpB2 (also named HSP101) is located in the vacuole between the plasma membrane and the parasitophorous vacuole membrane (PVM) that surrounds the parasite within the infected host23, 25 where it functions as core subunit of the Plasmodium translocon of exported proteins, PTEX26 [Fig. 1(A)]. A homolog of bacterial ClpA has not been identified in the genomes of different Plasmodium species sequenced thus far. The apicoplast is essential to parasite survival and hosts several prokaryotic biochemical pathways. By analogy with the roles played by Clp chaperones in animals, plants, fungi and bacteria in supporting cell viability, stress tolerance, or pathogenicity, it has been proposed that the Plasmodium Clp machinery plays an essential role in maintaining a functional apicoplastic proteome27; it is thus a prime drug target.
Figure 1.
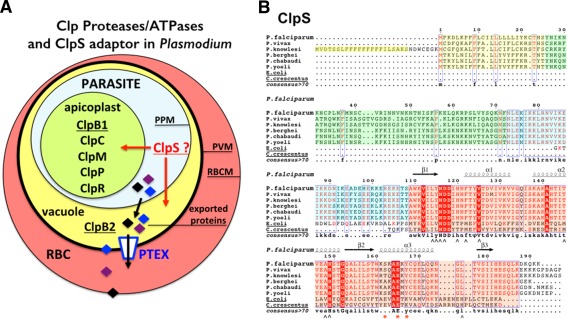
(A) Cellular localization of Clp chaperones and proteases in the parasite Plasmodium falciparum. A red blood cell (RBC) infected by a malaria parasite is shown. Within its host cell, the parasite is self‐contained in the parasitophorous vacuole. PPM, parasitic plasma membrane; PVM, parasitophorous vacuole membrane; RBCM, red blood cell membrane. The apicoplast contains most of the Plasmodium Clp chaperones and proteases (ClpB1, ClpC, ClpM, ClpP, and ClpR) while the ClpB2/Hsp101 protein, a core subunit of the Plasmodium translocon of exported proteins, localizes to the vacuole. No ClpA has been identified in Plasmodium. (B) Sequence alignments of ClpS‐like proteins from different Plasmodium and bacterial ClpS from E. coli and Ccre. In the proteins from the different Plasmodium species, the signal sequences are boxed in yellow. The conserved structural core in all ClpS is boxed in pink. The sequences boxed in green or blue respectively correspond to poorly or highly conserved sequences in Plasmodia; they are part of the predicted apicoplast targeting transit peptide. Secondary structure elements of P. falciparum ClpS are indicated. ∧ symbols indicate residues involved in N‐degron binding in bacterial ClpS structures; red asterisks indicate residues involved in specific binding of ClpS to the N‐terminal domain of ClpA in bacteria.
In this study, we report the crystal structure of a protein annotated as a putative apicoplastic ClpS‐like protein in the available Plasmodium genomes [Fig. 1(B)]. Our crystal structure reveals a protein closely related to the bacterial ClpS adaptors characterized in E. coli and Caulobacter crescentus (Ccre). Binding assays in solution also demonstrate that this plasmodial ClpS‐like protein binds a bona fide N‐degron and suggests the existence on a “N‐end rule pathway” in the parasitic plastid. This first crystal structure of a eukaryotic ClpS‐like adaptor also serves as template to describe the ClpS from other plastids such as plant or algae chloroplasts.
Results
Overall structure of ClpS from Plasmodium falciparum
We used a codon‐optimized synthetic gene encoding the putative ClpS‐like adaptor protein of P. falciparum (Pfal‐ClpS) to generate several expression constructs in order to express, purify and crystallize this protein. Despite all efforts, we were unable to obtain crystals of Pfal‐ClpS by itself. We resolved this by using carrier‐driven and surface entropy reduction (SER) crystallization techniques28 to crystallize a Maltose‐Binding Protein (MBP)‐ClpS fusion protein (Material and Methods). The construct crystallized encodes full‐length MBP and a fragment (residues N73‐K192) of ClpS that is conserved among all ClpS‐like proteins annotated in the six available Plasmodium genomes [Figs. 1(B) and 2(A)]. This fragment encompasses most of the predicted transit peptide (or apicoplast targeting signal) followed by the “core” ClpS domain that is present in bacteria and other eukaryotes. The Plasmodium ClpS‐like proteins share only 12% sequence identity, and 17–30% sequence similarity with bacterial ClpS.
Figure 2.
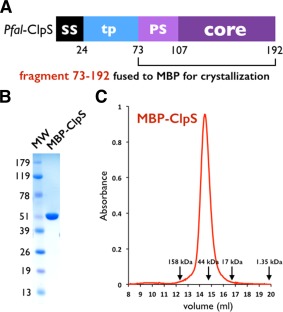
Domain organization of ClpS in Plasmodia. (A) The Plasmodium falciparum ClpS gene contains one signal sequence (SS) followed by the putative transit peptide (tp) for apicoplast targeting. A ∼30 residue long Plasmodium‐specific sequence (PS) is followed by the ∼70 residue long ClpS core [Fig. 1(B)] conserved between Plasmodium and bacteria. (B) SDS PAGE analysis of purified MBP‐ClpS used for structure determination. C: MBP‐ClpS is a monomer in solution. Size exclusion profile of MBP‐ClpS purified on a Superdex S200 HR 10/30 column. The elution volumes of molecular mass standards are indicated with arrows.
MBP‐ClpS was expressed in E. coli and purified as a monomer [Fig. 2(B,C)]. The 2.7 Å resolution crystal structure was solved using a combination of molecular replacement (MR) and single anomalous dispersion (SAD) as phasing methods using the structures of ClpS from E. coli 20, 21 and C. crescentus 11, 19 to guide model building (Table 1 and Material and Methods). The initial MR‐phased electron density maps at 2.7 Å resolution were of rather good quality, however, there were deviations from the MR search model. To resolve this, we used the unbiased phasing information obtained from a 3.1 Å resolution SAD dataset collected on a single crystal of selenium‐labeled protein. Electron density maps obtained using the anomalous phases enabled us to build 73 (T107‐S179) out of 120 residues of ClpS cloned in the construct successfully crystallized.
Table 1.
