

Abstract
The misidentification of a protein sample, or contamination of a sample with the wrong protein, may be a potential reason for the non‐reproducibility of experiments. This problem may occur in the process of heterologous overexpression and purification of recombinant proteins, as well as purification of proteins from natural sources. If the contaminated or misidentified sample is used for crystallization, in many cases the problem may not be detected until structures are determined. In the case of functional studies, the problem may not be detected for years. Here several procedures that can be successfully used for the identification of crystallized protein contaminants, including: (i) a lattice parameter search against known structures, (ii) sequence or fold identification from partially built models, and (iii) molecular replacement with common contaminants as search templates have been presented. A list of common contaminant structures to be used as alternative search models was provided. These methods were used to identify four cases of purification and crystallization artifacts. This report provides troubleshooting pointers for researchers facing difficulties in phasing or model building.
Keywords: protein purification artifacts, crystallization artifacts, YodA (metal‐binding lipocalin), YadF (carbonic anhydrase), reproducibility
Short abstract
Abbreviations
- CSGID
Center for Structural Genomics of Infectious Diseases
- GNAT
Gcn5‐related N‐acetyltransferase
- IMAC
immobilized metal affinity chromatography
- MAD
multi‐wavelength anomalous diffraction
- MR
molecular replacement
- MCSG
Midwest Center for Structural Genomics
- NYSGRC
New York Structural Genomics Research Consortium
- PDB
Protein Data Bank
- RMSD
root mean square deviation
- SAD
single‐wavelength anomalous dispersion
- SEC
size exclusion chromatography
- TEV
tobacco etch virus.
Introduction
Determination of the three‐dimensional structure of a protein by X‐ray crystallography is a laborious, multistep process. It is easy to imagine the disappointment of a researcher when this process is unsuccessful, even when “good” diffraction data is obtained. There are many reasons why this may happen, for example, when the collected diffraction data is produced from a crystal of a different protein than anticipated. In such situations, it is usually quite difficult to solve the structure. High resolution anomalous diffraction phases might allow for sequence determination from electron density maps, but in the case of molecular replacement (MR), it is essentially impossible to select an adequate model for phase determination, except through a brute force approach, that is, screening using an entire non‐redundant subset of the PDB.1
Based on our own experience and data in the literature, there are four main reasons why a protein other than the one anticipated (henceforth called an artifact) may be found in a crystal structure. First, the wrong protein is purified instead of the protein of interest (or is co‐purified with it). This may be a protein native to the expression host2, 3 (when recombinant expression systems such as Escherichia coli are used), or the wrong protein is purified from a natural source. Second, a protein preparation may be contaminated with an exogenous protein added during the production process, such as lysozyme or DNAse added during cell lysis, proteases used for tag cleavage, or proteases added for in situ proteolysis.3, 4 Third, if a recombinant protein is expressed as a fusion protein, only the fusion partner may crystallize.4 Fourth, human errors such as mislabeling of samples (unfortunately not an uncommon event in high‐throughput environments) may produce crystals of the wrong protein.
Contamination of a target protein purified from expression host or natural source
Strains of E. coli are the most common hosts for heterologous protein expression, and contamination of purified protein samples with an endogenous protein from these bacteria has been reported many times.2, 3 Most native bacterial contaminants are proteins that directly bind either to chromatography resins or to the recombinant protein of interest itself. Because some of the common native E. coli contaminant proteins show high affinity for divalent metal ions such as Zn2+, Cu2+, Ni2+, and Co2+, it is difficult to separate the target protein without at least trace amounts of the contaminant using immobilized metal affinity chromatography (IMAC) alone.2 In addition to the endogenous proteins and target protein, some accessory proteins are specifically expressed by some expression plasmids to facilitate recombinant expression. These include antibiotic resistance proteins, chaperones, repressors/activators, RNA polymerases, and proteases,3 and may become contaminants as well.
Purification of a protein from a natural source is also prone to purification artifacts: the wrong protein may be co‐purified with (or contaminate) a protein of interest, because purification may be based on subtle differences between the target protein and the remaining proteins present in the cell lysate.5 Moreover, the wrong protein may be co‐purified from a natural source when the target protein has a native binding partner, or when chaperones or chaperone‐like proteins nonspecifically interact with the protein of interest. In many cases, complete separation of such complexes is difficult or impossible.6
Exogenous proteins and fusion tags as crystallization artifacts
Exogenous proteins introduced during purification and crystallization may also lead to crystallization artifacts. For example, lysozymes and/or deoxyribonucleases (DNases) are commonly added to lysis buffers to improve protein extraction from cells. Both of these proteins, due to nonspecific interactions with either the protein of interest or a chaperone, may be present in purified fractions. Although crystallization of exogenous proteins instead of target proteins is quite rare, there are published examples. An exogenous lysozyme was reported to co‐crystallize in a 1:1:1 heterotrimeric complex with target proteins.4
Proteases such as thrombin, elastase, enterokinase, factor Xa, or TEV protease are added to remove fusion tags from the target protein and may be another source of exogenous protein. Proteases such as chymotrypsin or trypsin are also used for in situ proteolysis to trim the target proteins, or to generate separate domains to improve crystallization and/or diffraction.7 Both types of proteases are typically added in very small amounts, minimizing the risk of protease crystallization. However, this is still possible, for example, due to accidental addition of high amounts of protease or high crystallization propensity of the added protease.
Large fusion tags such as glutathione S‐transferase (GST), maltose binding protein (MBP), or N‐utilizing substance A (NusA) may also crystallize instead of the target protein when they are not removed prior to crystallization. Fusion tags may be kept intentionally because they may be crucial for protein solubility and stability, or to facilitate the crystallization of target proteins.8 Such tags may form the bulk of the ordered crystal lattice, if the target protein is degraded or is too disordered to form a well‐structured part of the lattice.9 If the tag is intentionally cleaved in situ or accidentally cleaved by a trace amount of contaminating protease, this may potentially increase the chance of its accidental crystallization instead of the target protein.7
Here, we present several crystallization artifacts that we have encountered in practice during the past several years. In our opinion, purification and crystallization artifacts are inevitable from time to time, even when best practices are followed. Therefore, straightforward and simple methods are required to discover artifacts as soon as possible before expensive resources have been invested and more serious troubleshooting is pursued. Using practical examples as illustration, we suggest and discuss various approaches for discovering crystallization artifacts at early stages of data collection and structure solution, and demonstrate that these methods are easy and effective. The presented approaches may save a lot of effort that would be otherwise spent on unsuccessful attempts to solve the structure of proteins different than the ones expected.
Results
Purification and crystallization of E. coli YodA instead of recombinant Sigma70 factor
E. coli stress response protein YodA was crystallized during attempts to determine the structure of Sigma70 factor from Planctomyces limnophilus DSM 3776 (MCSG target APC100648). SDS‐PAGE analysis of the protein obtained during purification of the selenomethionine derivative of Sigma70 factor showed bands corresponding to 23 kDa [Fig. 1(A)]. Since Sigma70 factor has a very similar molecular weight (20 kDa), it was assumed that the correct protein was purified. The protein was successfully crystallized and diffraction data were collected to 1.95 Å at the selenium absorption peak wavelength. Despite a very weak anomalous signal at this wavelength, a reasonable electron density map was obtained by SAD and initial model building was partly successful—the protein was built as a polyalanine model with only approximately 10% of side chains assigned. Because of the weakness of the SAD signal, we also attempted MR, but numerous attempts using the available PDB models (Sigma70 factor ortholog: 4CXF, fragments of catalase‐3 with sequence similar to Sigma70 factor cut according to BLAST comparison search: 4BIM, 4AJ9) all failed. After all these computational experiments, it became apparent that the electron density did not correspond to the sequence of the Sigma70 factor protein.
