

Abstract
Sin3A‐associated protein 30‐like (SAP30L) is one of the key proteins in a multi‐subunit protein complex involved in transcriptional regulation via histone deacetylation. SAP30L, together with a highly homologous SAP30 as well as other SAP proteins (i.e., SAP25, SAP45, SAP130, and SAP180), is an essential component of the Sin3A corepressor complex, although its actual role has remained elusive. SAP30L is thought to function as an important stabilizing and bridging molecule in the complex and to mediate its interactions with other corepressors. SAP30L has been previously shown to contain an N‐terminal Cys3His type zinc finger (ZnF) motif, which is responsible for the key protein–protein, protein–DNA, and protein–lipid interactions. By using high‐resolution mass spectrometry, we studied a redox‐dependent disulfide bond formation in SAP30L ZnF as a regulatory mechanism for its structure and function. We showed that upon oxidative stress SAP30L undergoes the formation of two specific disulfide bonds, a vicinal Cys29‐Cys30 and Cys38‐Cys74, with a concomitant release of the coordinated zinc ion. The oxidized protein was shown to remain folded in solution and to bind signaling phospholipids. We also determined a solution NMR structure for SAP30L ZnF that showed an overall fold similar to that of SAP30, determined earlier. The NMR titration experiments with lipids and DNA showed that the binding is mediated by the C‐terminal tail as well as both α‐helices of SAP30L ZnF. The implications of these results for the structure and function of SAP30L are discussed.
Keywords: SAP30L, Sin3A‐associated protein, zinc finger, disulfide, redox regulation, mass spectrometry, Fourier transform ion cyclotron resonance, NMR
Short abstract
PDB Code(s): 2N1U
Abbreviations
- ECD
electron capture dissociation
- ESI
electrospray ionization
- FT‐ICR
Fourier transform ion cyclotron resonance
- HDAC
histone deacetylase
- SAP
Sin3A‐associated protein
- SAP30L
Sin3A‐associated protein 30‐like
- ZnF
zinc finger
Introduction
Redox‐dependent protein modifications in cellular proteins provide mechanisms for the cells to respond to oxidative stress, caused by reactive oxygen species (ROS), via initiation of signal transduction pathways and induction of gene expression.1, 2, 3, 4 Many eukaryotic proteins utilize zinc finger (ZnF) motifs with reactive cysteine residues as redox switches to sense changes in cellular redox potential by reversible disulfide bond formation or other oxidative modifications. These modifications may induce conformational changes, which in turn affect protein activities. A ZnF is one of the most frequent structural motifs in regulatory proteins. ZnF proteins are typically involved in protein–nucleic acid and protein–protein interactions, although the concept of a zinc finger is vague and several different zinc binding motifs have been characterized.5 In a zinc finger, the Zn(II) ion is most frequently coordinated by four cysteine residues, having tetrahedral binding geometry.6 ZnF‐based redox switches are involved in many physiological events such as cell growth and differentiation, transcription, translation, signal transduction, and apoptosis.1, 2, 3, 4 For example, it has been shown that redox‐dependent modifications in histone deacetylases (HDACs) can directly affect their transcriptional repression function, most likely involving ZnF motifs.7, 8
Sin3A‐associated protein 30‐like (SAP30L) is one of the key proteins in a multi‐subunit protein complex, which is involved in transcriptional regulation through histone deacetylation.9, 10 Besides Sin3A, which functions as an important scaffold protein, the complex comprises of at least ten other proteins [Fig. 1(A)], two class I HDACs (HDAC1 and HDAC2), two histone‐binding proteins (RBBP4/RbAp46 and RBBP7/RbAp48) and, at least, seven Sin3A‐associated proteins (SAP25, SAP30, SAP30L, SAP45/SUDS3, SAP130, SAP180/ARID4B, and RBBP1/ARID4A), whose functions are largely unknown.9, 11, 12 The Sin3A complex regulates a wide variety of genes involved in a number of cellular processes. The complex is among the very few mammalian corepressor complexes that have been characterized so far. Sin3A is a large acidic protein involved in transcriptional repression, but it is unable to bind DNA directly and, therefore, requires the recruitment and interaction of other DNA binding proteins. SAP30L is the newest member of Sin3A‐associated proteins and shares 70% sequence identity with SAP30 (thus the name like) (Supporting Information Fig. S1). SAP30L was originally discovered from T84 cells induced to differentiate by transforming growth factor β (TGF‐β), and was further characterized to interact with DNA and histones 2A/2B as well as signaling phospholipids.11, 13 SAP30 and SAP30L (collectively known as SAP30 proteins) are evolutionarily highly conserved, and the genes of both proteins can be found from many different organisms. In previous studies, it has been suggested that SAP30 proteins have roles as stabilizing and bridging molecules in the Sin3A complex. Apart from directly binding to DNA and nuclear signaling lipids, they can also interact with several other corepressors and transcription factors.9, 11, 14 Previous experiments and truncation assays have indicated that the paired amphipatic helix 3/4 (PAH3/PAH4) regions in Sin3A are essential for gene repression.14 SAP30 and SAP30L have been shown to mainly associate with the PAH3 domain of Sin3A.14, 15, 16 They can also directly interact with HDAC1 and RBBP4 in addition to other transcriptional factors, such as transcription factor YY1 and the inhibitor of growth protein ING1b.16, 17, 18 Furthermore, SAP30 is known to interact with some viral proteins and affect protein expression.19, 20, 21
Figure 1.
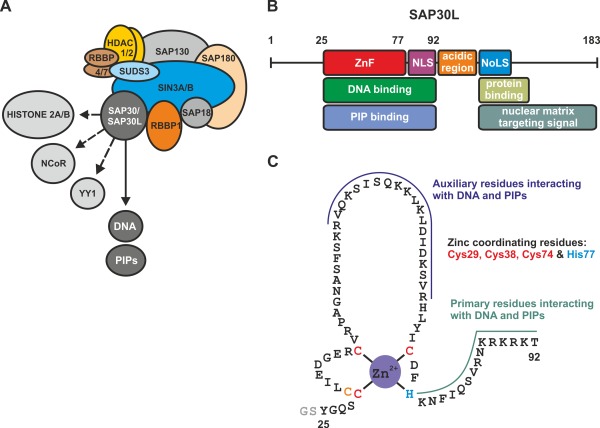
(A) Schematic representation of the multi‐component Sin3A corepressor complex. The complex comprises at least ten proteins (see text for details). Sin3‐associated protein 30 (SAP30) mediates the key DNA and phospholipid interactions of the complex and also associates with histone 2A/2B proteins as well as other transcription factors (e.g., NCoR and YY1; see text for details). (B) Known functional motifs in SAP30L. ZnF, a zinc finger motif; NLS, nuclear localization signal; NoLS, nucleolar localization signal; PIP, phosphatidylinositol phosphate. (C) The zinc finger motif of SAP30L. The expressed construct comprises residues 25–92 of the full‐length SAP30L. The construct also contains two additional N‐terminal residues (GS) from the expression vector (thrombin cleavage site).
As previously demonstrated, SAP30L has several structural motifs which have many different functions in the Sin3A complex [Fig. 1(B) and Supporting Information Fig. S1].10, 11 In addition to the DNA/phospholipid binding ZnF motif, SAP30L also contains an N‐terminal, poorly conserved low complexity region which is markedly shorter as compared to SAP30 (Supporting Information Fig. S1). The function of this region is presently unknown. SAP30L also contains a (polybasic) nuclear localization signal (NLS), which also mediates the DNA and phospholipid binding. This is followed by a central acidic region that contributes to histone 2A/2B interaction, and a C‐terminal nuclear matrix targeting signal (NMTS) along with a nucleolar localization signal (NoLS).22 The structural motifs downstream of NLS make up the C‐terminal domain of SAP30L, also known as a Sin3A interaction domain (SID), which is highly conserved (Supporting Information Fig. S1). The solution NMR structure of the SAP30 SID in complex with the Sin3A PAH3 domain of was recently reported.16
Both SAP30 and SAP30L have been previously characterized in two independent studies to contain a novel Cys3His type ZnF motif [Fig. 1(C) and Supporting Information Fig. S1) that is necessary for protein folding and function.11, 23 The ZnF motif is almost identical in SAP30 and SAP30L, containing exactly the same four zinc coordinating amino acid residues (Cys29, Cys38, Cys74 and His77 in SAP30L; for the corresponding residues in SAP30, see Supporting Information Fig. S1).11 The ZnF motif in both SAP30 and SAP30L is a key element for the repression activity of the Sin3A complex. The ZnF motif in SAP30L mainly comprises residues 25–77 (Fig. 1). The solution NMR structure for SAP30 ZnF (PDB entry 2KDP) indicated that it adopts a totally new fold, comprising two anti‐parallel β‐strands packed against a pair of α‐helices, and a well‐defined hydrophobic core.23 Although lacking any sequence similarity to other known zinc fingers, the fold bears a resemblance to the treble clef ZnF motif, the closest three‐dimensional structure being the ZnF of Drosophila transcription factor grauzone (PDB entry 1PZW).24, 25 Interestingly, both SAP30L and SAP30 also contain an additional cysteine residue, Cys30, next to Cys29. This cysteine residue is highly conserved through evolution.22 Previous mutation and NMR analyses have shown that this residue is not involved in the zinc ion binding.11, 23 However, the appearance of an “unliganded” cysteine residue in a zinc finger protein is highly unusual, especially nearby the coordinating residues. This residue could function as a backup coordinating residue for zinc in case of mutation or has a role in the redox‐regulation.26 Although the entire zinc finger structure is required for the DNA binding, the C‐terminal α‐helix and the following polybasic region are the key structural elements for the binding, as demonstrated by mutation and truncation analysis.11, 23 So far, SAP30 and SAP30L have not been shown to have any DNA sequence specificity, thereby functioning as a general DNA binding element.11, 16, 23
In this work, we characterized a redox‐dependent disulfide bond formation in SAP30L as a regulatory mechanism for its structure and function. The main hypothesis was that the ZnF motif in SAP30L works as a redox switch, which controls the DNA and phospholipid binding and affects the repression activity of the Sin3A complex. We employed high‐resolution native mass spectrometry (native MS) to study structural changes and phospholipid/metal ion binding in SAP30L upon simulated oxidative stress conditions. In recent years, native MS has emerged as a powerful tool for structural biology studies.27, 28 Native MS is based on electrospray ionization (ESI) which allows proteins and their ligand or metal complexes to be analyzed in native state. ESI in combination with a Fourier transform ion cyclotron resonance (FT‐ICR) technique29 is especially suited for the analysis of intact metalloproteins due to its high mass resolution and mass accuracy. This allows small structural changes, for example, disulfide bond formation or metal atom oxidation/reduction, to be directly analyzed. In addition, we also determined a solution NMR structure for the SAP30L ZnF motif and studied its phospholipid and DNA binding by chemical shift perturbation analysis.