X‐Ray Data Collection and Structure Refinement Statistics
| Data set | ALS 092813 8.3.1 | APS 080913 24‐ID‐C |
|---|---|---|
| Protein | Pfal ClpS | Pfal ClpS |
| Data collection statistics | Se‐SAD phasing | PDB ID 4O2X |
| Wavelength | 0.97965 Å | 0.9793 Å |
| Resolution (last shell) | 84.6–3.1 Å (3.27–3.10 Å) | 84.9–2.70 Å (2.77–2.70 Å) |
| Unique reflections | 29,226 (4,280) | 43,011 (3,128) |
| Completeness | 100% (100%) | 97.3% (96.4%) |
| I/σ (I) | 12.5 (2.4) | 15.8 (1.7) |
| Redundancy | 4.4 (4.5) | 2.7 (2.7) |
| Rsym | 6.8% (61.8%) | 4.0% (66.9%) |
| Rmeas | 5.9% (53.1%) | 4.9% (83.9%) |
| CC(1/2) | 99.8% (72.0%) | 99.9% (55.5%) |
| Space group | P61 | P61 |
| AU content | a = 97.7 Å, c = 298.8 Å | a = 97.9 Å, c = 298.2 Å |
| Solvent content | 2 molecules | 2 molecules |
| 62% | 62% | |
| Refinement statistics | ||
| Resolution | – | 84.9–2.70 Å |
| Reflections | – | 41,875 |
| Work set/test set | – | 39,780/2,095 |
| R free /R cryst | – | 23.9%/18.9% |
| B wilson | 91 Å2 | 78 Å2 |
| Protein atoms, ADP | – | 6,833, 89 Å2 |
| Solvent atoms, ADP | – | None |
| Others atoms, ADP | – | – |
| rmsd bonds | – | 0.009 Å |
| rmsd angles | – | 1.232° |
| Ramachandran analysis | ||
| Allowed regions | – | 93.8% |
| Generously allowed | – | 5.6% |
| Outliers | – | 0.6% |
R sym = ∑hkl∑i |I hkl,i‐<I hkl,i>|/∑hkl∑i |I hkl,i| where <I hkl,i> is the average intensity of the multiple hkl, i observations for symmetry‐related reflections.
R meas is the redundancy independent R‐factor.45
CC(1/2) percentage of correlation between intensities from random half‐datasets.46
R cryst = ∑|F obs‐F calc|/∑|F obs|. F obs and F calc are observed and calculated structure factors, R free is calculated from a set of randomly chosen reflections, and R cryst is calculated over the remaining reflections.
ADP is the atomic displacement parameter.
rmsd is the root‐mean square deviation from ideal geometry.
No refinement statistics are reported for the SAD data as it was only used to guide model building.
The asymmetric unit contains 2 molecules of MBP‐ClpS related by a noncrystallographic 2‐fold axis of symmetry [Fig. 3(A)]. The N‐terminus (N73‐V83) of Pfal‐ClpS adopts a rigid helical conformation (labeled αp on [Fig. 3(A)] that extends from the triple‐alanine linker at the C‐terminus of MBP (a fixed‐arm linker designed to promote rigidity and compaction of MBP‐fusion proteins for crystallization) and packs against its MBP fusion partner.28 These residues were reliably traced in the electron density maps, and likely correspond to a fragment of the transit peptide that might localize Pfal‐ClpS in the apicoplast. A disordered region (residues I84‐T107) had no electron density and is thus missing from the final model. This loop connects the N‐terminal αp to the compact, well‐folded, and rigid core of Pfal‐ClpS (residues S108‐E183). Amino acids encoding the complete MBP carrier protein and the core of ClpS were fully accounted for in the electron density map. The final structure was refined to final R cryst and R free factors of 18.9 and 23.9%, respectively, with good stereochemistry and electron density maps of excellent quality (Supporting Information Fig. S1).
Figure 3.
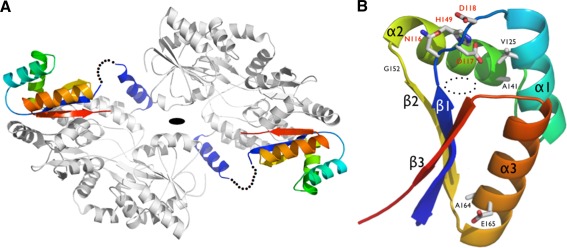
(A) Structure of the MBP‐ClpS fusion. The MBP carrier protein is colored in gray, while a rainbow pattern is used to color the ClpS from P. falciparum. The disordered N‐terminal region of ClpS is indicated by a dotted line. (B) General architecture of Pfal‐ClpS. Conserved residues are labeled. The conserved N116‐D117‐D118 and H149 residues that form the lid of the N‐end residue binding pocket observed in bacterial ClpS are labeled in red. Helices and strands are labeled accordingly to [Fig. 1(B)]. The dotted circles mark the position of the hydrophobic binding pocket.
Overall, Pfal‐ClpS adopts a globular and rigid fold comprising three α‐helices (α1–α3) connected to three antiparallel β‐strands (β1–β3) with a βααβαβ topology [Fig. 3(B)]. The β‐sheet packs against a curved layer made of the three helices, creating a pocket nested between strand β1 and helices α1 and α2. Each of the two ClpS copies present in the asymmetric unit tightly packs against one MBP molecule through the flat outer face of the β‐sheet [Fig. 3(A)]; the essentially hydrophobic interface buried at each ClpS/MBP carrier interface spans 1,500 Å2, representing a substantial amount (∼35%) of the total solvent accessible surface of the ClpS domain (4,360 Å2).
Structural comparison between plasmodial ClpS and its bacterial orthologs
The Plasmodium ClpS structure is essentially superimposable with the two apo bacterial ClpS structures from the bacteria C. crescentus (PDB 3GQ0) or E. coli (PDB 1LZW) with overall r.m.s.d values of 1.4 and 1.5 Å over the 72 residues aligned, respectively. Our comparison also reveals noticeable structural differences that are specific to the ClpS from plastid‐containing organisms. For example, the C‐terminus of helix α3 is longer in Ccre‐ClpS compared to Pfal‐ClpS, and the loop between strand β1 and helix α1 is shorter in bacterial ClpS compared to Plasmodium ClpS (Supporting Information Fig. S2A). With an r.m.s.d of 1.8 Å over the 65 residues aligned, the ClpS adaptor is also structurally related to the C‐terminal domain of ribosomal L7/L12 (PDB 1CTF)29 (Supporting Information Fig. S2B). In mammals, E3 ubiquitin ligases named N‐recognins, are capable of recognizing type 1 and type 2 N‐degrons,30 through different domains; one protein domain of these N‐recognins harbors ClpS‐like sequences and is responsible for the binding of type 2 substrates of the N‐end rule pathway.31
The N‐degron binding pocket of Plasmodium ClpS is not accessible
Akin to its bacterial homologs, the rigid scaffold of plasmodial ClpS harbors a preformed cavity for binding N‐end rule substrates. This pocket is mostly delineated with hydrophobic residues L114 (I45 in Ccre‐ClpS), Y115 (L46), I119 (T51), F122 (M53), V125 (V56), T145 (M75), A148 (V78), and L176 (L112) but also a tetrad of highly conserved polar residues: N116 (N47), D117 (D48), D118 (D49), and H149 (H79) (Fig. 4 and Supporting Information Fig. S1). However, this binding pocket is inaccessible compared with other bacterial ClpS structures where the pocket is open even in absence of substrate. The “closed” conformation is due to the rearrangement of the highly conserved N116‐D117‐D118 motif (“NDD motif”) between strand β1 and helix α1 [Fig. 4(A)] and the strictly conserved H149 on helix α2. The NDD loop seems to act as a latch, at the entrance of the putative N‐degron binding pocket.