Figure 1.
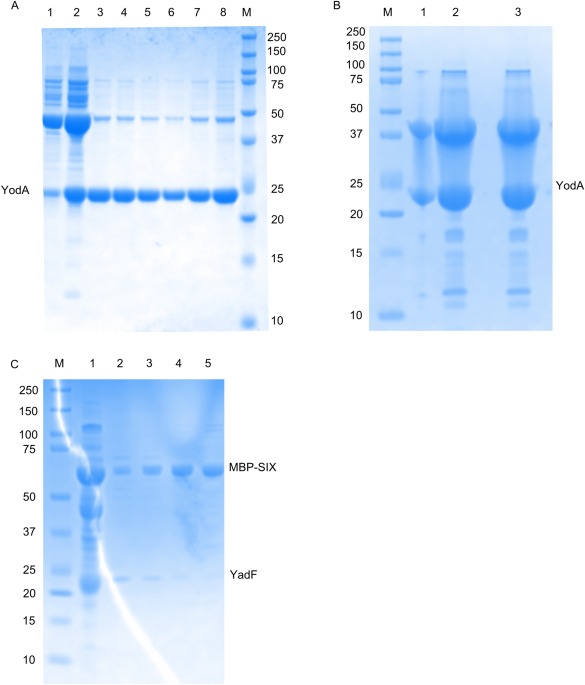
Purity of protein samples used for crystallization estimated by SDS‐PAGE. Protein samples after Superdex200 size‐exclusion chromatography (SEC) were separated by polyacrylamide gel electrophoresis on a 12% SDS‐PAGE gel and Coomassie‐stained. As a reference, for A (line 1,2) and C (line 1) samples of the protein preparation prior to SEC are also shown. The numbers on the sides of each gel indicate the molecular weight (MW) of the markers (M) in kDa; the numbers above gels are fraction numbers from SEC. (A) The final samples obtained after the presumed purification of Sigma70 (20 kDa). All fractions were pooled together, concentrated, and used for crystallization as described in the text. The band between 20 and 25 kDA was eventually identified as YodA. (B) The final samples obtained after the purification of presumed GNAT (21 kDa—MW including the N‐terminal His‐tag).The band between 20 and 25 kDa was eventually identified as YodA (23 kDa). (C) The final samples obtained after the purification of MBP‐survivin SIX (MBP‐SIX) fusion protein (60 kDa). Fractions 2–5 were pooled together, concentrated, and used for crystallization. The amount of contamination in the 23 kDa band (eventually identified as YadF [25 kDa]) was incorrectly assumed to be too small to seriously affect the suitability of the sample for crystallization.
A computational tool for recognizing sequence from electron density Fitmunk10 (available at http://kniahini.med.virginia.edu/fitmunk/server/and http://fitmunk.bitbucket.org/) was used, which assigned probable residue identities to some sidechains in the partially built initial structure. The partially assigned sequence was then used to perform a BLAST search against the NCBI NR database, which was sufficient to identify the protein as E. coli YodA. The query sequence obtained from Fitmunk was similar to the YodA sequence at 52% of positions. This result was independently confirmed using the three‐dimensional (3D) protein comparison PBDeFOLD service, which matched the initial model built using SAD electron density maps with the YodA structure 1OEE, with an RMSD of 0.92 Å for 185 out of 193 Cα atoms in chain A of the identified structure. After rebuilding the model with the corrected sequence, the structure was successfully refined further against 1.95 Å data and deposited to the PDB (4TNN). Data processing and refinement statistics are presented in Table S1, Supporting Information.
Purification and crystallization of E. coli YodA instead of recombinant Gcn5‐related N‐acetyltransferase
Similar to the previous case, the YodA protein was crystallized instead of Gcn5‐related N‐acetyltransferase (GNAT) from Staphylococcus aureus subsp. aureus COL (CSGID target IDP00844). SDS‐PAGE analysis of GNAT purification [Fig. 1(B)] showed two bands with around a 1:1 ratio of intensity. The additional band (at 37 kDa) was interpreted as a possible GNAT interaction partner, which hopefully could be identified in the crystallization experiment. The crystallization trials were set bearing in mind that the sample was heterogeneous and the contaminant protein might crystallize instead of or together with GNAT. The purified protein was crystallized and a dataset was collected at 1.5 Å. The diffraction pattern was successfully indexed but the anomalous signal from selenomethionine residues was very weak and the structure could not be solved using SAD. MR was not an option as there was no close homolog of GNAT in the PDB. Since we were aware that a different protein may have crystallized, the determined space group and unit cell parameters (P21, a = 40.05 Å, b = 65.09 Å, c = 41.46 Å, β = 117.8°) were used to search against the entire PDB for similar (within ±1%) unit cell dimensions, using a tool in HKL‐3000 for that purpose. This approach was successful, albeit simplistic due to the reasons described in the discussion below, and the 1OEJ deposit of YodA was found to have the best match of unit cell parameters (P21, a = 40.35 Å, b = 65.56 Å, c = 41.50 Å, β = 117.8°) to the crystallized protein. The structure was successfully refined using 1OEJ as a starting model, confirming the presence of a crystallization artifact. The presence of YodA in the sample used for crystallization was additionally confirmed by MALDI‐TOF mass‐spectrometry analysis. We assumed that lower band (around 23 kDa) is the GNAT protein, therefore only upper band (above 37 kDa) was sent for mass spectrometry analysis. The possible explanation for the presence of YodA protein in the upper band is the formation of intermolecular disulfide bridge between two YodA monomers that due to large amount of protein were not removed by standard amount of reducing agents during SDS‐PAGE electrophoresis. As described in discussion, we ultimately concluded that both bands observed on the SDS‐PAGE represented YodA protein.
Purification and crystallization of E. coli YadF instead of recombinant survivin SIX
E. coli stress response protein YadF was accidentally crystallized during attempts to determine the structure of survivin SIX protein from Xenopus laevis. The expression level of survivin SIX in a fusion construct with MBP was sufficient for purification, and SDS‐PAGE analysis of the final fraction obtained from size exclusion chromatography (SEC) showed that the purified sample contained trace amounts of a contaminating protein with a molecular weight between 20 and 25 kDa [Fig. 1(C)]. It was presumed that such a small amount of contaminant would not affect survivin crystallization, and the sample was used for crystallization trials. The presence of zinc ions in the crystals (expected because survivin SIX contains a zinc finger motif) was confirmed by X‐ray fluorescence emission spectroscopy, lending further support to the correct identity of the crystallized protein.