Results
SAP30L contains a ZnF motif similar to SAP30
In our previous work, we showed, by using native mass spectrometry, that SAP30L contains an N‐terminal ZnF, where zinc is coordinated by a novel Cys3His motif.11 We also showed, by mutation analysis, that Cys29, Cys38, Cys74, and His77 are the zinc coordinating amino acid residues. The full‐length SAP30L has 183 residues and at least six structural/functional motifs [Fig. 1(B)]. In our previous work, we used a ZnF construct which spans residues 1–92 and comprises all structural motifs of the N‐terminal domain of SAP30L [Fig. 1(C)]. However, since a largely unstructured N‐terminal region (residues 1–24) has no role in the DNA or the lipid binding,11 we produced a shorter construct that contains the residues 25–92, which includes both the ZnF and the NLS motifs. This polypeptide represents a minimal structural construct for the SAP30L ZnF that can autonomously fold and bind both DNA and phospholipids, and is essentially the same as the one used in the solution NMR structure determination for the highly homologous SAP30.23 For simplicity, this construct is hereafter referred to as SAP30L.
The ESI FT‐ICR mass spectrum of SAP30L measured in denaturing solution conditions showed a wide charge state distribution (6+ to 14+), typical for a denatured protein of that size [Fig. 2(A)]. The mass for the most abundant isotopologue was determined to be 8100.291 ± 0.002 Da, which agrees well with the mass calculated from the sequence without a coordinated zinc ion (8100.267 Da for C350H580N112O101S4). There were no signs of associated zinc ions (not even any trace signals) in the spectrum. This indicates that in these conditions SAP30L existed as an unfolded polypeptide in solution.30 To test whether the chelation of zinc from the structure causes the polypeptide to unfold, similar to that observed upon the denaturing solution conditions, a mass spectrum of SAP30L was also measured in 20 mM ammonium acetate (pH 6.8) in the presence of excess 1,10‐phenanthroline, a known zinc‐chelating agent [Fig. 2(B)]. The ESI FT‐ICR spectrum measured in these conditions was essentially the same as in the case of denaturing solution conditions [Fig. 2(A)], indicating an unfolded apoprotein in solution. This is not surprising given that the structural zinc sites are known to lose their tertiary structure upon zinc removal due to a small hydrophobic core. The experimental mass was determined to be 8100.289 ± 0.005 Da, indicating that all cysteine residues remained as free thiols, that is, no disulfide bonds or any other oxidative modifications were present in these conditions. EDTA, another zinc‐chelating agent, was also tested but a considerably slower chelation rate was observed. The improved zinc removal with 1,10‐phenanthroline over EDTA might be due to its ability to substitute zinc coordinating amino acids (structure similar to histidine) by forming a ternary complex with the protein and eventually removing the bound zinc ion. Therefore, we used 1,10‐phenantroline in all further experiments. The 1H,15N HSQC spectrum of zinc‐chelated SAP30L showed features typical for a disordered protein, as well (Supporting Information Fig. S2). The lack of a stable unique structure results in exchange on a fast time scale, which in turn leads to relatively sharp cross‐peaks occupying a narrow 1H region of the spectrum.
Figure 2.
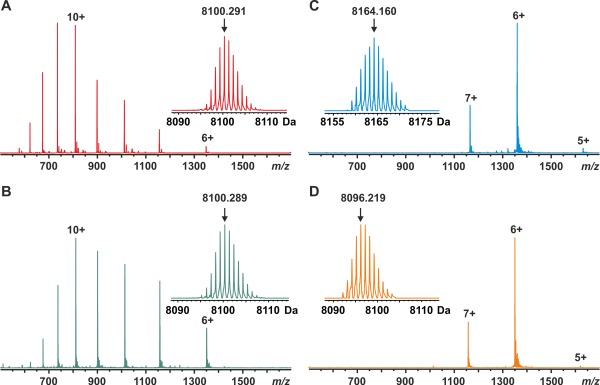
ESI FT‐ICR mass spectra of SAP30L (residues 25–92) measured in (A) MeCN/H2O/HOAc (49.5:49.5:1.0, v/v), (B) 20 mM NH4OAc buffer (pH 6.8) in the presence of 1,10‐phenanthroline (100‐fold molar excess), (C) 20 mM NH4OAc buffer (pH 6.8), and (D) 20 mM NH4OAc buffer after oxidation for 10 min with hydrogen peroxide (10‐fold molar excess). The numbers denote different charge states. The protein concentration was 1 µM in (A) and 5 µM in other measurements. Insets show the deconvoluted isotopic distributions of SAP30L. The arrows denote peaks representing the most abundant isotopologues with their masses indicated.
In contrast, a mass spectrum of SAP30L measured in near‐native solution conditions (20 mM ammonium acetate, pH 6.8) showed a narrow charge state distribution, centered around 6+ [Fig. 2(C)], with the mass being ∼64 Da higher as compared to the apoprotein, consistent with the association of one Zn2+ ion. The experimental mass, averaged over the three observed charge states, was 8164.160 ± 0.007 Da, which almost perfectly matches with the theoretical value of the holoprotein (8164.181 Da for C350H578N112O101S4Zn1). The mass was calculated by assuming an addition of one Zn2+ ion accompanied by the loss of two protons, causing a theoretical mass increment of 63.91 Da. In ZnF, zinc ion binding is always accompanied by the removal of two protons, resulting in a formally neutral “zinc complex.”31, 32 Moreover, the narrow charge state distribution having a considerably lower average charge and fewer charge states, as compared to the denatured/zinc‐chelated protein, indicated the presence of a fully folded protein in solution. Earlier NMR studies have shown that in the presence of zinc SAP30 folds very quickly to a compact, well‐folded structrure.23 This result is consistent with our earlier study11 of the SAP30L ZnF construct comprising residues 1–92, and indicates that the construct used here (residues 25–92) was equally capable of binding one zinc ion. No protein without zinc was observed, implying very high affinity. It must be noted that in these experiments, no extra zinc was added to the sample, thus the only source for zinc being the zinc salt added upon the protein expression. This also means that there was no loss of zinc from the structure, even during the extensive desalting of the protein samples prior to MS experiments.
The 1H,15N HSQC spectrum of holo‐SAP30L also showed characteristics of a well‐folded protein in solution with good signal dispersion and roughly equal cross peak intensities (Fig. 3). A standard assignment procedure led to an almost complete chemical shift assignment. The amide signals of seven residues (Gly23, Ser24, Phe47, Lys61, Val68, Arg69, and Tyr72) were missing from the HSQC spectrum, and Gly23 lacked Hα and Arg69 Hα and Hβ assignments. The residues Gly23 and Ser24 are not part of the original sequence but the result of the bacterial expression construct used. The slightly downfield Cβ shifts of cysteines 29, 38, and 74 (32.8, 31.3, and 33.3 ppm, respectively) compared to that of Cys30 (28.9 ppm) are in accordance with these three cysteines being coordinated to zinc.33
Figure 3.
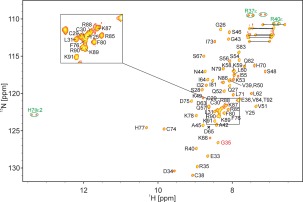
Assigned 1H,15N HSQC spectrum of holo‐SAP30L. Cross peaks marked in green originate from side chain N—H groups; they appear as doublets due to insufficient decoupling power outside the backbone amide 15N region. The cross‐peak of Gly35 is aliased from the lower 15N field, 101.9 ppm. Side chain NH2 resonances of Asn and Gln residues are connected with horizontal lines.
The three‐dimensional structure for SAP30L was determined by using distance and TALOS dihedral angle restraints employing an automated iterative structure calculation—NOE assignment procedure.34 The final ensemble of fifteen structures is shown in Figure 4(A) and the structural statistics are summarized in Table 1. The residues Ser28‐Arg85 form the core structure, while the N‐ and C‐terminal residues, Gly23‐Gln27 and Asn86‐Thr92, respectively, are disordered. The regular secondary structure elements are formed by residues Leu31‐Glu33 and Leu62‐Ile64, located in a short antiparallel β‐strand, and the residues Val51‐Ser56 and Asp75‐Ser85 located in the two α‐helices [Fig. 4(B)]. Thus, the majority of the residues are found in loops.