Figure 4.
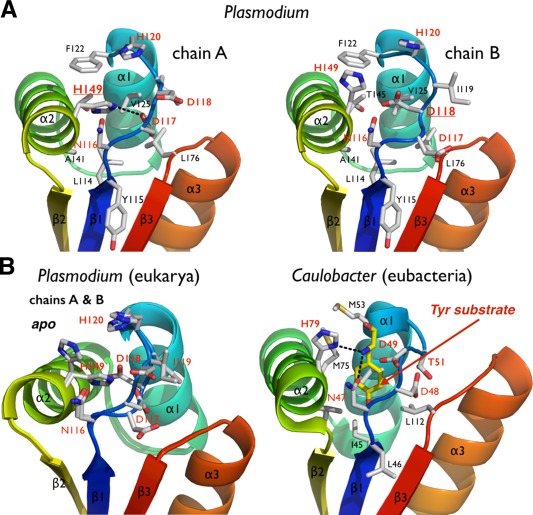
The putative N‐end residue binding site of Plasmodium ClpS. (A) The two copies of ClpS in the asymmetric unit display slightly different NDD loop conformations. (B) Comparison of the Plasmodium and bacterial N‐end degron binding pockets. Equivalent residues are labeled. The bacterial structure (Caulobacter) is shown bound to a tyrosine N‐end rule degron (PDB 3DNJ).11 For Plasmodium, the two chains observed in the asymmetric unit are superposed to emphasize the rigidity of the protein scaffold and the flexibility of the NDD loops.
The NDD loop is also flexible as it samples subtly different conformations in the two copies present in the asymmetric unit. Whereas H149 rotates inwards forming a hydrogen bond with D117 in chain A, these same residues (H149 and D117) project outward from the pocket in chain B. Additionally, D118 of chain A rotates outwards from the pocket, while in chain B it occupies the pocket [Fig. 4(A,B)]. Despite these differences between the two chains, the ClpS binding pocket is occluded. The equivalent histidine and aspartate residues in bacterial ClpS structures protrude outward from the pocket and are solvent‐exposed [Fig. 4(B)]; as a result, the opening is not occluded and the N‐degron binding pocket is accessible. Although Pfal‐ClpS appears to be in a closed state, the crystal structure still provides us with a detailed view of the protein surface and potential N‐degron binding site [Fig. 4(B,C)]. Removal of the side chains of H149 and H120 from chain A, or D118 and H120 from chain B, exposes a cavity with a volume large enough to accommodate an N‐end rule substrate (Fig. 5). The side chains of the largest bona fide N‐end residues, phenylalanine, tyrosine, leucine, and in principle tryptophan, can occupy the binding pocket of Pfal‐ClpS (Fig. 5).
Figure 5.
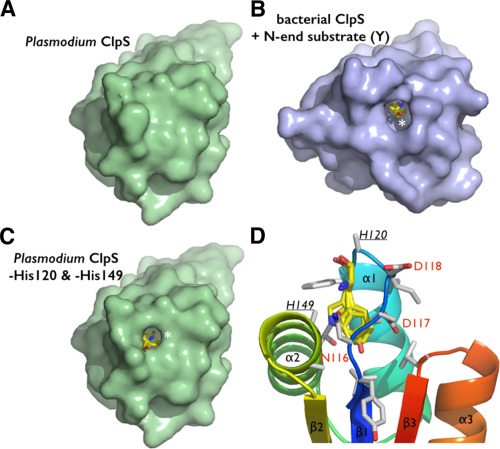
Steric adequacy and adaptability of the N‐degron specific pocket of Plasmodium ClpS. (A) Solvent accessible surface representation of the Pfal‐ClpS showing the absence of accessible binding cavity in contrast with the bacterial ClpS. (B) shown bound to a tyrosine N‐end degron (yellow sticks and white asterisk). (C) Side chains of residues H120 and H149 conceal a N‐degron binding pocket in Pfal‐ClpS. Solvent accessible surface representation of Pfal‐ClpS where side chains of residues H120 and H149 (underlined) have been omitted (all side chain atoms except the Cα where removed) is shown and reveals a pocket large enough to accommodate N‐end rule residues such as tyrosine (yellow sticks and white asterisk). (D) The Pfal‐ClpS binding pocket is shown with 4 distinct side chains (Y, F, W, and L) bound as observed in bacterial ClpS/peptide complexes (PDBs 3DNJ, 3G19, 3GQ1, and 3GW1).11, 12 Labeled residues delineate the hydrophobic pocket shown in the same orientation as in [Fig. 3(B)].
While residue H149 (in Plasmodium) is strictly conserved in all ClpS proteins (i.e. bacteria and plastids from plants, algae or Plasmodium) but is replaced by D or E in N‐recognins, H120 (in Plasmodium but also N‐recognins) is replaced by either T or N in most bacteria, non‐charged residues with polar groups both suited for hydrogen‐bonding.31, 32 Thus, this Plasmodium putative ClpS shares characteristics of bacterial ClpS adaptors and eukaryotic N‐recognins.
Examination of the crystal packing reveals that the entrance of the putative binding site is solvent‐exposed (facing solvent‐filled channels in the crystalline lattice). Although the NDD loop in each of the two chains sits against its MBP carrier protein, residues D117, D118, and, H149 are not engaged in interactions with the MBP chain. Thus each loop remains unconstrained and is free to adopt the two distinct conformation described in [Fig. 4(A,B)]. Besides these contacts between MBP and ClpS, there are no other direct molecular interactions between the NDD loop of ClpS and the MBP carrier protein (or another ClpS) from symmetry‐related neighboring molecule. In our opinion, this rules out the possibility that this “closed” conformation is induced by the presence of the MBP fusion partner. We would rather favor an indirect effect of crystal packing forces or more likely the crystallization conditions characterized by a high ionic strength and a low pH (pH = 5.0–5.5). For the bacterial ClpS, the binding pocket for the N‐end residue is preformed in the apo state, and minimal rearrangements can (are sufficient) to accommodate N‐end rule residues of various shapes and sizes. This conformational switching of the highly conserved “NDD‐H” tetrad observed in our Plasmodium ClpS structure could regulate N‐degron binding.