A diffraction data set was collected on the presumed survivin protein crystal to 2.7 Å at the zinc absorption peak wavelength and initial phases were obtained by SAD. Similar to the case with the presumed Sigma70 factor, an initial model was built, but very little of the survivin side chain sequence could be docked to the polyalanine chain to match the electron density. The sequence recognition mode of Fitmunk was used to identify sequence fragments in the electron density maps but no significant hits were identified in a BLAST search against the NCBI NR database when default parameters were used. However, a search against the smaller PDB database found a significant hit for YadF (because the PDB database is smaller, a hit may be significant even if it was insignificant when a larger database is used). In addition, a search of the NCBI CDD11 database identified the carbonic anhydrase superfamily motif in the density‐assigned sequence, which is characteristic of YadF. Because the initial model was fragmented, the protein could not be identified by structural alignment using any of the tested 3D protein comparison services (PDBeFOLD,12 DALI,13 and FatCat14).
The structure was successfully rebuilt using the sequence of YadF, and completed using PDB deposit 1I6P of YadF as a guide. The structure was refined and deposited to the PDB (4ZNZ, Table S1, Supporting Information). The difficulty in identifying the sequence by electron density was most likely due to the relatively low resolution compared with the Sigma70 case; when aligned, the sequence assigned from electron density map had similar residues at only 32% of positions. YadF is known to utilize zinc as a cofactor, and a zinc ion was identified in the YadF active site due to a peak in the anomalous difference map and a plausible binding environment. This explains the detection of zinc in the crystal by X‐ray fluorescence.
Sample mislabeling
During our work with hundreds of New York Structural Genomics Research Consortium (NYSGRC), Midwest Center for Structural Genomics (MCSG), and Center for Structural Genomics of Infectious Diseases (CSGID) targets, a few cases of mislabeled samples were encountered. For example, the protein labeled as NYSGRC target 021790 (hypothetical protein SMc00576 from Sinorhizobium meliloti) was successfully crystallized and diffraction data were collected to 1.8 Å at the selenium absorption peak wavelength. Initial phases were obtained by SAD using the strong anomalous signal from the incorporated selenomethionine. However, numerous attempts to build a model with the expected sequence were all unsuccessful. The PBDeFOLD service was run with the initial model but did not produce any significant hits, probably because the model was split in seven disconnected fragments. Manual assignment of residue types with distinct, identifiable electron density was attempted, and two targets on the list of current lab projects were found to have some sequence similarity to those deduced from the maps. In parallel, Fitmunk in sequence recognition mode (with subsequent refinement with REFMAC) was run twice to assign the amino acid sequence by density recognition. A BLAST search with the density‐assigned sequence unequivocally matched the protein NYSGRC‐022189 (trimethylamine methyltransferase from Sinorhizobium meliloti). The corrected sequence was successfully used for structure refinement. Since a higher resolution dataset (1.56 Å) was subsequently obtained, it was used for further structure refinement and deposited to the PDB (4YYC, Table S1, Supporting Information). As a final validation of the artifact, MALDI‐TOF mass spectrometry analysis was performed to confirm the identification of the protein in the sample used for crystallization.
Structures of common contaminants suitable for MR
The methods for contaminant identification described in the case studies above are suitable when either experimental phasing is possible, or there is a structure in the PDB of the protein crystallized with very similar unit cell parameters. To extend the available troubleshooting tools to MR cases, we have compiled a list of structures of the most common purification and crystallization artifacts, based on the literature2, 3, 15, 16 and our own experience. The list includes proteins native to E. coli that were previously reported to be common contaminants during IMAC purification, common expression tags, proteases typically used for in situ proteolysis, and exogenous proteins known to be used in protein production.3, 15 All of these targets (or highly similar homologs) are of known structure. The structures which represent a distinct conformational states, suitable for MR are provided in Table 1.
Table 1.
Known Structures of Proteins that Have Been Identified as Common Purification and Crystallization Artifacts
| Name of the protein | Molecular weight (kDa) | PDB ID | |
|---|---|---|---|
| Affinity, solubility, anti‐aggregation tags | Maltose‐binding protein (MBP) | 43 | 1LLS, 1MPB, 3PUW, 3SEU, 4KYC |
| Glutathione‐S‐transferase (GST) | 24 | 4ECB | |
| Thioredoxin (Trx) | 11 | 1F6M, 2AJQ, 2H73, 4HU9, 4X43 | |
| N‐Utilization substance (NusA) | 55 | 1U9L,a 1WCN,b 2KWP,c 4MTNd | |
| Small ubiquitin related modifier 1 (SUMO1) | 12 | 2UYZ, 1Z5S, 4WJQ, 2IO2 | |
| Haloalkane dehalogenase | 33 | 4E46 | |
| E. coli native proteins | Metal‐binding lipocalin (YodA) | 25 | 1OEJ, 4TNN |
| Carbonic anhydrase (YadF) | 25 | 2ESF | |
| Ferric uptake regulator (Fur) | 16 | 2FU4 | |
| cAMP‐regulatory protein (CRP) | 24 | 1CGP, 2CGP, 2GZW, 3FWE, 3HIF, 3N4M, 3QOP, 4FT8, 4HZF, 4I0A, 4I0B, 4N9H, 4N9I | |
| Glucosamine‐6‐phosphate synthase (GlmS) | 67 | 4AMV, 1JXA, 3OOJ, 2J6H | |
| Glycogen synthase (GlgA) | 53 | 2QZS | |
| Component 1 of the 2‐oxoglutarate dehydrogenase complex (ODO1) | 105 | 2JGD | |
| Component E2 of dihydrolipoamide succinyltransferase (ODO2) | 44 | 1C4T | |
| Formyl transferase (YfbG, ArnA) | 46 | 1U9J, 1YRW, 1Z7E, 2BLN, 4WKG | |
| Cu/Zn‐superoxide dismutase (Cu/Zn‐SODM) | 16 | 1ESO | |
| Chloramphenicol‐O‐acetyl transferase (CAT) | 26 | 1Q23 | |
| Host factor‐I protein (Hfq) | 11 | 3VU3 | |
| Proteases | Tobacco etch virus (TEV) | 28 | 1LVM |
| Rhinovirus 3C protease | 48 | 1CQQ | |
| SUMO protease C‐terminal domain | 26 | 2HL9 | |
| Enterokinase | 26 | 1EKB | |
| Trypsin | 26 | 3UY9 | |
| Chymotrypsin | 26 | 1GGD | |
| Thrombin (active form) | 36 | 3SQE, 1MH0, 4H6T | |
| Thermolysin | 60 | 4D9W | |
| Proteinase K | 40 | 3DVS | |
| Pepsin | 41 | 5PEP | |
| Neutrophil elastase | 29 | 5ABW | |
| LysN Peptidyl‐Lys metalloendopeptidase | 44 | 1GE7 | |
| Lysyl endopeptidase | 28 | 4NSY | |
| Factor Xa | 55 | 1KIG | |
| Exogenous proteins | Lysozyme | 16 | 4TWS, 4PRQ, 1AKI |
| DNase protein | 31 | 2A40 |
Listed deposits were selected from crystal structures in PDB to represent distinct conformational states of the given protein.
Crystal structure of the C‐terminal fragment of NusA from E. coli.
NMR structure of the C‐terminal fragment of NusA from E. coli.
NMR structure of the N‐terminal fragment of NusA from E. coli.
Homologous structure from P. limnophilus.