Figure 4.
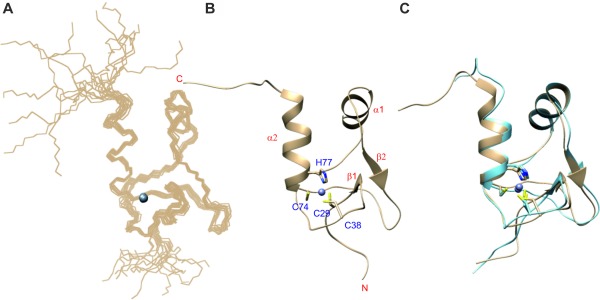
Solution NMR structure of SAP30L. (A) Ensemble of 15 lowest‐energy structures. (B) Ribbon structure with secondary structure elements and zinc binding site highlighted. (C) Overlay of the best matching structures of SAP30L (tan) and SAP30 (light blue) NMR ensembles. Superposed are the backbone atoms of the residues in the secondary structures, omitting the C‐terminal half of α2.
Table 1.
Structural Statistics of the SAP30L Zinc Finger NMR Ensemble
| Structural restraints | |||
| Distance restraints | 924 | ||
| short–range |i–j| ≤ 1 | 508 | ||
| medium–range 1 < |i–j| <5 | 123 | ||
| long–range |i–j| ≥ 5 | 293 | ||
| zinc coordination | 12 | ||
| TALOS dihedral restraints | 88 (44 ϕ, 44 ψ) | ||
| Average AMBER energy (kcal mol−1) | −1905.3 ± 132.5 | ||
| Agreement with experimental data | |||
| Number of NOE violations >0.3 Å | 0 | ||
| Average RMS deviations from distance restraints (Å) | 0.046 ± 0.004 | ||
| Average RMS deviations from ideal covalent geometry | |||
| Bonds (Å) | 0.0104 ± 0.0001 | ||
| Angles (°) | 2.138 ± 0.030 | ||
| Ramachandran plot regions (%) | |||
| Favored regions | 87.5 | ||
| Allowed regions | 10.1 | ||
| Disallowed regions | 2.4 | ||
| Ensemble RMSD (Å)a | |||
| Backbone | 0.37 ± 0.09 | ||
| Heavy | 0.99 ± 0.08 | ||
Residues 27–83.
To investigate how mobile these long loops are, we acquired 15N relaxation data and calculated the product of longitudinal and transverse relaxation rates, R 1 R 2. This quantity reveals local mobility.35 Low R 1 R 2 values are indicative of fast pico‐ to nanosecond time scale mobility, and high R 1 R 2 values are associated with slow micro‐ to millisecond exchange broadening. Several outliers are present in the R 1 R 2 plot of SAP30L (Supporting Information Fig. S3). The terminal residues exhibit low R 1 R 2 values, consistent with disordered tail regions. The residues Ser46, Ser50, Lys53, Ile55, Lys66, His70, and Ile73 have high R 1 R 2 values, indicative of exchange broadening. The neighboring residues Phe47, Lys61, Val68, Arg69, and Tyr72, for which no cross‐peaks were observed in the 1H,15N HSQC spectrum, are most probably mobile in the same time scale, undergoing exchange that results in extreme line broadening.
The overall three‐dimensional structure of SAP30L is remarkably similar to that of SAP30 [Fig. 4(C)]. Overlay of the residues in their secondary structures (including only the first turn of the second α‐helix, the residues Asp75‐Phe80) for the best matching pair of structures in the ensembles gives a backbone atom RMSD of 0.58 Å. The minor differences are found in the length of the second α‐helix, which is, on average, three residues longer in SAP30L, and around the residues Val39 (Gly77 in SAP30) and Arg69‐His70 (Arg107‐His108), where the Cα traces differ by more than 2 Å. Also, the aromatic rings of His70 and His108 are in a perpendicular orientation in the two structures. It is likely that the differences at both locations originate from the sequence differences. The valine residues Val39 and Val68 in SAP30L correspond to Gly77 and Ala106 in SAP30. Via these minor modifications, the larger side chains can be accommodated.
SAP30L undergoes formation of two disulfide bonds upon oxidative stress
As ZnF proteins are known to be redox‐regulated, we sought to investigate the structural and functional roles of redox‐dependent modifications in SAP30L.36, 37 In our earlier study, we observed disulfide formation upon selective serine/alanine mutations of the zinc coordinating amino acid residues of SAP30L.11 However, the functional role of these disulfide bonds remained elusive, as the mutated proteins quickly degraded in solution. To simulate oxidative stress conditions in vitro, SAP30L was treated with a 10‐fold molar excess of hydrogen peroxide (H2O2) for 10 minutes at room temperature. A mass spectrum of the oxidized SAP30L [Fig. 2(D)] was very similar to that observed with the holoprotein [Fig. 2(C)]. However, the experimental mass, determined to be 8096.219 ± 0.005 Da, suggested that two disulfide bonds had been formed with a concomitant release of the coordinated zinc ion (calc. 8096.236 Da for C350H576N112O101S4). The experimental isotopic distribution suggested the presence of a fully oxidized protein, that is, no free thiols were present in the structure. SAP30L has four cysteines, two adjacent Cys29 and Cys30, as well as Cys38 and Cys74, from which three (Cys29, Cys38, and Cys74) are involved in the zinc ion binding.11 Because these four cysteine residues are in close proximity in the folded protein structure, it is plausible that only minor conformational changes are required for their formation. No zinc binding was detected with the oxidized protein. Moreover, no other oxidative modifications were detected, even up to millimolar peroxide concentrations (data not shown).
Interestingly, the charge state distribution of the oxidized SAP30L was essentially the same as observed for the zinc‐complexed holo‐SAP30L [Fig. 2(C,D)]. This result suggests that the oxidized SAP30L remained folded in solution or, at least, had a very similar solvent‐accessible surface area as compared to holo‐SAP30L.38 This is an intriguing finding, since zinc fingers are known to unfold in the absence of a coordinated zinc ion. To analyze the structure further, we attempted to acquire an 1H,15N HSQC spectrum for the oxidized SAP30L. In contrast to apo‐ or the holoproteins, no reasonable signals could be detected in the conditions similar to the native MS experiments, and thus the structure of the oxidized SAP30L remains to be studied in our future endeavors with this protein.
To determine the reversibility of the oxidation, cysteine reduction was performed by incubating the oxidized protein samples at room temperature with 1 mM dithiothreitol (DTT), which resulted in the complete reduction of the two disulfide bonds and instantaneous re‐coordination of zinc. The protein reduction was fully reversible and the appearance of the mass spectrum following DTT treatment was essentially the same as that observed for the holoprotein (data not shown). This shows that the folding of SAP30L triggered by the zinc binding and its oxidation with a concomitant zinc release are fully reversible processes. Parallel incubations of the oxidized SAP30L in a reducing buffer with an excess amount of 1,10‐phenanthroline did not reduce the protein, nor did it have any effect on the charge state distribution, suggesting that the reduction is only possible in the presence of free zinc ions in solution, and the reduced cysteines are only transiently populated in the folded protein structure.
To further examine which of the four cysteines had been paired upon oxidation, in‐solution trypsin and on‐line pepsin digestions were performed. Two different proteases were tested in order to validate their performance in different redox conditions. While trypsin specifically cleaves after lysine and arginine residues, pepsin is less specific and many peptide bonds can be cleaved at various rates. Supporting Information Figure S4 shows a deconvoluted ESI FT‐ICR spectrum for the tryptic digest of holo‐SAP30L, obtained in non‐reducing conditions. All identified tryptic peptides can be found in Supporting Information Table SI. Among the identified peptides, we observed six of them to have disulfide bonds even if the cysteine residues were not initially oxidized. We found the N‐terminal peptide [1–15] to have an intramolecular disulfide bond, Cys29‐Cys30, even with very short digestion times. The two other peptides, [1–27] and [1–28], also contained the same disulfide bond as well as one free cysteine (Cys38). These observations are in line with our earlier study that showed an instantaneous disulfide formation between the adjacent Cys29 and Cys30 upon protein degradation.11 Cys38 was also detected to form a disulfide bond with itself, thus making the two disulfide‐linked peptides, [16–27 + 16–27/28]. However, Cys74 did not form any disulfide bonds (it appeared as free thiol in the peptides [48–56] and [48–63]). These oxidized peptides should be considered as auto‐oxidation products under nonreducing conditions. Therefore, we also performed trypsin digestion in reducing conditions. Figure 5(A) shows an ESI‐FT‐ICR spectrum obtained from a tryptic digest of holo‐SAP30L in the presence of DTT (for the deconvoluted spectrum, see Supporting Information Fig. S4). A total of 22 specific tryptic peptides were identified in these conditions, accounting for 100% sequence coverage (Table 2). No disulfide‐containing peptides were detected, including the tryptic peptide [1–15] with adjacent cysteine residues. Yet, formation of intramolecular disulfide bonds were observed after a short‐term storage, even in reducing conditions, indicating that an oxidized form is favored in small tryptic peptides having free thiol groups.
Figure 5.