Plasmodium ClpS binds and discriminates N‐end rule substrates
By homology with the holo Ccre‐ClpS structure (PDB 3DNJ),11 the N116‐D117‐D118 and His149 specificity determinants necessary to bind the α amino group of the N‐terminal aminoacid are present in Pfal‐ClpS (Supporting Information Fig. S4); in particular, residue N116 is in position to establish the hydrogen bond with the α amino group of the N‐degron. To test whether Pfal‐ClpS could bind and discriminate N‐degrons, we synthesized several fluorescently labeled peptides harboring different N‐terminal residues (Phenyalanine, Tryptophan, Isoleucine, and Alanine) and measured their binding to Pfal‐ClpS using fluorescence polarization18 (Fig. 6 and Material and Methods). For this purpose, we purified the same ClpS fragment used for structure determination expressed as a cleavable MBP fusion protein [Fig. 6(A)]. The purified protein is monomeric in solution [Fig. 6(B)]. As shown in [Fig. 6(C)], we measured quantifiable binding for the Phe substrate, a bona fide type 2 N‐degron, and no binding for the Ala and Ile substrates (which are not type 2 N‐degrons). The K d value of 635 ± 85 nM determined for Pfal‐ClpS binding to the Phe substrate [Fig. 6(D)] is in the range of values reported for E. coli‐ClpS (∼250 nM–1.5 μM)5 and Ccre‐ClpS (∼150 nM–500 nM).11 Based on these results, Pfal‐ClpS is capable of binding and discriminating type 2 N‐degrons. Interestingly, we did not detect measurable binding for the Trp peptide, described as a type 2 N‐degron in bacterial systems.
Figure 6.
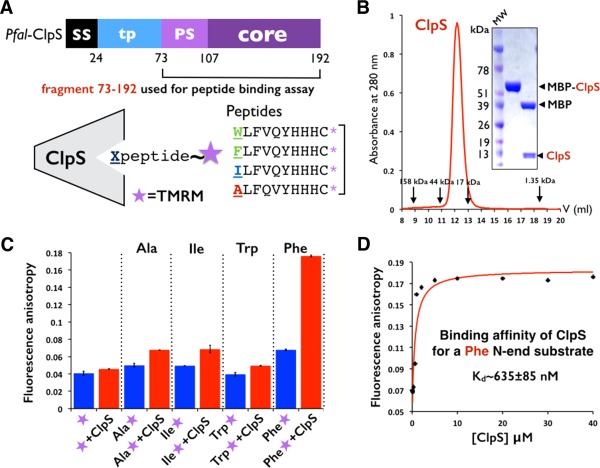
Plasmodium ClpS binds and discriminates N‐end rule substrates. (A) The N73‐K192 fragment of Pfal ClpS was cleaved from MBP and used to measure its affinity towards synthetic peptides labeled with the fluorescent probe TMRM. (B) ClpS is a monomer in solution. Size exclusion profile of MBP‐ClpS purified on a Superdex S75 HR 10/30 column. The elution volumes of molecular mass standards are indicated. SDS PAGE analysis of purified ClpS used to measure peptide binding. (C) Fluorescence anisotropy changes with labeled peptides shows that Plasmodium ClpS distinguishes the N‐end residue Phenylalanine from other residues such as Alanine, Isoleucine, and even Tryptophan. Error bars represent the standard deviation over three independent trials. (D) Plasmodium ClpS binds the N‐end rule peptide beginning with Phenylalanine with submicromolar affinity. Titration curve quantifying the binding of ClpS to the Phenylalanine peptide as assayed by change in fluorescence anisotropy. The Kd value is an average of three independent experiments.
When compared with the bacterial ClpS, the binding pocket of the adaptor from Plasmodium has dimensions, chemical properties and a shape compatible with N‐degron binding; more explicitly the Plasmodium and the two bacterial ClpS whose structures are available are absolutely identical when it comes to the presence of the so‐called “NDD and H” tetrad. Previous studies have shown that mutation of the conserved asparagine11 or of the two aspartate33 residues in E. coli resulted in loss of substrate interaction. However, two residues are noticeably different: F122 (corresponding to M53/M40 in Ccre/Ecoli) and T145 (M75/M62) (Supporting Information Fig. S4). These two methionines are highly conserved in most bacterial ClpS while ClpS from other lineages (i.e., plastid from plants and algae, cyanobacteria, and green sulfur bacteria) diverge at these positions (especially for M53 on helix α1).31, 32 In E. coli this flexible methionine side chain serves as specificity gatekeeper, excluding β‐branched aminoacids such as Isoleucine but not its isostere, leucine; modest changes in the N‐degron binding pocket could easily account for differences in recognition of bulky hydrophobic N‐end rule residues between prokaryotes and eukaryotes.11, 34 It is thus possible that the differences observed in the case of Pfal‐ClpS reflect subtle variations in the N‐end rule specific to this organism. It is worth mentioning that minor flipping of a few sidechains (i.e., D117 and D118, H120, and H149) are sufficient to expose the entry of the putative N‐degron binding site [Figs. 4(A,B) and 5]. However since crystallographic structures usually represent snapshots of the most stable conformations that proteins can adopt, it is currently difficult to assess if the “latch” we describe herein does play a role in regulating peptide binding.