The possibility of identifying crystallization artifacts by MR, which is carried out using predefined set of structures of possible artifacts as templates, was tested using subset of the deposits representing the proteins described above. The structures of native E. coli proteins and two selected solubilization tags (MBP and NusA) were used to run MR for the diffraction datasets described in the cases above: Sigma70 (YodA), GNAT (YodA), survivin SIX (YadF), and NYSGRC‐021790 (NYSGRC‐022189). As the dataset for the NYSGRC protein is known not to yield the structure of any of the proteins listed in Table 1, this dataset served as a negative control. The best solution for each template structure vs. each dataset was subjected to 5 cycles of rigid body and 15 cycles of restrained REFMAC refinement, and the average of overall R‐factors from the final refinement from all templates of a given protein are shown in Figure 2. The exact R‐factor values for each template that was used are listed on http://www.bioreproducibility.org/pages/protein_purification_artifacts/mr_results webpage. The only combinations of template and dataset with acceptable R‐factor values (i.e., R‐factor <0.3) were YodA for the Sigma70 and GNAT datasets, and YadF for the survivin dataset. In other words, the structures were only determined in cases where the correct protein model was used as a template, and there were no other false positives.
Figure 2.
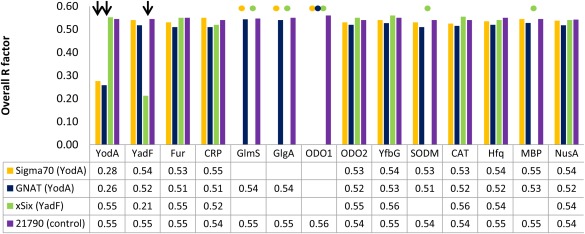
Overview of MR experiments run with diffraction data collected from crystallization artifacts: Sigma70 factor (YodA), GNAT (YodA), survivin SIX or xSix (YadF), and NYSGRC‐021790 (as a negative control). The MR method was applied to each test case, using as templates selected structures of purification artifacts listed in Table S2, Supporting Information. Because the identities of the artifact proteins were known before running MR experiments, only the structures of E. coli native proteins and two selected affinity/solubility tags were used as templates. All PDB deposits listed in Table S2, Supporting Information for a given purification artifact were tested—the R‐factors reported on the figure are mean of R‐factor values of MR experiments run for each template corresponding to a given artifact. The overall R‐factors used for success‐failure determination are calculated after 15 cycles of REFMAC refinement of the best MR solution (determined by MOLREP) for each template‐dataset pair. Successful identification of an artifact is marked with arrow and a failure of MOLREP to produce a MR solution is marked with a dot. For all templates, chain A was selected for MR.
Discussion
Successful, high‐yield production of pure, soluble protein is of paramount importance for most research projects using biochemical and/or biophysical methods, including protein crystallography. Many parameters influence the robustness of subsequent experiments, and suboptimal values of these parameters may significantly impair data reproducibility and outcomes of projects. Ideally, a protein sample of good quality should be relatively pure, be free of soluble and insoluble aggregates, have a verified identity, and maintain its biological activity (if it is present and measurable). That is why at each step of protein production, appropriate quality control procedures should be employed to ensure that the correct proteins are purified, and that the resulting protein samples are of the highest quality. This maximizes the probability that the experiments involving them are credible and reproducible.
Many methods can provide data for validation of protein identity and purity, depending on the protein and available resources, such as SDS‐PAGE, analytical SEC, mass spectrometry, enzymatic activity assays, peptide sequencing, and immunoblotting. Unfortunately, these validation methods sometimes do not conclusively establish protein identity and/or are not used because they are not feasible, available, or possible. Furthermore, even if protein identity is verified, robust validation is not always feasible for each subsequent repetition of the purification procedure. Often one will employ full validation procedures while establishing the best protocol for purification, but will use simpler, more cost‐effective methods such as SDS‐PAGE or SEC for routine purification afterward.3, 17
However, individual expression and purification experiments differ. It is presumed that for the most part, repetitions of a protocol will lead to similar results; however, it is possible that small, unforeseen deviations from a protocol may lead to the production of contaminants or even the wrong protein. Some research techniques are more sensitive to potential contamination than others. Protein crystallization is relatively robust to contamination, due to the self‐selection of molecules incorporated into the crystal lattice, but the method cannot be considered as wholly unaffected by or resistant to all contamination.
In this work, we describe four case studies of crystallization experiments where the proteins found in the determined structures turned out to be artifacts. One case was the result of sample contamination with a native E. coli protein, two were the result of purification of the wrong protein, and one was a consequence of mislabeling of the sample.
Protein production traps and pitfalls that resulted in purification/crystallization artifacts
In the case of survivin, the SDS‐PAGE analysis revealed that sample was heterogeneous. However, the contamination was relatively minor, and the sample purity was considered acceptable for crystallization. Nevertheless the contaminant (YadF) showed much higher crystallization propensity than the target protein (survivin). Our observations indicate that when the target protein sample contains protein contaminants at much lower concentrations, this does not automatically rule out the possibility of obtaining the crystal structure of the target protein. In fact, bulk crystallization itself has long been used as an industrial purification procedure.18 Therefore, it is not always time and cost effective to purify a protein for each experiment to the highest purity levels, but lower protein purity levels do increase the likelihood of crystallizing an artifact.
In the case of GNAT the ratio of the presumed target protein to the contaminant was about 1:1, and crystallization trials were set up in the hope that the correct protein will crystallize regardless of the contamination (or that an interaction partner will be identified by crystallization of a complex). Structure determination attempts revealed that YodA crystallized instead of the expected GNAT, and we concluded that under the growth conditions used, the GNAT protein was not measurably expressed. Specifically, when the SDS‐PAGE results were revisited, the measured apparent mass of the observed protein (23 kDa) deviated slightly from the calculated mass of the His‐tagged GNAT construct (21 kDa).
In the case of Sigma70 factor, presumably unfavorable cell culture growth conditions caused low or blocked levels of recombinant protein production, and concomitant transcriptional activation of native stress proteins, including YodA.3 The very similar mass of the two proteins (Sigma70 and YodA) resulted in the failed expression being recognized as successful, and the purified sample was used for crystallization trials.
A number of experimental conditions can increase the probability of purification of an unintended protein—for example, if the yield of expression of the target protein is low, and too much affinity resin is used relative to the amount of the target protein. A lower than anticipated amount of the target protein leaves many unoccupied sites on the resin, which may promote unspecific binding of contaminants even if they have lower affinity for the resin. It is difficult to precisely estimate the amount of the protein of interest in the cell lysate (e.g., from SDS‐PAGE results), when the masses of the intended protein and native E. coli proteins such as YodA, are very similar. Although in many cases this can be mitigated by carefully adjusting the binding and wash protocols, this optimization may be overlooked when generic purification protocols are used without adjustment. Another cause of an excess of unoccupied resin might be an incompetent affinity tag, which may be (for example) occluded by the target protein, making it inaccessible to the binding site on the resin.19
Accidental mislabeling of protein samples, although rare, may also lead to crystallization of the wrong protein as described above. Even if these errors should be theoretically eliminated by good laboratory practices, in practice they occasionally do happen, even in systems with automated controls.