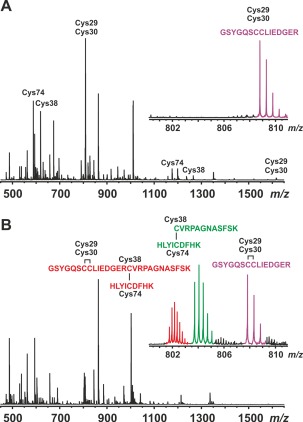
ESI FT‐ICR mass spectra of tryptic digests of (A) holo‐SAP30L (with DTT) and (B) oxidized SAP30L. The peptides containing free cysteines have been labeled in A according to the residue number of the cysteine(s). The insets in A and B show isotopic distributions for the ions corresponding to the reduced and oxidized tryptic peptide [1–15] (2+ ion), and oxidized, disulfide‐linked peptides [16–27 + 48–56] (3+ ion) and [1–27 + 48–56] (4+ ion); these peptide ions were subjected for further ECD‐MS/MS analysis (see, Supporting Information Fig. S5 and text for details).
Table 2.
Identified Tryptic Peptides for Holo‐ and Oxidized SAP30L
| No.a | Mass, exp. (Da)b | Mass, theor. (Da)b | Error (ppm) | Residues | Sequencec |
|---|---|---|---|---|---|
| holo‐SAP30L | |||||
| 1 | 529.3340 | 529.3336 | 0.76 | 28–31 | RVQK |
| 2 | 561.3127 | 561.3122 | 0.89 | 32–36 | SISQK |
| 3 | 602.3286 | 602.3275 | 1.74 | 40–44 | LDIDK |
| 4 | 689.4087 | 689.4072 | 2.18 | 32–37 | SISQKK |
| 5 | 843.5076 | 843.5066 | 1.19 | 38–44 | LKLDIDK |
| 6 | 862.4674 | 862.4661 | 1.51 | 57–63 | NFIQSVR |
| 7 | 944.5290 | 944.5291 | −0.11 | 40–47 | LDIDKSVR |
| 8 | 971.6018 | 971.6015 | 0.31 | 37–44 | KLKLDIDK |
| 9 | 1174.5613 | 1174.5594 | 1.62 | 48–56 | HLYICDFHK |
| 10 | 1185.7100 | 1185.7081 | 1.60 | 38–47 | LKLDIDKSVR |
| 11 | 1235.6101 | 1235.6081 | 1.62 | 16–27 | CVRPAGNASFSK |
| 12 | 1313.8041 | 1313.8031 | 0.76 | 37–47 | KLKLDIDKSVR |
| 13 | 1391.7116 | 1391.7092 | 1.72 | 16–28 | CVRPAGNASFSKR |
| 15 | 1615.6617 | 1615.6607 | 0.62 | 1–15 | GSYGQSCCLIEDGER |
| 16 | 2019.0169 | 2019.0149 | 0.99 | 48–63 | HLYICDFHKNFIQSVR |
| 22 | 2833.2641 | 2833.2582 | 2.08 | 1–27 | GSYGQSCCLIEDGERCVRPAGNASFSK |
| 23 | 2930.5968 | 2930.5926 | 1.43 | 48–70 | HLYICDFHKNFIQSVRNKRKRKT |
| 24 | 2945.5402 | 2945.5334 | 2.31 | 40–63 | LDIDKSVRHLYICDFHKNFIQSVR |
| 26 | 2989.3613 | 2989.3593 | 0.67 | 1–28 | GSYGQSCCLIEDGERCVRPAGNASFSKR |
| 27 | 3186.7203 | 3186.7125 | 2.45 | 38–63 | LKLDIDKSVRHLYICDFHKNFIQSVR |
| 28 | 3314.8165 | 3314.8074 | 2.75 | 37–63 | KLKLDIDKSVRHLYICDFHKNFIQSVR |
| 31 | 4769.6908 | 4769.6868 | 0.84 | 32–70 | SISQKKLKLDIDKSVRHLYICDFHKNFIQSVRNKRKRKT |
| oxidized SAP30L | |||||
| 1 | 529.3327 | 529.3336 | −1.70 | 28–31 | RVQK |
| 2 | 561.3114 | 561.3122 | −1.42 | 32–36 | SISQK |
| 3 | 602.3270 | 602.3275 | −0.83 | 40–44 | LDIDK |
| 4 | 689.4067 | 689.4072 | −0.73 | 32–37 | SISQKK |
| 5 | 843.5062 | 843.5066 | −0.47 | 38–44 | LKLDIDK |
| 6 | 862.4657 | 862.4661 | −0.46 | 57–63 | NFIQSVR |
| 7 | 944.5294 | 944.5291 | 0.41 | 40–47 | LDIDKSVR |
| 8 | 971.6012 | 971.6015 | −0.30 | 37–44 | KLKLDIDK |
| 10 | 1185.7066 | 1185.7081 | −1.27 | 38–47 | LKLDIDKSVR |
| 12 | 1313.8017 | 1313.8031 | −1.07 | 37–47 | KLKLDIDKSVR |
| 14 | 1613.6443 | 1613.6450 | −0.43 | 1–15 | GSYGQSCCLIEDGER (ox) |
| 17 | 2408.1505 | 2408.1518 | −0.54 | 16–27 + 48–56 | CVRPAGNASFSK + HLYICDFHK (ox) |
| 19 | 2564.2520 | 2564.2529 | −0.35 | 16–28 + 48–56 | CVRPAGNASFSKR + HLYICDFHK (ox) |
| 29 | 4003.7842 | 4003.7862 | −0.51 | 1–27 + 48–56 | GSYGQSCCLIEDGERCVRPAGNASFSK + HLYICDFHK (2ox) |
| 30 | 4159.8839 | 4159.8873 | −0.85 | 1–28 + 48–56 | GSYGQSCCLIEDGERCVRPAGNASFSKR+ HLYICDFHK (2ox) |
| 32 | 4848.2383 | 4848.2418 | −0.71 | 1–27 + 48–63 | GSYGQSCCLIEDGERCVRPAGNASFSK + HLYICDFHKIQSVR (2ox) |
For the peptides observed for holo‐SAP30L without DTT, see Supporting Information Table SI.
Monoisotopic mass. In case that the identified peptide appeared in multiple different charges states, an average value has been reported.
Oxidized cysteines in the sequences have been indicated by (ox) or (2ox).
From the tryptic digest of the oxidized protein [Fig. 5(B)], we identified 16 specific tryptic peptides (Table 2 and Supporting Information Fig. S4). These peptides accounted for 90% sequence coverage for the oxidized SAP30L, lacking a few C‐terminal residues. This is most likely due to a high amount of preferable cleavage sites in the polybasic NLS region (7 arginines and 10 lysines), resulting in many low‐mass tryptic peptides. Six of the identified peptides contained one or two disulfide bonds. Among the peptides, we found the N‐terminal peptide [1–15] to have an intramolecular disulfide bond between Cys29 and Cys30. This peptide was further subjected to ECD‐MS/MS measurement, proving that the disulfide bond is, indeed, formed between the two adjacent cysteinyl residues (i.e., a vicinal disulfide). The ECD spectrum of [1–15]2+ ion showed multiple z‐ions up to the formed disulfide bond (Supporting Information Fig. S5). We also found two disulfide‐linked peptides, [16–27/28 + 48–56], both having a disulfide bond between Cys38 and Cys74. Additionally, we detected three larger disulfide‐linked peptides, [1–27/28 + 48–56] and [1–27 + 48–63], having both disulfide bonds. The ECD spectra measured for [1–27 + 48–56]4+ and [16–27 + 48–56]3+ ions further confirmed these peptides (Supporting Information Fig. S5). As electron capture mainly targets disulfide bonds in polypeptides,39 and it is the only bond keeping these peptide chains together, the main ions in the ECD‐MS/MS spectra resulted from the preferential cleavages between the two sulfur atoms with limited backbone cleavages. The peptides corresponding to any other possible (mixed) disulfide bonds between the four cysteine residues, or disulfide‐linked peptide dimers were not observed, indicating that SAP30L is specifically oxidized to form the two specific disulfide bonds, Cys29‐Cys30 and Cys38‐Cys74.
The on‐line pepsin digestion yielded essentially the same results. Supporting Information Figure S6 shows deconvoluted ESI FT‐ICR mass spectra for holo‐ and the oxidized SAP30L digested with immobilized pepsin. A complete list of all identified peptic peptides can be found in Supporting Information Table SII. The sequence coverage was 100% in both cases. From the pepsin digest of holo‐SAP30L, we identified all cysteine‐containing peptides as free thiols. Thus, it is evident that low pH used in on‐line pepsin digestion is sufficient to keep cysteine thiols protonated, which likely prevents their auto‐oxidation in the absence of a reducing agent. A total of 19 peptic peptides were identified for the holoprotein, accounting for 100% sequence coverage. The mass spectrum measured for the pepsin digest of the oxidized SAP30L was markedly different as compared to the holoprotein. All peptides containing free cysteines were absent, indicating that the disulfide bonds are formed before digestion and oxidation does not occur at low pH. Five of the peptides were identified to contain disulfide bonds; the peptides [1–13] and [1–25] confirmed again the vicinal disulfide bond between the adjacent Cys29 and Cys30, while the three larger disulfide‐linked peptides confirmed the other disulfide bond between Cys38 and Cys74. Therefore, it seems that pepsin could be preferred over trypsin for identifying redox‐active disulfide bonds in proteins since even in the absence of a reducing agent, no auto‐oxidation or mixed disulfide bonds were observed following digestion. However, in contrast to trypsin, pepsin digestion is rather unspecific and this might pose a problem in peptide identification for larger proteins without subsequent MS/MS analysis.