The apicoplastic Plasmodium ClpS as a template to model other eukaryotic ClpS‐like adaptor proteins
Analysis of plasmodial ClpS protein sequences with PATS 35 and PlasmoDP 36 reveals the presence of an apicoplast transit peptide, suggesting that ClpS localizes to this organelle where most of the Clp machinery functions. In plants, ClpS1 is a conserved substrate selector for the chloroplastic Clp machinery,32 where it functions in association with the ClpC/P chaperone/protease complex to control protein degradation37 in association with another recently characterized factor, ClpF.38The two plastids, apicoplast and chloroplast, share a common “plastid” ancestor; thus our plasmodial ClpS structure can serve as a template to model the ClpS1 from plastid containing organisms such as plants and algae but also the phylogenetically related ClpS1/S2 from Cyanobacteria.32
Despite the arsenal of Clp chaperones and proteases present in Plasmodium, there is no ClpA, the cognate effector chaperone of ClpS in bacteria. Two Clp AAA+ chaperones, ClpB1 and ClpC, are also targeted to the apicoplast. In plants, ClpS1 functions in association with the ClpC/PR chaperone/protease complex to control protein degradation.39 Consistently, previous studies have shown that ClpS1 and S2 interact with the ClpC chaperone in the cyanobacteria Synechococcus.40 The most noticeable feature of the bacterial ClpA/ClpS complex structure is the presence of the highly conserved ExAExK ClpA‐binding motif in ClpS20, 21; this corresponds to residues S162 through Y167 in Pfal‐ClpS, with the alanine‐glutamate motif A164‐E165 (corresponding to A63‐E64 in Ecoli‐ClpS) being the only conserved residues between bacteria, Plasmodium, chloroplastic and cyanobacterial ClpS (Supporting Information Fig. S5). In the bacteria B. subtilis, ClpC has also been shown to interact with the adaptor MecA41; although MecA is structurally unrelated to ClpS, it also binds to the same “hot spot” adaptor‐binding surface on the N‐domain of ClpC using a conserved glutamate residue.41 Analysis of the surface of Plasmodium ClpS reveals similar shape and electrostatics features (Supporting Information Fig. S5). It is thus likely that in the apicoplast of the malaria parasite, ClpS regulates protein turnover in association with the ClpC/PR chaperone/protease complex23 although the apicoplastic ClpR component has been shown to be inactive.24
Conclusion
The 2.7 Å resolution crystal structure of a Plasmodium ClpS represents the first crystal structure of a eukaryotic ClpS adaptor protein. Our biochemical data indicate that the N‐degron binding properties (affinity and specificity) of plasmodial ClpS are overall similar to those of bacterial ClpS, thus supporting its involvement in the regulation of protein turnover in the parasite. This plasmodial ClpS structure thus represents a template to describe plastid‐derived ClpS1 adaptors from the chloroplast of photosynthetic plants and algae and the evolutionarily related ClpS1/S2 from cyanobacteria. Based on the presence of an apicoplast targeting sequence, it is likely that Pfal‐ClpS localizes to this organelle and functions in association with the ClpC chaperone.
Based mostly on computational analysis, over 500 Plasmodium proteins (from a total of about 1,300–1,500 proteins) are predicted to localize to this essential plastid. Chemical rescue experiments have shown that isoprenoid biosynthesis is an essential function of this organelle.42, 43 Protein maturation and proteolysis in plant plastids have been thoroughly studied using proteomic and genetic methods44 revealing the complexities of the “N‐terminone” in the stroma of plant chloroplasts where the possibility for a chloroplastic N‐end rule has recently been discussed.45 However, although the apicoplast from Plasmodium is a clearly identified target for the development of novel parasite‐specific drugs, its proteome (and consequently “N‐terminome”) remains largely uncharacterized, let alone the rules governing its proteostasis (i.e., lifetime of its proteins). This is largely due to the difficulties associated with the growth and genetic manipulation of Plasmodium as an obligate intracellular blood parasite. Our structure and preliminary peptide‐binding analysis suggests that the machinery required for a “N‐end rule”‐like proteostatic pathway is present in the plastid of the parasite. Establishing the idiosyncrasies of a possible N‐end rule in the apicoplast of Plasmodium will necessitate more systematic measurements. Our structure of ClpS from Plasmodium falciparum, the etiologic agent of malaria, open new venues for the design of novel antimalarial drugs aimed at disrupting parasite‐specific, protein quality control pathways in an organelle unique to this class of pathogenic eukaryotes.
Materials and Methods
Protein expression and purification
A synthetic gene (DNA2.0 Inc.) encoding residues S25‐K192 of Plasmodium falciparum ClpS‐like adaptor protein (Pfal‐ClpS: PF3D7_1320100) was designed after codon optimization for its expression in E. coli as a C‐terminal octa‐histidine fusion protein (residues M1 through H24 correspond to the signal sequence). Initial expression and purification trials yielded an unstable protein prone to aggregation. We further engineered our expression constructs by removing residues S25‐K70 corresponding to most of the putative transit peptide that might target this protein to the apicoplast. Thus, a shorter construct corresponding to the conserved core (residues N73‐K192) of all ClpS‐like proteins annotated in the different Plasmodium genomes, was cloned into a pRSF vector (Novagen) to be expressed as an N‐terminal fusion with Maltose Binding Protein (MBP) and harboring cleavable histidine tags at both ends. A third construct lacking residues S25‐T107 corresponding to a strict “bacterial‐like” ClpS core (residues S108‐K192) was also generated guided by sequence alignments with bacterial ClpSs. This first generation of vectors enabled removal of the MBP fusion partner and the histidine tags with thrombin.
The core (residues N73‐K192) of Pfal‐ClpS fused to MBP was expressed in C43(DE3) E. coli cells grown in 2× LB media at 37°C until cultures reached OD600 = 0.6, protein expression was then induced with 0.8 mM IPTG and cells allowed to grow for 5 more hours. Cells were harvested, washed in 150 mM KCl and 20 mM Tris‐HCl pH = 7.8, and stored at −80°C until processing. Thawed cell pellets were resuspended in lysis buffer (500 mM NaCl, 20 mM Tris‐HCl pH = 7.8, 15% glycerol) supplemented with 2.8 mM β‐mercaptoethanol (β‐ME), 0.2 mM of phenylmethylsulfonyl fluoride (PMSF), and 1 tablet of EDTA‐free protease inhibitor cocktail (Roche). All subsequent steps were carried out at 4°C or on ice. Cells were disrupted by three passes through a C‐3 Emulsiflex (Avestin) pressurized at 15.000 psi. The lysate was clarified by centrifugation at 25.000g for 1 h, after which the resulting supernatant corresponding to the total soluble extract was applied onto a gravity‐flow column (BioRad) packed with 5–10 mL of Cobalt‐NTA IMAC resin (Qiagen). Non‐specifically bound bacterial proteins were subsequently washed from the column using Cobalt‐A wash buffer (12.5 mM imidazole, 500 mM NaCl, 20 mM Tris‐HCl pH = 7.8, 10% glycerol, 2.8 mM β‐ME, and 0.2 mM PMSF). The protein was eluted from the column with Cobalt‐B buffer (125 mM imidazole, 500 mM NaCl, 20 mM Tris‐HCl pH = 7.8, 10% glycerol, 2.8 mM β‐ME, and 0.2 mM PMSF). The IMAC eluate was concentrated to 0.5–1 mL using a 50 kDa cutoff centricon, and desalted using a PD‐10 desalting column (BioRad) equilibrated in 300 mM NaCl, 20 mM Tris‐HCl pH = 7.8, 5% glycerol, 2.8 mM β‐ME, and 0.2 mM PMSF, to remove imidazole. Following desalting, the protein was treated with thrombin for 24 h at 4°C (0.25 units enzyme/mg of protein) to remove the histidine purification tag and the MBP fusion partner. Following thrombin treatment, ClpS was purified by cation exchange chromatography (cIEX) on a CaptoS HiTrap column (GE Healthcare); this step quantitatively removed MBP. The final purification step consisted in a size exclusion chromatography (SEC) on a Superdex 75 HR10/30 (GE Healthcare) equilibrated in 150 mM NaCl, 20 mM Tris‐HCl pH = 8.0, 10% glycerol and 14 mM β‐ME. Full‐length ClpS protein was not stable upon cleavage from its fused MBP expression partner; although the core (N73‐K192) and the minimalistic (S108‐K192) constructs were somehow stable once released from the MBP fusion expression partner, they did not yield crystals.