Identification of purification/crystallization artifacts from diffraction data
We propose several methods that may be used for identification of purification/crystallization artifacts if routine structure determination is unsuccessful and crystallization of the wrong protein is suspected. These methods may minimize the effort needed for structure solution by expediting artifact identification and directing attention toward optimization of protein production methods rather than simply troubleshooting.
Lattice parameter search against PDB
Given that unit cell dimensions comprise up to six independent parameters, taken together these parameters are a surprisingly good discriminant for the identity of a crystal form. Searching against the PDB relies on the idea of finding previously solved structure(s) with the same unit cell dimensions (±1%) as the crystal under question. If a close match is found, it is very probable that it could be of the same protein as the possible artifact, which may be confirmed by MR. This type of search can be performed by the HKL‐3000 system or by the advanced online search tool of the RCSB PDB.
This approach was successfully applied in the case of the attempted GNAT structure determination. The fact that the unit cell parameters closely matched those of YodA was discovered shortly after diffraction data collection, which saved futile effort on structure solution. Unfortunately, this method was not used in the case of survivin SIX. Since in retrospect it was shown that this approach would have detected the YadF artifact in the crystal lattice, it would have saved the effort spent on finding the artifact by SAD phasing, multiple attempts at model building, and sequence recognition with subsequent BLAST and PDB searches. As searching the PDB for unit cell dimensions is very simple and only requires indexing of the diffraction data, we propose it should be used routinely as the first troubleshooting step when initial attempts to solve a structure have failed, especially when the possibility of crystallization artifacts was recognized in the protein production process (e.g., significant contaminant bands noted in SDS‐PAGE).
In some ways this approach is a simplistic one. In particular it assumes that standard crystallographic conventions are strictly obeyed. This unfortunately is not always the case, and thus this approach does not guarantee that the artifact is recognized even if it is present in the PDB. To make this method more robust, proper standardization or/and reduced Niggli cells20 should be used. Nevertheless, identification of crystallization artifact using this simple approach is possible and can be done using readily available tools.
The unit cell parameters check did not help in the case of Sigma70, where YodA crystallized with unit dimensions significantly different than those of any other YodA structure in the PDB. However, it is possible that we were not the first to crystallize YodA protein with the unit cell parameters reported herein. Therefore we would like to encourage researchers to deposit structures of artifact proteins, or in other words, to publish “negative results” on web pages and/or in various repositories (e.g., the PDB), especially if these artifacts were crystallized with unit cell parameters not reported previously. An increase in the size of the library of crystallization artifact structures deposited to the PDB can potentially make troubleshooting of new artifacts easier, and save much effort for those who are new to such cases.
Sequence and/or fold identification from a partial model built based on experimental phasing
Additional possibilities emerge if initial experimental phases can be obtained (e.g., from incorporated selenomethionine, intrinsic metal ions, or heavy metal derivatives) and a partial model can be built. With a comparatively good quality model, one can employ 3D structure similarity services such as PDBeFold, Dali, or FatCat in order to find a structure with similar fold and identify a potential artifact or a homolog. In the current study, PDBeFold server was shown to retrieve the correct match (YodA) in the case of Sigma70 factor, but in the case of survivin SIX, the model was too fragmented to find any significant match for the contaminant (YadF). Therefore, this approach might be insufficient if only a poor model has been built or if there is no structure of the artifact protein or its homolog in the PDB.
Sequence identification using the electron density map, as implemented by Fitmunk, is more straightforward and effective in our experience than manual assignment of sequence to well‐ordered fragments of electron density. Obviously, the accuracy of a sequence assignment is affected by the diffraction data resolution and quality of the electron density maps. Even if the sequence can be assigned correctly to only a small fraction of the protein, it is usually possible to use bioinformatics tools to identify one or a small number of possible proteins. For example, in the case of survivin SIX (YadF), there was only approximately 30% similarity of the density‐assigned sequence to that of YadF, yet the identification of the protein was straightforward when the PDB sequence database was used. In the case when only the side chain topology is assigned by Fitmunk, it is advisable to use smaller sequence databases (e.g., PDB) or a sequence profile database (such as CDD). A search against a large sequence database may provide more false positives when the recognition rate is low, because many more hits may be matched at random. Still, a match to the correct protein should be found, but because the significance of a match is calculated relative to the database size, it may be considered insignificant. Use of sequence profile databases partially mitigates this problem, as profile‐based searches are more sensitive to relationships between distantly similar sequences.21
MR with common contaminants as search templates
The structures of common contaminants, applicable fusion tags, and exogenous additives (Table 1) may be used as templates for MR if the cell dimensions screening failed to produce a positive result and experimental phases are not available, or the sequence or fold identification did not work. Since the list of templates is extensive and MR methods are sensitive to changes in protein conformation, it is advisable to use automated MR pipelines such as AMPLE,22 MRBUMP,23 or Balbes.24 These programs can also be used to test multiple structures of each common contaminant available in the PDB, and employ various methods of template modification, such as different side‐chain truncation schemes. In our experience, it is also beneficial to employ different refinement protocols after MR, such as “jelly‐body” refinement in REFMAC for refinement of low‐resolution or low phase quality data. As shown above, this method unequivocally identified all three purification artifacts described herein.
Materials and Methods
Protein purification, crystallization, and structure determination
Purification and crystallization of YodA instead of Sigma70 factor
The Plim_1876 gene encoding Sigma70 factor from Planctomyces limnophilus DSM 3776 was cloned into the pMCSG48 (modified pMCSG7,25 which has 6xHis‐Tag replaced with 8xHis‐Tag fused with NusA solubilization tag) expression vector. The Sigma70 factor clone was obtained from the MCSG (target APC100648). The protein was overexpressed in E. coli BL21‐CodonPlus(DE3)‐RILP (Stratagene, La Jolla, CA) grown in SelenoMethionine expression media (Molecular Dimensions, Suffolk, UK). The cells were grown at 37°C to an OD600 of 3. The temperature was then decreased to 16°C, 100 mg of L‐selenomethionine was added per 1 L of media in each flask, and the culture was induced with 0.5 mM IPTG and grown overnight. Cells were harvested by centrifugation, and the pellet was frozen afterward at −80°C. The pellet was thawed at room temperature and resuspended in buffer A, containing 50 mM Tris pH 7.5, 500 mM NaCl, 5 mM imidazole, 10 mM 2‐mercaptoethanol, and a protease inhibitor coctail (cOmplete Mini, EDTA‐free, Roche, Basel, Switzerland). The pellet was homogenized and disrupted with a high pressure homogenizer (EmulsiFlex‐C3, Avestin Inc. Ottawa, Canada). The cell lysate was centrifuged at 35,000 rpm at 4°C for 40 min. The supernatant was loaded onto a gravity‐fed chromatography column containing 5 mL nickel‐nitrilotriacetic acid agarose charged resin (Qiagen, Venlo, The Netherlands), previously equilibrated with buffer A. The column was then washed with buffer A supplemented with 10 mM imidazole. Finally, protein was eluted with buffer A supplemented with 300 mM imidazole. The His‐tag was removed by TEV protease cleavage; 0.2 mg of protease was added to the eluted sample five times every 4 hours. The sample was dialyzed in buffer B containing 50 mM Tris pH 7.5, 250 mM NaCl, and 10 mM 2‐mercaptoethanol. After dialysis, the sample was concentrated with centrifugal filters (10 kDa Amicon Ultra‐15 Centrifugal Filter Units, EMD Millipore, Merck, Darmstadt, Germany) and loaded onto an XK 16/60 Superdex200 column attached to an AKTA FPLC gel filtration system (GE Healthcare, Little Chalfont, UK) at 4°C, previously equilibrated with buffer B at a flow rate of 1.5 mL/min. The fractions containing the target protein were combined and concentrated in centrifugal filters. Protein purity was confirmed by SDS‐PAGE. The protein was crystallized using 0.3 µL of 9.6 mg/mL protein mixed with 0.3 µL of 2.5M ammonium sulfate, 0.1M Bis‐Tris propane pH 7.0, and equilibrated against 1.5M NaCl in MRC 2 drop 96 well crystallization plate (Swissci, Neuheim, Switzerland). Crystals were harvested and vitrified in liquid nitrogen; no cryoprotectant was used. Diffraction data were collected at the wavelength of 0.9786 Å at the 21‐ID‐G beam line of the Life Sciences Collaborative Access Team (LS‐CAT) of the Advanced Photon Source (APS).