Both native and oxidized SAP30L are able to bind phospholipids
We next sought to investigate whether the oxidized SAP30L is also able to bind phospholipids and compared its affinity and specificity to that observed with the holoprotein. Cell signaling phospholipids are known to mediate DNA binding of SAP30L by targeting the same binding site with nucleic acids. In our previous work, we showed that nuclear phospholipids can regulate chromatin association and therefore decrease repression activity of SAP30L.11 We also showed that the polybasic NLS region is indispensable for the lipid binding and lipids can therefore displace the bound DNA. First, we tested the lipid binding with three phosphatidylinositol monophosphates (PIPs) by using native mass spectrometry. Initially, the lipid binding experiments were made without heating the samples. In these experiments, no protein–lipid complexes were observed at the beginning of the experiment. After ∼30 minutes, increasing signals for lipid complexes were observed which reached a constant value in an extended measurement time (data not presented). We suspect this is due to the self‐association of the lipid molecules into micelles at room temperature, and the increasing protein–lipid complex signals during a prolonged measurement was caused by a slow dissociation of the micelles into monomers that are then able to bind to the protein. Pure lipids measured in organic solvent (acetonitrile) or in ammonium acetate buffer showed peaks from monomers to dimers and higher oligomers with lipid concentrations of 20 µM or greater. To prevent time‐dependent complex formation, all further experiments were made with preheating the lipid solutions at 50°C for 15 min, which resulted in a constant amount of protein–lipid complexes from the beginning of the experiment. Similar time‐dependent complex formation has been previously detected for phosphatidylcholine and phosphatidylglycerol binding to the fusion peptide P294.40
The lipid binding experiments showed a 1:1 complex formation with the holo‐SAP30L (Supporting Information Fig. S7). No higher lipid binding stoichiometries were observed, indicating that SAP30L binds the lipids in a monomeric form. The K d values for the PIP binding to holo‐SAP30L were determined to be 60–80 μM (Table 3), with an affinity order of PI(5)P ≥ PI(4)P > PI(3)P. The determined values cannot be directly compared, as there are no other quantitative data available but the affinity order is qualitatively similar to that observed in our previous work with SAP30L [1–92] construct.11 The NMR titration experiments of the highly homologous SAP30 with PI(5)P, reported previously, suggested a weak binding but without an estimate of the binding affinity.23
Table 3.
Equilibrium Dissociation Constants (K d)a for Phospholipid Binding to SAP30L Based on Native Mass Spectrometry
| Protein | Phospahtidylinositol n‐phosphate, PI(n)P | ||
|---|---|---|---|
| PI(5)P | PI(4)P | PI(3)P | |
| Holo‐SAP30L | 61 ± 4 µM | 66 ± 4 µM | 83 ± 6 µM |
| Oxidized SAP30L | 16 ± 1 µM | 22 ± 1 µM | 32 ± 2 µM |
From 70 consequtive measurements.
The binding of PI(5)P to SAP30L was also characterized by NMR in order to map the residues involved. The chemical shift perturbations were monitored in a series of 1H,15N HSQC spectra acquired from a sample containing PI(5)P to SAP30L concentration ratios of 4:1, 3:1, 2:1, and 1:1 (Supporting Information Fig. S8). A reverse titration was carried out because of an aggregation phenomenon occurring in the direct titration with PI(5)P concentration exceeding that of SAP30L. The cross‐peaks shifted approximately linearly with an increasing concentration of the ligand, indicative of low, micro‐ to millimolar binding affinity that is consistent with the native ESI‐MS experiments (Table 3). The chemical shift changes were observed in the structurally delimited region, namely for the residues in the C‐terminal tail, the second half of the second α‐helix, the beginning and end of the first α‐helix and the residues flanking this helix (Supporting Information Fig. S8). This region does not overlap with the zinc‐coordination site.
To examine whether the oxidized SAP30L was still able to bind phospholipids, we titrated the oxidized protein with PI(5)P, which had the highest affinity toward holo‐SAP30L. Surprisingly, native mass spectra indicated that PI(5)P can also bind to the oxidized SAP30L (Supporting Information Fig. S7), having approximately four times higher affinity as compared to holoprotein. The binding was also observed to occur with the other lipids, PI(3)P and PI(4)P. The determined K d values have been reported in Table 3. The tested PIPs have highly similar structures, only differing by the location of the distal phosphate group. They are also the most similar structures compared to the sugar‐phosphate chain of DNA.41 The lipid binding to the oxidized SAP30L further supports the finding that it adopts a folded structure in solution, as suggested based on the native MS data.
We then attempted to examine whether the oxidized SAP30L would have markedly lower or even no measurable affinity to DNA, as would be expected based on the earlier results of the other redox‐regulated repressors and transcription factors. Unfortunately, all attempts to detect DNA binding with native ESI‐MS, either with the oxidized or holo‐SAP30L, failed since protein signals completely disappeared upon adding the 8‐bp DNA to the protein sample. However, we could detect signals from the DNA itself, suggesting that the protein–DNA complex was not efficiently ionized or the protein aggregation had occurred (data not presented). By using NMR, however, we were able to map chemical shift perturbations induced by the binding of the 8‐bp DNA to the holoprotein (Supporting Information Fig. S8). Protein aggregation compelled the use of a reverse titration in this case. The binding affinity was notably higher for DNA than for PI(5)P, given that the cross‐peaks did not significantly shift after an equimolar concentration ratio had been reached. A pattern of perturbations similar to that with PI(5)P were observed, namely, the most affected residues located in the C‐terminal region as well as in the first α‐helix. No attempts were made to characterize the DNA binding to the oxidized SAP30L.
Discussion
The SAP30 family of proteins is highly conserved through species and evolution, especially their N‐terminal zinc finger motif.22 From the sequence alignment (Supporting Information Fig. S1) it is clear that the zinc coordinating cysteine and histidine residues are exceptionally conserved. Using mass spectrometry, we examined if the ZnF in SAP30L can be redox‐regulated through reversible disulfide bond formation, which may have important implications for its structure and function. The N‐terminal ZnF motif of SAP30L is responsible for the DNA‐ and phospholipid‐mediated interactions as well as transcriptional repression. In contrast, the C‐terminal region is involved in protein–protein interactions, including Sin3A association, although most of the interactions have not been studied in detail.16 It has been shown that the disruption of the zinc‐coordinating structure by zinc removal or mutations of the coordinating amino acid residues abolishes the DNA binding and eventually causes degradation of the protein.11 In the reduced form, zinc is coordinated by a CysCys‐X8‐Cys‐X35‐Cys‐X2‐His motif (the coordinating residues are underlined).
On the basis of the present results, the ZnF motif of SAP30L undergoes a redox‐dependent disulfide bond formation upon oxidative stress. The two specific disulfide bonds, Cys29‐Cys30 and Cys38‐Cys74, are formed, involving Cys30, the non‐coordinating but a highly conserved cysteine residue. The disulfide bond between the two adjacent cysteines is particularly interesting. Two adjacent cysteines form a well‐known, redox‐active Cys‐Cys protein motif, which has been frequently reported to occur, especially in ribonucleases.42, 43, 44 The disulfide bond between two adjacent cysteine residues (i.e., a vicinal disulfide bond) is referred to in the literature as a “forbidden” disulfide, involving an eight‐membered ring structure in which the peptide bond typically adopts a highly distorted, non‐planar trans conformation.43, 44, 45, 46 Vicinal disulfides have been reported, for example, in insecticidal atracotoxins,47 the Helicobacter pylori heat shock protein A48 and the human replication protein A (RPA).49 The formation of a vicinal disulfide bond in RPA alters the direction of the backbone and causes a strain in the peptide chain.44
Interestingly, the native MS data suggested that the oxidized SAP30L retains a folded structure in solution,50 which is further supported by the observed affinity toward the PIP ligands. This may have important implications in relation to its function in the Sin3A complex. In the absence of a three‐dimensional structure for the oxidized SAP30L, we modeled the two disulfide bonds into the NMR structure of holo‐SAP30L. The disulfide‐bonded structure was generated by removing the zinc ion and rotating the torsional angles of the cysteine residues to be able to make the two disulfide bonds, followed by an energy‐minimization of the resulting structure by using the GROMOS96 implementation (∼500 deepest descents, no restraints for atom movement) of Swiss‐PdbViewer 4.1.0.51 The modeled structure (see Supporting Information SAP30L_ox.pdb) shows that the formation of these two disulfide bonds is plausible without any major conformational changes in the overall structure (the backbone atom RMSD was 0.68 Å between the two structures). The peptide bond in the eight‐memberd ring of the vicinal Cys29‐Cys30 adopts a clearly distorted trans‐conformation (ψ = 103.2°), consistent with the other reported vicinal disulfide bonds.45, 46 Therefore, the zinc center in SAP30L forms a redox switch, which, upon oxidative stress, releases the coordinated zinc ion with a concomitant formation of the two specific disulfide bonds, a vicinal Cys29‐Cys30 and Cys38‐Cys74 (Fig. 6).
Figure 6.
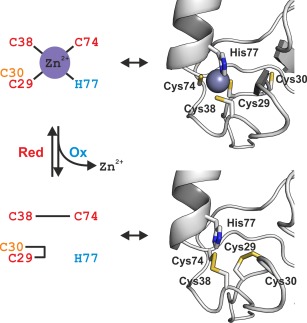
Schematic representation of the redox switch in SAP30L N‐terminal zinc finger motif (left). Upon oxidative stress, the two specific disulfide bonds, a vicinal Cys29‐Cys30 and Cys38‐Cys74, are formed. The zinc sites in both the zinc‐complexed holoprotein (the NMR structure; PDB entry 2N1U) and the oxidized SAP30L (the modeled structure; see text for details) have been presented as cartoon models (right). In the holoprotein, the zinc coordination is perfectly tetrahedral. In the oxidized form, the vicinal Cys29‐Cys30 forms an eight‐membered ring with the ring peptide bond adopting a highly distorted trans‐conformation.