We thus recloned all corresponding constructs (full‐length, core and minimalistic plasmodial ClpS) into a modified MBP construct (pMal‐E vector)28 where the MBP protein carries five SER point mutations (A83D, A84K, A1733, A174N, A240K, A360E, A363K, and A364D) and a rigid AAA linker that connects to the protein of interest (ClpS). We attempted crystallization of the entire MBP‐ClpS fusion protein after cleavage of the C‐terminal octahistidine purification tag with thrombin. The purification process was identical. The final protein MBP‐ClpS was purified by SEC on a Superdex 200 HR10/30 (GE Healthcare) equilibrated in 100 mM KCl, 10% glycerol, 20 mM Tris‐HCl pH = 8.0 and 14 mM β‐mercaptoethanol and used in crystallization trials. This strategy proved successful and yielded diffracting crystals in the case of the MBP‐ClpS core construct (residues N73‐K192).
Crystallization
High‐throughput crystallization trials were performed with a robotic nanoliter Mosquito workstation in hanging‐drop setups using the vapor diffusion technique at 4°C with protein concentrated at 25 mg/mL. Six commercial screens from Qiagen (AmSO4, PEGs, ComPAS, Classics, JCSG, and PACT crystallization suites) were screend to identify initial crystallization conditions. Reservoir volumes were 100 μL and three hanging drops (drop volume of 150 nL), corresponding to three different protein‐to‐well solution ratios (2:1, 1:1, and 1:2), were setup for each condition screened. Suitable crystals grew in a reservoir containing 1.8–2M ammonium sulfate (pH = 5–5.5) and 1.6–2.1M sodium chloride. The final optimized crystals were obtained in large hanging drops (well volume of 1 mL and drop volumes ranging from 1 to 5 μL). They belong to hexagonal space group P61 (a = 97.9 Å and c = 298.2 Å) with 2 molecules related by a noncrystallographic 2‐fold axis of symmetry in the asymmetric unit and a solvent content of 63%.
Structure determination and refinement
An initial 2.7 Å resolution native data set was collected on beamline 24‐ID‐C at the Advanced Photon Source at the Argonne National Laboratory and 8.3.1 at the Advanced Light Source at the Lawrence Berkeley National Laboratory. Crystals were cryoprotected in mother liquor supplemented with 20–25% ethylene glycol or glycerol. Data were indexed, scaled, and reduced using XDS.46 The structure could be solved by MR in Phaser 47 using the structures of MBP (PDB 3WAI) and bacterial ClpS (PDBs 1MG9, 1MBX, and 3DNJ) as search probes and templates for structure‐guided sequence alignments. Despite the overall good quality of the initial maps phased by MR, we also obtained experimental phases using SAD of selenium data collected on another crystal diffracting at 3.1 Å; each MBP‐ClpS fusion protein contains 6 methionines (not including methionine 1) and all of them are contained in the MBP carrier protein and could be located. SAD phases were obtained using ShelX in CCP4 followed by density modification in DM. The quality of the SAD phases determined at 3.1 Å resolution was such that automatic rebuilding in BUCCANEER yielded an entire backbone trace for ClpS together with a fully rebuilt MBP carrier protein. Refinement was carried out against the native data set in Phenix 48 and model building performed using COOT.49 Rigid body refinement combined with simulated annealing torsion and TLS refinement yielded the final model.
Peptide binding assay and polarization anisotropy measurements
Four decapeptides peptides (A LFVQYHHHC, I LFVQYHHHC, F LFVQYHHHC, and W LFVQYHHHC) were synthesized and purified by HPLC to ≥ 98.0% purity by Genscript; the choice of the sequence x‐LFVQY‐z was based on the previous work published by Wang et al.18 Each peptide harbors a C‐terminal cysteine suited for its conjugation to the thiol‐reactive fluorescent probe, tetramethylrhodamine‐5‐maleimide (TMRM) purchased from Life Technologies. In a 150 μL, labeling reaction, TMRM (3 mM) and either one of the four peptides (3 mM) were incubated in the dark at room temperature for 24 h. The reaction was quenched with the addition of TCEP at a final concentration of 1 mM, 25% acetonitrile and 100 mM Hepes‐NaOH at pH = 7.0. After incubation, the TMRM‐labeled peptide was purified by reverse phase chromatography HPLC in buffers A (0.1% TFA) and B (100% acetonitrile and 0.1% TFA) using a steady gradient (20–60%) of buffer B at a flow rate of 10 mL/min. The eluted fractions were analyzed by MALDI‐TOF to determine the extent of labeling. Fractions containing the TMRM‐labeled peptide were pooled and lyophilized. The lyophilized product was resuspended in 100 μL of 100% DMSO and quantified by spectrophotometry. Labeled peptides were repurified by HPLC on a C18 column to eliminate excess unreacted dye. After lyophilization, labeled peptides were analyzed by MALDI‐TOF mass spectrometry and dissolved in DMSO at final concentrations ranging from 0.5 to 1 mM for storage.
For anisotropy fluorescence measurements, we expressed the cleavable version of the MBP‐ClpS construct used for crystallization (fragment N73‐K192). Protein was purified as described by cobalt IMAC followed by SEC and cation exchange chromatography. The ClpS fragment was then severed from its MBP carrier using thrombin. The proteolytic digest was then repurified by cation exchange to separate MBP from the ClpS fragment. The protein was prepared in a buffer containing 400 mM KCl, 5% glycerol, 20 mM Hepes‐NaOH pH = 7.5 and 0.1 mM TCEP. Labeled‐peptide and protein concentrations were determined by UV absorption spectroscopy using extinction coefficients values calculated from their sequence (ε peptides = 1490 M−1 cm−1 and ε protein = 15470 M−1 cm−1). All fluorescence measurements were carried out at 25°C in 400 mM KCl, 5% glycerol, 20 mM Hepes‐NaOH pH = 7.5 and 0.1 mM TCEP. For bulk measurements, TMRM‐labeled peptide (at 40 nM final concentration) was incubated in presence of excess purified unlabeled ClpS (at 40 μM final concentration).