Purification and crystallization of YodA instead of GNAT
The SACOL0519 from Staphylococcus aureus subsp. aureus COL gene was cloned into the pMCSG725 expression vector. The protein was overexpressed in E. coli BL21‐CodonPlus(DE3)‐RILP cells (Stratagene, La Jolla, CA) grown in M9 SeMet High‐Yield Growth Medium. The cells were grown at 37°C to an OD600 of approximately 0.6, followed by induction of protein expression with 1 mM IPTG and incubation overnight with shaking at 16°C. Cells were harvested, and the protein was purified as described previously.26 The resin with bound protein was washed with buffer containing 30 mM imidazole and the protein was eluted from the column with 250 mM imidazole. Crystals were grown at 16°C using vapor diffusion and a sitting drop setup. The crystallization drops were a 1:1 mixture of protein solution at 15 mg/mL concentration and the precipitant solution from the well (25% PEG 3350, 0.1M Bis–Tris pH 5). Crystals were harvested and vitrified in liquid nitrogen; no cryoprotectant was used. Diffraction data were collected at 0.9787 Å and 0.9786 Å wavelength, respectively, at the 21‐ID‐F and 21‐ID‐G LS‐CAT beamlines of the APS.
Purification and crystallization of YadF instead of survivin SIX
The 398454 gene encoding the survivin SIX (Uniprot ID: Q804H7) protein from Xenopus laevis was cloned into the pMCSG13 vector25 and overexpressed in E. coli BL21‐CodonPlus(DE3)‐RIL (Stratagene, La Jolla, CA). Cells were grown at 37°C in LB medium and induced at OD600 of 1.0 with 0.15 mM IPTG. After induction, ZnCl2 was added to a final concentration of 80 μM to the medium to facilitate survivin folding under conditions of induced overexpression. Cells were grown overnight at 18°C with shaking. After harvesting, cells were lysed in buffer A containing 50 mM Tris pH 8.0, 200 mM NaCl, 0.5 mM tris(2‐carboxyethyl)phosphine (TCEP), and 5 mM imidazole. The NiNTA resin was washed with buffer A containing 10 mM imidazole, and protein was eluted with buffer A containing 250 mM imidazole. Purified protein was separated on Superdex200 column in a buffer containing 50 mM Tris pH 7.9, 200 mM NaCl, and 0.5 mM TCEP, and fractions containing MBP‐SIX protein (MW 60 kDa) were pooled together and concentrated to 7.4 mg/mL. The protein was crystallized by mixing in 1:1 ratio the MBP‐SIX protein solution with 60% v/v Tacsimate pH 7.0 and 0.1M Bis–Tris propane pH 7.0. Crystals were grown by the sitting‐drop vapor‐diffusion method. Crystals were harvested, cryoprotected in mother liquor supplemented with 30% (vol/vol) ethylene glycol and vitrified in liquid nitrogen. A fluorescence spectrum was recorded on the 19‐ID beamline of the Structural Biology Center (SBC) at the APS. Diffraction data were collected at a wavelength of 1.278 Å on the LS‐CAT 21‐ID‐D beamline of the APS.
Crystallization of NYSGRC‐022189 instead of NYSGRC‐021790
The protein was cloned, expressed, and purified at NYSGRC with standard protocols as described elsewhere.27, 28 In brief, the mttB gene (locus tag SMc04330) encoding for trimethylamine methyltransferase from Sinorhizobium meliloti 1021 was cloned into pSGC‐His vector (which is a modified pMCSG7 vector with SspI restriction site replaced with BseRI and with added SacB expression cassette for negative selection screening) and overexpressed in E. coli BL21‐CodonPlus(DE3)‐RIL grown on PASM‐Semet media. The protein was purified by Ni‐IMAC and gel filtration. The purified protein was concentrated to 14 mg/mL and stored in 20 mM HEPES pH 7.5, 150 mM NaCl, 10% glycerol, 0.1% NaN3, and 0.5 mM TCEP. TEV protease was added to the protein right before crystallization trials for in situ His‐tag cleavage with 1:200 protein‐to‐TEV protease mass ratio. The crystallization was set up with the sitting‐drop vapor‐diffusion method by mixing 0.2 μL of protein with 0.2 μL of screening solution and using 1.5M NaCl as reservoir liquid. Diffraction quality crystals were grown in MCSG Suite 1 conditions #33 (32% w/v PEG 4K, 0.1M Tris HCl pH 7.0 and 0.2M calcium acetate). Crystals were harvested and vitrified in liquid nitrogen; no cryoprotectant was added. The first diffraction dataset (resolution of 1.8 Å) was collected at a wavelength of 0.97891 Å on the LS‐CAT 21‐ID‐D beamline of the APS. The second dataset (resolution of 1.56 Å) was collected at a wavelength of 1.078 Å on the same beamline.
Data processing, structure solution, model building, and refinement
Data reduction and scaling was done with HKL‐2000/HKL‐3000.29 MOLREP30 as implemented in HKL‐3000 was used for all MR calculations. Phasing by single wavelength anomalous diffraction was performed with HKL‐3000 coupled with SHELX,31 MLPHARE, DM, and other CCP4 programs.32, 33 Model building attempts were performed with Buccaneer,34 ARP/wARP,35 and RESOLVE36 as implemented in HKL‐3000. The structures were refined with REFMAC.37 Coot38, 39 was used for the visualization of electron density maps and manual inspection and correction of the atomic models. TLS groups for the deposited structures were determined with the TLS Motion Determination Server.40 MolProbity41 and PDB validation tools were used for structure validation.
Sequence recognition based on electron density map with Fitmunk
Partial models built from initial and improved electron density maps were used as input to Fitmunk running in a topology recognition mode. Fitmunk, when run without a specified sequence, assigns a topologically representative residue type to each amino acid position based on electron density corresponding to a side chain. In topology recognition mode, the program does not try to differentiate between: Asn, Asp and Leu; Arg and Lys; Glu and Gln, Phe and Tyr; and Thr and Val. In the YadF (Survivin SIX) case, the initial model had four fragments; therefore, all permutations of unique sequence fragments (24 sequences in total) were used to perform a BLAST search of the NCBI NR or PDB sequence databases. All fragment permutations identified the same protein and it was enough to use the sequence of the longest fragment, which comprised 75% of residues. All other cases had initial model built as one fragment. The top hit in a BLAST search was then used as a sequence for further rebuilding. It has to be noted that search method which uses permutations of sequence fragments is unsuitable for very fragmented models. Firstly, because the large number of resulting sequences would be prohibitive to do manual BLAST searches. Secondly, the reliability of sequence assignment in very fragmented models may by low.