From the phylogenetic analysis, it can be deduced that all SAP30/SAP30L proteins contain the same cysteines, suggesting an evolutionary pressure to conserve these residues.22 Interestingly, the oxidized SAP30L was also able to bind phospholipids that can affect the DNA binding (chromatin association). We have shown that both the ZnF and C‐terminal polybasic NLS motif in SAP30L mediate the lipid binding.11 A similar polybasic region has been found in the PHD1 zinc finger of the Sin3A‐associated protein Pf1.41, 52, 53 In our previous study, it was shown that treating cells with hydrogen peroxide increased the concentration of phosholipids, which led to the relocalization of SAP30L to the cytoplasm.11 Therefore, the disulfide‐mediated redox‐regulation may have a key role in the function of SAP30 family proteins. Such redox‐regulation has been reported for a number of other transcriptional regulators. For example, in transcription factor OxyR54 the formation of disulfide bonds causes a major conformational change that leads to the altered DNA binding. In chaperone Hsp33,55, 56 the oxidation of protein leads to the formation of two disulfide bonds and a concomitant loss of the coordinated zinc ion. The oxidation followed by dimerization leads to the active form of the chaperone. Similarly, in human RPA the oxidation leads to the formation two specific disulfide bonds that regulate the DNA binding.49, 57, 58 Moreover, the reversible disulfide bond formation in several HDACs, directly affecting their repressor function, has also been reported.7, 8 For example, a novel regulatory mechanism mediated by the oxidation of both HDAC4, a class II histone deacetylase, and DnaJb5, a heat shock protein 40, has been reported, related to the attenuation of cardiac hypertrophy.8 SAP30L also contains a nuclear localization signal and the oxidation of the protein might cause conformational changes sufficient to impair cell signaling, similar to the Yap1 protein.59
The observed redox‐regulation of SAP30L suggests a more complex model for its transcriptional activity, depending on the oxidation status of the cells. How this affects the association with chromatin and histones as well as other interacting proteins within the Sin3A complex is presently not understood and will be studied in our future endeavors. It has been previously reported that HDAC1, which is part of the Sin3A complex, is involved in carcinogenesis through a redox‐mediated regulatory mechanism, possibly involving similar disulfide bond formation in response to the oxidative stress.60 Interestingly, it has also been proposed that SAP30 protein may act as a tumor suppressor as well as a viral transmission cofactor, where redox‐regulation may have a role by destabilizing or stabilizing the repressor complex.19, 61
Materials and Methods
Protein production and purification
To produce a SAP30L ZnF motif, an expression construct15 for the full‐length human SAP30L in pGEX‐4T1 vector was modified to produce the residues 25–92 in Escherichia coli Rosetta (DE3) cells. The protein production was carried out in LB broth supplemented with 50 µg mL−1 carbenicillin, 34 µg mL−1 chloramphenicol, and 20 µM ZnCl2. The production was induced with 1 mM isopropyl β‐d−1‐thiogalactopyranoside for 16 h at 28°C. The cell lysate was treated with DNase and RNase, 5 µg mL−1 each, in the presence of 0.2% Triton X‐100, and the GST‐SAP30L fusion protein was recovered from the cleared lysate in the presence of 1 mM dithiothreitol (DTT) using a prepacked glutathione sepharose column (GE Healthcare). The bound fusion protein was eluted with a reduced glutathione, dialysed against a PBS buffer, and digested with thrombin for 16 h at 20°C. Thrombin and GST were removed by benzamidine and glutathione affinity columns (GE Healthcare), and SAP30L was further purified by cation exchange (Resource S column; GE Healthcare) and size exclusion chromatography (Superdex 75 column; GE Healthcare). The purified proteins contain two additional N‐terminal residues (Gly‐Ser) from the thrombin cleavage site. For NMR studies, incorporation of stable isotopes (13C and/or 15N) was carried out as described earlier,62 except that the M9 medium was additionally supplemented with 0.5 g L−1 labelled ISOGRO powder (Sigma–Aldrich) and 20 µM ZnCl2, and pH of the medium was adjusted to 6.8.
Lipid and DNA materials
Dioctaonyl phosphatidylinositol monophosphates (PIPs), bearing the distal phosphate group at the 3‐, 4‐, or 5‐position of the inositol ring (named hereafter as PI(3)P, PI(4)P and PI(5)P, respectively), were purchased from Echelon Biosciences (Salt Lake City, UT) and used without further purification. An eight‐base pair, self‐annealing DNA duplex (5′‐GCGGCCGC) was purchased from Sigma–Aldrich (St. Louis, MO) and used as received.
Protease digestions
For the disulfide bond analysis, 150 µL of SAP30L in 20 mM NH4OAc (of 5 µM protein) was incubated with TPCK‐treated trypsin (Sigma–Aldrich, St. Louis, MO) at a protease‐to‐substrate ratio of 1:5 to 1:40 (w/w) at room temperature from 15 min up to overnight. To quench the reaction, the digest solutions were mixed with 150 µL of MeCN containing 1% HOAc and directly analyzed without chromatographic separation. On‐line pepsin digestion was performed by using a previously described protocol.63 The numbering used for the proteolytic peptides refers to the amino acid sequence of the used SAP30L construct rather than the full‐length protein.
Mass spectrometry
Sample preparation
The protein samples were desalted for mass spectrometry by using PD‐10 desalting columns (Amersham Biosciences, Uppsala, Sweden), equilibrated in advance with 20 mM ammonium acetate (NH4OAc) buffer (pH 6.8) and further concentrated with 3000 MWCO centrifugal filter devices (Amicon Ultra; Millipore, Billerica, MA). Protein concentrations were determined spectrophotometrically from the absorbance at 280 nm by using sequence‐derived extinction coefficients (2560 M −1 cm−1 for the native/denatured, and 2800 M −1 cm−1 for the oxidized protein). The protein samples were further diluted with MeCN/H2O/HOAc (49.5:49.5:1.0, v/v) for denaturing or 20 mM NH4OAc (pH 6.8) for native ESI‐MS experiments. Zinc chelation was obtained using EDTA or 1,10‐phenanthroline and in vitro oxidation was carried out with hydrogen peroxide (H2O2).
ESI FT‐ICR MS experiments
All mass spectrometric experiments were performed on a 4.7‐T hybrid quadrupole FT‐ICR mass spectrometer (Bruker Apex‐Qe; Bruker Daltonics, Billerica, MA), equipped with an ESI source (Apollo‐II; Bruker Daltonik GmbH, Bremen, Germany). The instrument was also equipped with an indirectly heated hollow dispenser cathode for ECD‐MS/MS. The samples were infused directly to the ion source by using a syringe pump at a flow rate of 1.5 µL min−1 with N2 gas serving as nebulizing and drying gas. The ESI‐generated ions were externally accumulated in a hexapole ion trap for 0.1 s and transferred to the Infinity ICR cell for trapping, excitation and detection. Up to 512 coadded time‐domain transients (512‐kWord each) were fast Fourier transformed prior to magnitude calculation and external mass calibration with respect to the ions of an ES Tuning Mix (Agilent Technologies, Santa Clara, CA). All data were acquired and processed by using Bruker XMASS 7.0.8 software. Charge‐deconvolution was accomplished by using a built‐in Tcl‐script. The peptides from trypsin/pepsin digestions were identified based on their monoisotopic masses by using GPMAW 7.10 software (Lighthouse Data, Odense, Denmark). For ECD‐MS/MS experiments, the precursor ions were mass‐selectively accumulated in the collision cell (quadrupole isolation window of 5 m/z units) and irradiated with low‐energy electrons (cathode bias 0.6 V and irradiation time 0.2 s) inside the ICR cell. All masses of the intact proteins have been reported for the most abundant isotopologue, averaged over all detected charge states, while masses for the proteolytic peptides have been reported as monoisotopic masses.
Lipid and DNA binding experiments
The solid phospholipids (PIPs) were dissolved in 20 mM NH4OAc to the concentration of 1.5 mM and kept frozen until used. The lipid binding experiments were conducted by mixing small aliquots of the lipid and protein stock solutions to obtain a final concentration of 5 µM for both SAP30L and a PIP ligand and directly analyzed. As low lipid concentrations as possible were used in the binding experiments, in order to prevent lipid self‐association, as well as nonspecific association with the protein. Before mass spectrometric measurements, the protein–lipid solutions were heated at 50°C for 15 min to dissociate possible lipid micelles (see, Results for details). The binding affinity for each lipid was calculated from an average ESI‐MS intensity (I) ratio R of the protein–lipid complex PL to the free protein P (R = I PL/I P) from ∼70 consecutive measurements.64 The equilibrium dissociation constant (K d) was obtained from the equation K d = [P][L]/[PL] (1), where [P], [L], and [PL] are the equilibrium concentrations of the free protein, the free lipid and the protein–lipid complex, respectively. [PL] can be obtained from the equation [PL] = R[P]0/(1 + R) (2), in which [P]0 is the initial protein concentration. Taking into account that [P] = [P]0 – [PL] and [L] = [L]0 – [PL], one can obtain K d from the equation (1). The approximation I PL/I P ≈ [PL]/[P] is valid if one assumes there is the same ionization and transmission efficiencies for PL and P, which is typically true if the ligand is much smaller than the protein. All ion source parameters were carefully optimized in order to avoid unintentional dissociation of the weak noncovalent complexes within the high‐pressure regions. The DNA binding experiments were performed by mixing small aliquots of protein and DNA stock solutions and further diluted with 20 mM NH4OAc (pH 6.8) to obtain a final concentration of 5 µM for both SAP30L and the DNA duplex.