For titration, the Phenylalanine TMRM‐labeled peptide was incubated at a final fixed concentration of 40 nM with purified unlabeled ClpS present at final concentrations ranging from 12.5 nM to 40 μM. Polarization anisotropy was measured at 25°C using a multifrequency fluorimeter (Photon Technology International) and analyzed using KaleidaGraph (Synergy Software). Under the conditions used in our study, anisotropy is a measure of the rotational mobility of the protein bound to the fluorescent peptide. We excited the fluorophore with plane‐polarized light at 543 nm and measured emission at 575 nm at polarizations both parallel (I‖) and perpendicular (I⊥) to the excitation light. We simultaneously monitored total intensity to ensure that the quantum efficiency of the fluorophore was independent of the protein‐peptide complex. Equilibrium dissociation constants (K d) were determined using a quadratic binding model of 1:1 binding, as previously described.50 Curve fitting with this model makes no assumptions of the relative concentrations of labeled peptide and ClpS.
Accession Numbers
The atomic coordinates and structure factors corresponding to the structure determined in this study were deposited on 2013‐12‐17 and released on 2014‐12‐24 and are accessible at the Protein Data Bank (http://www.rcsb.org/pdb http://www.rcsb.org/pdb/explore.do?structureId=4o2x) under the accession code 4O2X. Electron density was validated by the PDB and the Electron Density Server of the Uppsala University.
Supporting information
Supporting Information
Acknowledgments
The authors are very grateful to Dr George Meigs and James Holton for support at beamline 8.3.1 at the Advanced Light Source at the Lawrence Berkeley National Laboratory. They thank M. Capel, K. Rajashankar, N. Sukumar, J. Schuermann, I. Kourinov, and F. Murphy at NECAT beamlines 24‐ID at the Advanced Photon Source at the Argonne National Laboratory. They wish to thank Lisa Johnson in Dr David Eisenberg's laboratory for her expertise and help with peptide labeling and purification. They thank Dr Margot Quinlan for the use of her fluorimeter. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare no conflict of interest.
Competing Interests: The Authors have declared that no competing interests exist.
Author contributions: Performed the experiments: APAY AK CLV DC PFE. Designed the experiments: APAY AK CLV DC PFE. Contributed reagents/materials/analysis: APAY AK CLV DC PFE. Analyzed the data: APAY CLV DC PFE. Wrote the article: APAY AK PFE.
References
- 1. Mogk A, Schmidt R, Bukau B (2007) The N‐end rule pathway for regulated proteolysis: prokaryotic and eukaryotic strategies. Trends Cell Biol 17:165–172. [DOI] [PubMed] [Google Scholar]
- 2. Dougan DA, Truscott KN, Zeth K (2010) The bacterial N‐end rule pathway: expect the unexpected. Mol Microbiol 76:545–558. [DOI] [PubMed] [Google Scholar]
- 3. Striebel F, Kress W, Weber‐Ban E (2009) Controlled destruction: AAA+ ATPases in protein degradation from bacteria to eukaryotes. Curr Opin Struct Biol 19:209–217. [DOI] [PubMed] [Google Scholar]
- 4. Yu AY, Houry WA (2007) ClpP: a distinctive family of cylindrical energy‐dependent serine proteases. FEBS Lett 581:3749–3757. [DOI] [PubMed] [Google Scholar]
- 5. Erbse A, Schmidt R, Bornemann T, Schneider‐Mergener J, Mogk A, Zahn R, Dougan DA, Bukau B (2006) ClpS is an essential component of the N‐end rule pathway in Escherichia coli . Nature 439:753–756. [DOI] [PubMed] [Google Scholar]
- 6. Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA (2003) Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX‐recognition signals. Mol Cell 11:671–683. [DOI] [PubMed] [Google Scholar]
- 7. Dougan DA, Mogk A, Zeth K, Turgay K, Bukau B (2002) AAA+ proteins and substrate recognition, it all depends on their partner in crime. FEBS Lett 529:6–10. [DOI] [PubMed] [Google Scholar]
- 8. Dougan DA, Reid BG, Horwich AL, Bukau B (2002) ClpS, a substrate modulator of the ClpAP machine. Mol Cell 9:673–683. [DOI] [PubMed] [Google Scholar]
- 9. Baker TA, Sauer RT (2006) ATP‐dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem Sci 31:647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kirstein J, Moliere N, Dougan DA, Turgay K (2009) Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat Rev Microbiol 7:589–599. [DOI] [PubMed] [Google Scholar]
- 11. Wang KH, Roman‐Hernandez G, Grant RA, Sauer RT, Baker TA (2008) The molecular basis of N‐end rule recognition. Mol Cell 32:406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roman‐Hernandez G, Grant RA, Sauer RT, Baker TA (2009) Molecular basis of substrate selection by the N‐end rule adaptor protein ClpS. Proc Natl Acad Sci USA 106:8888–8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Varshavsky A (1996) The N‐end rule: functions, mysteries, uses. Proc Natl Acad Sci USA 93:12142–12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Varshavsky A (2011) The N‐end rule pathway and regulation by proteolysis. Protein Sci 20:1298–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tobias JW, Shrader TE, Rocap G, Varshavsky A (1991) The N‐end rule in bacteria. Science 254:1374–1377. [DOI] [PubMed] [Google Scholar]
- 16. Dougan DA, Micevski D, Truscott KN (2012) The N‐end rule pathway: from recognition by N‐recognins, to destruction by AAA+proteases. Biochim Biophys Acta 1823:83–91. [DOI] [PubMed] [Google Scholar]
- 17. Hou JY, Sauer RT, Baker TA (2008) Distinct structural elements of the adaptor ClpS are required for regulating degradation by ClpAP. Nature Struct Mol Biol 15:288–294. [DOI] [PubMed] [Google Scholar]
- 18. Wang KH, Oakes ES, Sauer RT, Baker TA (2008) Tuning the strength of a bacterial N‐end rule degradation signal. J Biol Chem 283:24600–24607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schuenemann VJ, Kralik SM, Albrecht R, Spall SK, Truscott KN, Dougan DA, Zeth K (2009) Structural basis of N‐end rule substrate recognition in Escherichia coli by the ClpAP adaptor protein ClpS. EMBO Rep 10:508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zeth K, Ravelli RB, Paal K, Cusack S, Bukau B, Dougan DA (2002) Structural analysis of the adaptor protein ClpS in complex with the N‐terminal domain of ClpA. Nat Struct Biol 9:906–911. [DOI] [PubMed] [Google Scholar]
- 21. Guo F, Esser L, Singh SK, Maurizi MR, Xia D (2002) Crystal structure of the heterodimeric complex of the adaptor, ClpS, with the N‐domain of the AAA+ chaperone, ClpA. J Biol Chem 277:46753–46762. [DOI] [PubMed] [Google Scholar]