Selection of representative models for MR
The templates for MR provided in Table 1 were selected to represent distinct conformational states of the given protein using the following procedure. First, all crystal structures of a template were identified on the basis of BLAST sequence clustering provided by RCSB PDB website. Clusters formed by sequences with 95% identity to a template were used. For most cases, all individual protein chains present in the sequence cluster were selected for further analysis. In few cases (lysozyme, thrombin, thermolysin, trypsin), the number of deposits in a cluster was larger than 100. Therefore, instead of all chains in a cluster, the highest resolution structure for each unique space group and unique crystal lattice parameters (within 1% tolerance) was selected for further analysis. These selected chains were then clustered on the basis of pairwise RMSD values calculated using SSM12 superposition algorithm using the complete‐linkage clustering method. For each cluster that comprised structures that had their pairwise RMSD lower than 1 Å a medoid chain (chain that had lowest pairwise RMSDs to all remaining chains in a cluster) was selected as representative for the cluster and presented in Table 1, unless otherwise noted. If individual chains from one deposit were present in distinct clusters, a deposit is listed once.
Conclusions
In most laboratories, protein production is the starting point for further project development. We suspect that purification artifacts happen more frequently than reported in the literature, because these cases do not address primary research goals and are considered to be more of a problem to overcome rather than a result to report. In most of the cases described in this article, only a slim amount of evidence had indicated potential problems with the subsequent experiments, but they were not striking in any case. Except in the case of GNAT, the crystallization of an artifact was completely unexpected.
Even a very pure protein sample will contain at least trace amounts of contaminant proteins, which may affect not only crystallographic but also functional studies. While crystallography may be very robust both during crystallization and structure solution to impure protein samples, one should more closely scrutinize the outcomes of other methods. During functional studies even trace amounts of active contaminants have the potential to produce a false positive result, particularly when previously uncharacterized proteins are screened by broad activity assays. If the protein seems to be pure, an error may be very difficult to detect, since, unlike in crystallography, the presence of an artifact may be very difficult to visualize. This is especially important for proteins of unknown function, where, the first detected activity may be the consequence of the presence of a contaminant.
When experiments are carried out in high throughput mode, high throughput does not always mean high output. It may be reasonable to spend more time or sacrifice more resources to troubleshoot unsuccessful or unexpected results of an experiment. Overall, credible and repetitive results are much more important than the time savings on a particular experiment. Experimental protocols must be adjusted according to the resources available in a laboratory. Usually stricter protocols reduce the chances of obtaining an artifact, however they generally cost more and are more time and material consuming. If some procedures are less strict, one should keep in mind that a chance of obtaining an artifact is much greater.
Even if crystallization artifacts could potentially be avoided, they still happen in practice. We suggest that issues of experimental irreproducibility in a number of scientific fields, prominently discussed in several recent articles,42, 43 may be partly related to undetected purification artifacts. Herein, several straightforward computational approaches for the detection of purification/crystallization artifacts have been described, which we hope will significantly reduce the time spent on troubleshooting whenever such cases are encountered, and increase the overall awareness of potential pitfalls and artifacts. The methods presented here allow for quick identification of the artifact during and just after data collection—potentially saving lots of effort. Nevertheless, when resources permit one should also consider experimental methods of verifying the identity of the crystallized sample after data collection. Mass spectrometry analysis of the crystal can ensure the presence and integrity of a crystallized protein. An additional benefit of this method is confirmation of possible ligands, binding partners, or modification of amino acids (like oxidation of cysteines). Verifying protein mass could be useful not only when the chance of obtaining artifact is high, but also as a procedure to search for protein regions that may degrade during crystallization if the built protein model is incomplete and lacks certain protein regions or domains. Deviations from molecular weight can be also detected by SDS‐PAGE analysis of crystallized protein sample, which due to cost effectiveness can be done routinely.
Just before submitting revised manuscript, we encountered another case of crystallization of unexpected protein. The HaloTag7 used for enhancing protein solubility crystallized instead of the desired protein. By applying a method suggested in this work—searching PDB with unit cell parameters and subsequent MR, it was possible to quickly discover this artifact. This further shows that unfortunate issues described in this article are common in laboratory work and solutions presented here to detect them are useful.
The list of PDB codes that can be used to detect crystallization artifacts using MR and detailed data about MR experiments run with diffraction data collected from crystallization artifacts are also available here: http://www.bioreproducibility.org/pages/protein_purification_artifacts. Diffraction experiments were deposited to BD2K http://www.proteindiffraction.org/server.
Authors Contribution
EN, OG, KBH, KAM, IGS and EZ planned and performed all experiments. EN, OG, KBH, KAM, PJP, IGS, MC and WM analyzed data and wrote the manuscript.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
We thank Matthew D. Zimmerman and David Cooper for discussions and for critical reading of the manuscript. The diffraction data collection was performed at SBC‐CAT and LS‐CAT beamlines at the Advanced Photon Source, Argonne National Laboratory. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE‐AC02‐06CH11357. Use of LS‐CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri‐Corridor (Grant 085P1000817).
The authors declare that there is no conflict of interest.
Description of Supporting Information material: Summary of data collection and refinement statistics for the deposited structures and the list of deposits used to identify crystallization artifacts by MR. Filename: Supplementary Materials
Structural data are available in PDB database under accession numbers 4TNN, 4YYC, and 4ZNZ.
This work focuses on a particular difficulty that may occur as a result of accidental purification or contamination of the sample with a protein different than the protein of interest. Examples where the incorrect protein species were purified and/or crystallized are presented, and procedures to quickly rule out the possibility that the crystals obtained in crystallization experiments are the effect of purification artifacts are described.