NMR spectroscopy
Resonance assignment and solution structure for SAP30L
The NMR assignment was performed by using standard triple resonance experiments HNCA, HN(CO)CA, HNCACB, HN(CO)CACB for the backbone and H(CCO)NH, C(CO)NH, HCCH‐COSY (separate spectra for aliphatic and aromatic regions), (HB)CB(CGCD)HD and (HB)CB(CGCDCE)HE for the side‐chains. All NMR spectra were recorded on a Varian INOVA 800 MHz spectrometer equipped with a cryogenic probe head. All experiments were conducted at 25°C in 20 mM Bis‐Tris buffer with 30 mM NaCl, 7.5% D2O, 0.5 mM ZnCl2, and 0.04% NaN3 (pH 6.0). The distance restraints for automatic NOE assignment–structure calculation with the program Cyana 2.134 were derived from 1H,15N NOESY‐HSQC and 1H,13C NOESY‐HSQC. Distance restraints were added for Zn−Sγ (2.25 − 2.35 Å), Zn−Nδ1 (1.95 − 2.05 Å) and Sγ−Cβ (3.25 − 3.65 Å) and the zinc coordination was constrained to be approximately tetrahedral. Out of 300 structures generated with Cyana, 30 with the lowest target function were subsequently energy‐minimized in vacuum with AMBER 12.65 The 15 lowest‐energy structures were selected to represent the SAP30L structure in solution. The NMR chemical shift and structural data have been deposited under the accession numbers 25575 (BMRB) and 2N1U (PDB).
Lipid and DNA binding experiments
The phospholipid binding to 60 µM 15N‐labeled SAP30L was studied by monitoring chemical shift perturbations in the 1H,15N HSQC spectra acquired at 25°C. The protein in a buffer containing 30 mM NaCl, 20 mM Bis‐Tris‐HCl, 7.5% D2O, 60 µM ZnCl2, 0.05% NaN3 (pH 6.0) was mixed with PI(5)P to obtain 1:1, 2:1, 3:1, and 4:1 PI(5)P to SAP30L concentration ratios. To avoid protein aggregation and precipitation at low ligand‐to‐protein ratios, the titration was conducted in a reversed fashion, that is, the sample with a 4:1 PI(5)P to SAP30L ratio was measured first, and 75% of this sample was added to 25% of PI(5)P‐free sample to obtain a 3:1 ratio, and so forth. The DNA binding of holo‐SAP30L was studied in a similar fashion using a self‐annealing oligonucleotide (5′‐GCGGCCGC). As compared to the phospholipid titration experiments, an additional ligand‐to‐protein concentration ratio of 0.5:1 was included here. All chemical shift deviations were calculated from Δδ = (ΔδH2 + (0.154 ΔδN)2)1/2.
Supporting information
Supporting Information
Acknowledgment
The FT‐ICR MS instrument is a technology platform of Biocenter Finland and part of the core services of Structural Biology Center, a Biocenter Kuopio core facility.
References
- 1. Barford D (2004) The role of cysteine residues as redox‐sensitive regulatory switches. Curr Opin Struct Biol 14:679–686. [DOI] [PubMed] [Google Scholar]
- 2. Brandes N, Schmitt S, Jakob U (2008) Thiol‐based redox switches in eukaryotic proteins. Antioxid Redox Signal 11:997–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maret W (2006) Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid Redox Signal 8:1419–1441. [DOI] [PubMed] [Google Scholar]
- 4. Wouters MA, Fan SW, Haworth NL (2010) Disulfides as redox switches: from molecular mechanisms to functional significance. Antioxid Redox Signal 12:53–91. [DOI] [PubMed] [Google Scholar]
- 5. Krishna SS, Majumdar I, Grishin NV (2003) Structural classification of zinc fingers. Nucleic Acids Res 31:532–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Laitaoja M, Valjakka J, Jänis J (2013) Zinc coordination spheres in protein structures. Inorg Chem 52:10983–10991. [DOI] [PubMed] [Google Scholar]
- 7. Doyle K, Fitzpatrick FA (2010) Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J Biol Chem 285:17417–17424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, Vatner SF, Sadoshima J (2008) A redox‐dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 133:978–993. [DOI] [PubMed] [Google Scholar]
- 9. Grzenda A, Lomberk G, Zhang J‐S, Urrutia R (2009) Sin3: master scaffold and transcriptional corepressor. Biochim Biophys Acta 1789:443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McDonel P, Costello I, Hendrich B (2009) Keeping things quiet: roles of NuRD and Sin3 co‐repressor complexes during mammalian development. Int J Biochem Cell Biol 41:108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Viiri KM, Jänis J, Siggers T, Heinonen TYK, Valjakka J, Bulyk ML, Mäki M, Lohi O (2009) DNA‐binding and ‐bending activities of SAP30L and SAP30 are mediated by a zinc‐dependent module and monophosphoinositides. Mol Cell Biol 29:342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shiio Y, Rose DW, Aur R, Donohoe S, Aebersold R, Eisenman RN (2006) Identification and characterization of SAP25, a novel component of the mSin3 corepressor complex. Mol Cell Biol 26:1386–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lindfors K, Viiri KM, Niittynen M, Heinonen TY, Mäki M, Kainulainen H (2003) TGF‐β induces the expression of SAP30L, a novel nuclear protein. BMC Genom 4:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Laherty CD, Billin AN, Lavinsky RM, Yochum GS, Bush AC, Sun J‐M, Mullen T‐M, Davie JR, Rose DW, Glass CK, Rosenfeld MG, Ayer DE, Eisenman RN (1998) SAP30, a component of the mSin3 corepressor complex involved in N‐CoR‐mediated repression by specific transcription factors. Mol Cell 2:33–42. [DOI] [PubMed] [Google Scholar]
- 15. Viiri KM, Korkeamäki H, Kukkonen MK, Nieminen LK, Lindfors K, Peterson P, Mäki M, Kainulainen H, Lohi O (2006) SAP30L interacts with members of the Sin3A corepressor complex and targets Sin3A to the nucleolus. Nucleic Acids Res 34:3288–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xie T, He Y, Korkeamäki H, Zhang Y, Imhoff R, Lohi O, Radhakrishnan I (2011) Structure of the 30‐kDa Sin3‐associated protein (SAP30) in complex with the mammalian Sin3A corepressor and its role in nucleic acid binding. J Biol Chem 286:27814–27824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuzmichev A, Zhang Y, Erdjument‐Bromage H, Tempst P, Reinberg D (2002) Role of the Sin3‐histone deacetylase complex in growth regulation by the candidate tumor suppressor p33ING1. Mol Cell Biol 22:835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang NE, Lin C‐H, Lin Y‐S, Yu WCY (2003) Modulation of YY1 activity by SAP30. Biochem Biophys Res Commun 306:267–275. [DOI] [PubMed] [Google Scholar]
- 19. Le May N, Mansuroglu Z, Léger P, Josse T, Blot G, Billecocq A, Flick R, Jacob Y, Bonnefoy E, Bouloy M (2008) A SAP30 complex inhibits IFN‐β expression in rift valley fever virus infected cells. PLoS Pathog 4:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sichtig N, Körfer N, Steger G (2007) Papillomavirus binding factor binds to SAP30 and represses transcription via recruitment of the HDAC1 co‐repressor complex. Arch Biochem Biophys 467:67–75. [DOI] [PubMed] [Google Scholar]
- 21. Teittinen KJ, Grönroos T, Parikka M, Junttila S, Uusimäki A, Laiho A, Korkeamäki H, Kurppa K, Turpeinen H, Pesu M, Gyenesei A, Rämet M, Lohi O (2012) SAP30L (Sin3A‐associated protein 30‐like) is involved in regulation of cardiac development and hematopoiesis in zebrafish embryos. J Cell Biochem 113:3843–3852. [DOI] [PubMed] [Google Scholar]
- 22. Viiri KM, Heinonen TY, Mäki M, Lohi O (2009) Phylogenetic analysis of the SAP30 family of transcriptional regulators reveals functional divergence in the domain that binds the nuclear matrix. BMC Evol Biol 9:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. He Y, Imhoff R, Sahu A, Radhakrishnan I (2009) Solution structure of a novel zinc finger motif in the SAP30 polypeptide of the Sin3 corepressor complex and its potential role in nucleic acid recognition. Nucleic Acids Res 37:2142–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grishin NV (2001) Treble clef finger—a functionally diverse zinc‐binding structural motif. Nucleic Acids Res 29:1703–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liew CK, Crossley M, Mackay JP, Nicholas HR (2007) Solution structure of the THAP domain from Caenorhabditis elegans C‐terminal binding protein (CtBP). J Mol Biol 366:382–390. [DOI] [PubMed] [Google Scholar]
- 26. Bae J‐B, Park J‐H, Hahn M‐Y, Kim M‐S, Roe J‐H (2004) Redox‐dependent changes in RsrA, an anti‐sigma factor in Streptomyces coelicolor: zinc release and disulfide bond formation. J Mol Biol 335:425–435. [DOI] [PubMed] [Google Scholar]