- 22. Roman‐Hernandez G, Hou JY, Grant RA, Sauer RT, Baker TA (2011) The ClpS adaptor mediates staged delivery of N‐end rule substrates to the AAA+ ClpAP protease. Mol Cell 43:217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. El Bakkouri M, Pow A, Mulichak A, Cheung KL, Artz JD, Amani M, Fell S, de Koning‐Ward TF, Goodman CD, McFadden GI, Ortega J, Hui R, Houry WA (2010) The Clp chaperones and proteases of the human malaria parasite Plasmodium falciparum . J Mol Biol 404:456–477. [DOI] [PubMed] [Google Scholar]
- 24. El Bakkouri M, Rathore S, Calmettes C, Wernimont AK, Liu K, Sinha D, Asad M, Jung P, Hui R, Mohmmed A, Houry WA (2013) Structural insights into the inactive subunit of the apicoplast‐localized caseinolytic protease complex of Plasmodium falciparum . J Biol Chem 288:1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. AhYoung AP, Koehl A, Cascio D, Egea PF (2015) Structural mapping of the ClpB ATPases of Plasmodium falciparum: targeting protein folding and secretion for antimalarial drug design. Protein Sci 24:1508–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Koning‐Ward TF, Gilson PR, Boddey JA, Rug M, Smith BJ, Papenfuss AT, Sanders PR, Lundie RJ, Maier AG, Cowman AF, Crabb BS (2009) A newly discovered protein export machine in malaria parasites. Nature 459:945–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ngansop F, Li H, Zolkiewska A, Zolkiewski M (2013) Biochemical characterization of the apicoplast‐targeted AAA+ ATPase ClpB from Plasmodium falciparum . Biochem Biophys Res Commun 439:191–195. [DOI] [PubMed] [Google Scholar]
- 28. Moon AF, Mueller GA, Zhong X, Pedersen LC (2010) A synergistic approach to protein crystallization: combination of a fixed‐arm carrier with surface entropy reduction. Protein Sci 19:901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leijonmarck M, Liljas A (1987) Structure of the C‐terminal domain of the ribosomal protein L7/L12 from Escherichia coli at 1.7 A. J Mol Biol 195:555–579. [DOI] [PubMed] [Google Scholar]
- 30. Matta‐Camacho E, Kozlov G, Li FF, Gehring K (2010) Structural basis of substrate recognition and specificity in the N‐end rule pathway. Nature Struct Mol Biol 17:1182–1187. [DOI] [PubMed] [Google Scholar]
- 31. Lupas AN, Koretke KK (2003) Bioinformatic analysis of ClpS, a protein module involved in prokaryotic and eukaryotic protein degradation. J Struct Biol 141:77–83. [DOI] [PubMed] [Google Scholar]
- 32. Nishimura K, Asakura Y, Friso G, Kim J, Oh SH, Rutschow H, Ponnala L, van Wijk KJ (2013) ClpS1 is a conserved substrate selector for the chloroplast Clp protease system in Arabidopsis. Plant Cell 25:2276–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ninnis RL, Spall SK, Talbo GH, Truscott KN, Dougan DA (2009) Modification of PATase by L/F‐transferase generates a ClpS‐dependent N‐end rule substrate in Escherichia coli . EMBO J 28:1732–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xia Z, Webster A, Du F, Piatkov K, Ghislain M, Varshavsky A (2008) Substrate‐binding sites of UBR1, the ubiquitin ligase of the N‐end rule pathway. J Biol Chem 283:24011–24028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zuegge J, Ralph S, Schmuker M, McFadden GI, Schneider G (2001) Deciphering apicoplast targeting signals–feature extraction from nuclear‐encoded precursors of Plasmodium falciparum apicoplast proteins. Gene 280:19–26. [DOI] [PubMed] [Google Scholar]
- 36. Foth BJ, Ralph SA, Tonkin CJ, Struck NS, Fraunholz M, Roos DS, Cowman AF, McFadden GI (2003) Dissecting apicoplast targeting in the malaria parasite Plasmodium falciparum. Science 299:705–708. [DOI] [PubMed] [Google Scholar]
- 37. Nishimura K, van Wijk KJ (2015) Organization, function and substrates of the essential Clp protease system in plastids. Biochim Biophiys Acta 1847:915–930. [DOI] [PubMed]
- 38. Nishimura K, Apitz J, Friso G, Kim J, Ponnala L, Grimm B, van Wijk KJ (2015) Discovery of a unique Clp component, ClpF, in chloroplasts: a proposed binary ClpF‐ClpS1 adaptor complex functions in substrate recognition and delivery. Plant Cell 27:2677–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nishimura K, van Wijk KJ (2015) Organization, function and substrates of the essential Clp protease system in plastids. Biochim Biophys Acta 1847:915–930. [DOI] [PubMed] [Google Scholar]
- 40. Stanne TM, Pojidaeva E, Andersson FI, Clarke AK (2007) Distinctive types of ATP‐dependent Clp proteases in cyanobacteria. J Biol Chem 282:14394–14402. [DOI] [PubMed] [Google Scholar]
- 41. Wang F, Mei Z, Qi Y, Yan C, Hu Q, Wang J, Shi Y (2011) Structure and mechanism of the hexameric MecA‐ClpC molecular machine. Nature 471:331–335. [DOI] [PubMed] [Google Scholar]
- 42. Yeh E, DeRisi JL (2011) Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood‐stage Plasmodium falciparum . PLoS Biol 9:e1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu W, Herrera Z, Ebert D, Baska K, Cho SH, DeRisi JL, Yeh E (2015) A chemical rescue screen identifies a Plasmodium falciparum apicoplast inhibitor targeting MEP isoprenoid precursor biosynthesis. Antimicrob Agents Chemother 59:356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van Wijk KJ (2015) Protein maturation and proteolysis in plant plastids, mitochondria, and peroxisomes. Ann Rev Plant Biol 66:75–111. [DOI] [PubMed] [Google Scholar]
- 45. Rowland E, Kim J, Bhuiyan NH, van Wijk KJ (2015) The Arabidopsis chloroplast stromal N‐terminome: complexities of amino‐terminal protein maturation and stability. Plant Physiol 169:1881–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kabsch W (2010) Xds. Acta Cryst D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Cryst 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Echols N, Headd JJ, Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner RD, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2011) The Phenix software for automated determination of macromolecular structures. Methods 55:94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 50. Zalevsky J, Grigorova I, Mullins RD (2001) Activation of the Arp2/3 complex by the Listeria acta protein. Acta binds two actin monomers and three subunits of the Arp2/3 complex. J Biol Chem 276:3468–3475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information