References
- 1. Chu CK, Feng LL, Wouters MA (2005) Comparison of sequence and structure‐based datasets for nonredundant structural data mining. Proteins 60:577–583. [DOI] [PubMed] [Google Scholar]
- 2. Bolanos‐Garcia VM, Davies OR (2006) Structural analysis and classification of native proteins from E. coli commonly co‐purified by immobilised metal affinity chromatography. Biochim Biophys Acta 1760:1304–1313. [DOI] [PubMed] [Google Scholar]
- 3. Gräslund S, Nordlund P, Weigelt J, Hallberg BM, Bray J, Gileadi O, Knapp S, Oppermann U, Arrowsmith C, Hui R, Ming J, dhe‐Paganon S, Park HW, Savchenko A, Yee A, Edwards A, Vincentelli R, Cambillau C, Kim R, Kim SH, Rao Z, Shi Y, Terwilliger TC, Kim CY, Hung LW, Waldo GS, Peleg Y, Albeck S, Unger T, Dym O, Prilusky J, Sussman JL, Stevens RC, Lesley SA, Wilson IA, Joachimiak A, Collart F, Dementieva I, Donnelly MI, Eschenfeldt WH, Kim Y, Stols L, Wu R, Zhou M, Burley SK, Emtage JS, Sauder JM, Thompson D, Bain K, Luz J, Gheyi T, Zhang F, Atwell S, Almo SC, Bonanno JB, Fiser A, Swaminathan S, Studier FW, Chance MR, Sali A, Acton TB, Xiao R, Zhao L, Ma LC, Hunt JF, Tong L, Cunningham K, Inouye M, Anderson S, Janjua H, Shastry R, Ho CK, Wang D, Wang H, Jiang M, Montelione GT, Stuart DI, Owens RJ, Daenke S, Schütz A, Heinemann U, Yokoyama S, Büssow K, Gunsalus KC (2008) Protein production and purification. Nature Meth 5:135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu W, MacGrath SM, Koleske AJ, Boggon TJ (2012) Lysozyme contamination facilitates crystallization of a heterotrimeric cortactin‐Arg‐lysozyme complex. Acta Cryst F68:154–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Khan NA (2004) HPLC of peptides and proteins: methods and protocols, edited by Marie‐Isabel Aguilar (methods in molecular biology, volume 251, series editor J. M. Walker). Humana Press, Totowa, NJ, 2003, 413 pp, ISBN: 0‐86903‐977‐3. Biomed Chromatogr 18:475. [Google Scholar]
- 6. Kato K, Shinohara H, Goto S, Inaguma Y, Morishita R, Asano T (1992) Copurification of small heat shock protein with alpha B crystallin from human skeletal muscle. J Biol Chem 267:7718–7725. [PubMed] [Google Scholar]
- 7. Dong A, Xu X, Edwards AM, et al. (2007) In situ proteolysis for protein crystallization and structure determination. Nat Methods 4:1019–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smyth DR, Mrozkiewicz MK, McGrath WJ, Listwan P, Kobe B (2003) Crystal structures of fusion proteins with large‐affinity tags. Protein Sci 12:1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hassell AM, An G, Bledsoe RK, et al. (2007) Crystallization of protein‐ligand complexes. Acta Cryst D63:72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Porebski PJ, Cymborowski M, Pasenkiewicz‐Gierula M, Minor W (2016). Fitmunk: improving protein structures by accurate, automatic modelling of side chain conformations. Acta Cryst D. In Press. doi:10.1107/S2059798315024730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marchler‐Bauer A, Derbyshire MK, Gonzales NR, et al. (2014) CDD: NCBI's conserved domain database. Nucleic Acids Res 43:D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krissinel E, Henrick K (2004) Secondary‐structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst D60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 13. Holm L, Rosenström P (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ye Y, Godzik A (2004) FATCAT: a web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res 32:W582–W585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berman HM (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lohkamp B, Dobritzsch D (2008) A mixture of fortunes: the curious determination of the structure of Escherichia coli BL21 Gab protein. Acta Cryst D64:407–415. [DOI] [PubMed] [Google Scholar]
- 17. Kim Y, Babnigg G, Jedrzejczak R, et al. (2011) High‐throughput protein purification and quality assessment for crystallization. Methods 55:12–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Judge RA, Johns MR, White ET (1995) Protein purification by bulk crystallization: the recovery of ovalbumin. Biotechnol Bioeng 48:316–323. [DOI] [PubMed] [Google Scholar]
- 19. Majorek KA, Kuhn ML, Chruszcz M, Anderson WF, Minor W (2014) Double trouble‐Buffer selection and His‐tag presence may be responsible for nonreproducibility of biomedical experiments. Protein Sci 23:1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grosse‐Kunstleve RW, Sauter NK, Adams PD (2003) Numerically stable algorithms for the computation of reduced unit cells. Acta Cryst A60:1–6. [DOI] [PubMed] [Google Scholar]
- 21. Rychlewski L, Jaroszewski L, Li W, Godzik A (2000) Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci 9:232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bibby J, Keegan RM, Mayans O, Winn MD, Rigden DJ (2012) AMPLE: a cluster‐and‐truncate approach to solve the crystal structures of small proteins using rapidly computed ab initio models. Acta Cryst D68:1622–1631. [DOI] [PubMed] [Google Scholar]
- 23. Keegan RM, Winn MD (2008) MrBUMP: an automated pipeline for molecular replacement. Acta Cryst D64:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Long F, Vagin AA, Young P, Murshudov GN (2008) BALBES: a molecular‐replacement pipeline. Acta Cryst D64:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eschenfeldt WH, Stols L, Millard CS, Joachimiak A, Donnelly MI (2009) A family of LIC vectors for high‐throughput cloning and purification of proteins. Meth Mol Biol High Throughput Protein Expr Purif 498:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Majorek KA, Kuhn ML, Chruszcz M, Anderson WF, Minor W (2013) Structural, functional, and inhibition studies of a Gcn5‐related N‐acetyltransferase (GNAT) superfamily protein PA4794: a new C‐terminal lysine protein acetyltransferase from Pseudomonas aeruginosa. J Biol Chem 288:30223–30235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sauder MJ, Rutter ME, Bain K, Rooney I, Gheyi T, Atwell S, Thompson DA, Emtage S, Burley SK (2008) High throughput protein production and crystallization at NYSGXRC. Meth Mol Biol 426:561–575. [DOI] [PubMed] [Google Scholar]
- 28. Almo SC, Garforth SJ, Hillerich BS, Love JD, Seidel RD, Burley SK (2013) Protein production from the structural genomics perspective: achievements and future needs. Curr Opin Struct Biol 23:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Macromol Crytallogr 276:307–326. [DOI] [PubMed] [Google Scholar]
- 30. Vagin A, Teplyakov A (2010) Molecular replacement with MOLREP. Acta Cryst D66:22–25. [DOI] [PubMed] [Google Scholar]
- 31. Sheldrick GM (2008) A short history of SHELX. Acta Cryst A64:112–122. [DOI] [PubMed] [Google Scholar]
- 32. Cowtan K (1994) Jnt CCP4/ESF–EACBM. Protein Crystallogr 34–38. [Google Scholar]
- 33. Winn MD, Ballard CC, Cowtan KD, et al. (2011) Overview of the CCP4 suite and current developments. Acta Cryst 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cowtan K (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Cryst D62:1002–1011. [DOI] [PubMed] [Google Scholar]
- 35. Langer G, Cohen SX, Lamzin VS, Perrakis A (2008) Automated macromolecular model building for X‐ray crystallography using ARP/wARP version 7. Nat Protoc 3:1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Terwilliger T (2003) SOLVE and RESOLVE: automated structure solution, density modification and model building. J Synchro Radiat 11:49–52. [DOI] [PubMed] [Google Scholar]
- 37. Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum‐likelihood method. Acta Cryst D53:240–255. [DOI] [PubMed] [Google Scholar]
- 38. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Cryst D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 40. Painter J, Merritt EA (2006) TLSMD web server for the generation of multi‐group TLS models. J Appl Cryst 39:109–111. [Google Scholar]
- 41. Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Cryst D66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Collins FS, Tabak LA (2014) Policy: NIH plans to enhance reproducibility. Nature 505:612–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Prinz F, Schlange T, Asadullah K (2011) Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov 10:712 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information