- 27. Heck AJR (2008) Native mass spectrometry: a bridge between interactomics and structural biology. Nat Methods 5:927–933. [DOI] [PubMed] [Google Scholar]
- 28. Hopper JTS, Robinson CV (2014) Mass spectrometry quantifies protein interactions—From molecular chaperones to membrane porins. Angew Chem Int Ed 53:14002–14015. [DOI] [PubMed] [Google Scholar]
- 29. Marshall AG, Hendrickson CL, Jackson GS (1998) Fourier transform ion cyclotron resonance mass spectrometry: a primer. Mass Spec Rev 17:1–35. [DOI] [PubMed] [Google Scholar]
- 30. Kaltashov IA, Eyles SJ (2002) Studies of biomolecular conformations and conformational dynamics by mass spectrometry. Mass Spec Rev 21:37–71. [DOI] [PubMed] [Google Scholar]
- 31. Fabris D, Hathout Y, Fenselau C (1999) Investigation of zinc chelation in zinc‐finger arrays by electrospray mass spectrometry. Inorg Chem 38:1322–1325. [DOI] [PubMed] [Google Scholar]
- 32. Simonson T, Calimet N (2002) CysxHisy‐Zn2+ interactions: thiol vs. thiolate coordination. Proteins 49:37–48. [DOI] [PubMed] [Google Scholar]
- 33. Kornhaber G, Snyder D, Moseley H, Montelione G (2006) Identification of zinc‐ligated cysteine residues based on 13Cα and 13Cβ chemical shift data. J Biomol NMR 34:259–269. [DOI] [PubMed] [Google Scholar]
- 34. López‐Méndez B, Güntert P (2006) Automated protein structure determination from NMR spectra. J Am Chem Soc 128:13112–13122. [DOI] [PubMed] [Google Scholar]
- 35. Kneller JM, Lu M, Bracken C (2002) An effective method for the discrimination of motional anisotropy and chemical exchange. J Am Chem Soc 124:1852–1853. [DOI] [PubMed] [Google Scholar]
- 36. Lee S‐H, Maret W (2001) Redox control of zinc finger proteins: mechanisms and role in gene regulation. Antioxid Redox Signal 3:531–534. [Google Scholar]
- 37. Ilbert M, Graf PCF, Jakob U (2006) Zinc center as redox switch—new function for an old motif. Antioxid Redox Signal 8:835–846. [DOI] [PubMed] [Google Scholar]
- 38. Kaltashov IA, Mohimen A (2005) Estimates of protein surface areas in solution by electrospray ionization mass spectrometry. Anal Chem 77:5370–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zubarev RA, Kruger NA, Fridriksson EK, Lewis MA, Horn DM, Carpenter BK, McLafferty FW (1999) Electron capture dissociation of gaseous multiply‐charged proteins is favored at disulfide bonds and other sites of high hydrogen atom affinity. J Am Chem Soc 121:2857–2862. [Google Scholar]
- 40. Li Y, Heitz F, Le Grimellec C, Cole RB (2005) Fusion peptide−phospholipid noncovalent interactions as observed by nanoelectrospray FTICR−MS. Anal Chem 77:1556–1565. [DOI] [PubMed] [Google Scholar]
- 41. Viiri K, Mäki M, Lohi O (2012) Phosphoinositides as regulators of protein‐chromatin interactions. Sci Signal 5:pe19. [DOI] [PubMed] [Google Scholar]
- 42. Lima WF, Wu H, Nichols JG, Manalili SM, Drader JJ, Hofstadler SA, Crooke ST (2003) Human RNase H1 activity is regulated by a unique redox switch formed between adjacent cysteines. J Biol Chem 278:14906–14912. [DOI] [PubMed] [Google Scholar]
- 43. Kim B‐M, Schultz LW, Raines RT (1999) Variants of ribonuclease inhibitor that resist oxidation. Protein Sci 8:430–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Park C, Raines RT (2001) Adjacent cysteine residues as a redox switch. Protein Eng 14:939–942. [DOI] [PubMed] [Google Scholar]
- 45. Carugo O, Čemažar M, Zahariev S, Hudáky I, Gáspári Z, Perczel A, Pongor S (2003) Vicinal disulfide turns. Protein Eng 16:637–639. [DOI] [PubMed] [Google Scholar]
- 46. Hudáky I, Gáspári Z, Carugo O, Čemažar M, Pongor S, Perczel A (2004) Vicinal disulfide bridge conformers by experimental methods and by ab initio and DFT molecular computations. Proteins 55:152–168. [DOI] [PubMed] [Google Scholar]
- 47. Wang X, Connor M, Smith R, Maciejewski MW, Howden MEH, Nicholson GM, Christie MJ, King GF (2000) Discovery and characterization of a family of insecticidal neurotoxins with a rare vicinal disulfide bridge. Nat Struct Mol Biol 7:505–513. [DOI] [PubMed] [Google Scholar]
- 48. Loguercio S, Dian C, Flagiello A, Scannella A, Pucci P, Terradot L, Zagari A (2008) In HspA from Helicobacter pylori vicinal disulfide bridges are a key determinant of domain B structure. FEBS Lett 582:3537–3541. [DOI] [PubMed] [Google Scholar]
- 49. Men L, Roginskaya M, Zou Y, Wang Y (2007) Redox‐dependent formation of disulfide bonds in human replication protein A. Rapid Commun Mass Spectrom 21:2743–2749. [DOI] [PubMed] [Google Scholar]
- 50. Baldwin RL, Rose GD (2013) Molten globules, entropy‐driven conformational change and protein folding. Curr Opin Struct Biol 23:4–10. [DOI] [PubMed] [Google Scholar]
- 51. Guex N, Peitsch MC (1997) SWISS‐MODEL and the Swiss‐Pdb viewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723. [DOI] [PubMed] [Google Scholar]
- 52. Gozani O, Karuman P, Jones DR, Ivanov D, Cha J, Lugovskoy AA, Baird CL, Zhu H, Field SJ, Lessnick SL, Villasenor J, Mehrotra B, Chen J, Rao VR, Brugge JS, Ferguson CG, Payrastre B, Myszka DG, Cantley LC, Wagner G, Divecha N, Prestwich GD, Yuan J (2003) The PHD finger of the chromatin‐associated protein ING2 functions as a nuclear phosphoinositide receptor. Cell 114:99–111. [DOI] [PubMed] [Google Scholar]
- 53. Kaadige MR, Ayer DE (2006) The polybasic region that follows the plant homeodomain zinc finger 1 of Pf1 is necessary and sufficient for specific phosphoinositide binding. J Biol Chem 281:28831–28836. [DOI] [PubMed] [Google Scholar]
- 54. Lee C, Lee SM, Mukhopadhyay P, Kim SJ, Lee SC, Ahn W‐S, Yu M‐H, Storz G, Ryu SE (2004) Redox regulation of OxyR requires specific disulfide bond formation involving a rapid kinetic reaction path. Nat Struct Mol Biol 11:1179–1185. [DOI] [PubMed] [Google Scholar]
- 55. Barbirz S, Jakob U, Glocker MO (2000) Mass spectrometry unravels disulfide bond formation as the mechanism that activates a molecular chaperone. J Biol Chem 275:18759–18766. [DOI] [PubMed] [Google Scholar]
- 56. Graumann J, Lilie H, Tang X, Tucker KA, Hoffmann JH, Vijayalakshmi J, Saper M, Bardwell JCA, Jakob U (2001) Activation of the redox‐regulated molecular chaperone hsp33—a two‐step mechanism. Structure 9:377–387. [DOI] [PubMed] [Google Scholar]
- 57. Park J‐S, Wang M, Park S‐J, Lee S‐H (1999) Zinc finger of replication protein A, a non‐DNA binding element, regulates its DNA binding activity through redox. J Biol Chem 274:29075–29080. [DOI] [PubMed] [Google Scholar]
- 58. Li W, Bottrill AR, Bibb MJ, Buttner MJ, Paget MSB, Kleanthous C (2003) The role of zinc in the disulphide stress‐regulated anti‐sigma factor RsrA from Streptomyces coelicolor . J Mol Biol 333:461–472. [DOI] [PubMed] [Google Scholar]
- 59. Wood MJ, Storz G, Tjandra N (2004) Structural basis for redox regulation of Yap1 transcription factor localization. Nature 430:917–921. [DOI] [PubMed] [Google Scholar]
- 60. Kato T, Shimono Y, Hasegawa M, Jijiwa M, Enomoto A, Asai N, Murakumo Y, Takahashi M (2009) Characterization of the HDAC1 complex that regulates the sensitivity of cancer cells to oxidative stress. Cancer Res 69:3597–3604. [DOI] [PubMed] [Google Scholar]
- 61. Sironi E, Cerri A, Tomasini D, Sirchia SM, Porta G, Rossella F, Grati FR, Simoni G (2004) Loss of heterozygosity on chromosome 4q32–35 in sporadic basal cell carcinomas: evidence for the involvement of p33ING2/ING1L and SAP30 genes. J Cutan Pathol 31:318–322. [DOI] [PubMed] [Google Scholar]
- 62. Leppänen V‐M, Tossavainen H, Permi P, Lehtiö L, Rönnholm G, Goldman A, Kilpeläinen I, Pihlajamaa T (2007) Crystal structure of the N‐terminal NC4 domain of collagen IX, a zinc binding member of the laminin‐neurexin‐sex hormone binding globulin (LNS) domain family. J Biol Chem 282:23219–23230. [DOI] [PubMed] [Google Scholar]
- 63. Marie G, Serani L, Laprévote O (2000) An on‐line protein digestion device for rapid peptide mapping by electrospray mass spectrometry. Anal Chem 72:5423–5430. [DOI] [PubMed] [Google Scholar]
- 64. Sun J, Kitova EN, Wang W, Klassen JS (2006) Method for distinguishing specific from nonspecific protein−ligand complexes in nanoelectrospray ionization mass spectrometry. Anal Chem 78:3010–3018. [DOI] [PubMed] [Google Scholar]
- 65. Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ (2005) The Amber biomolecular simulation programs. J Comput Chem 26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information