

Abstract
Mitochondria are considered as the primary source of reactive oxygen species (ROS) in nearly all eukaryotic cells during respiration. The harmful effects of these compounds range from direct neurotoxicity to incorporation into proteins producing aberrant molecules with multiple physiological problems. Phenylalanine exposure to ROS produces multiple oxidized isomers: tyrosine, Levodopa, ortho‐Tyr, meta‐Tyr (m‐Tyr), and so on. Cytosolic phenylalanyl‐tRNA synthetase (PheRS) exerts control over the translation accuracy, hydrolyzing misacylated products, while monomeric mitochondrial PheRS lacks the editing activity. Recently we showed that “teamwork” of cytosolic and mitochondrial PheRSs cannot prevent incorporation of m‐Tyr and l‐Dopa into proteins. Here, we present human mitochondrial chimeric PheRS with implanted editing module taken from EcPheRS. The monomeric mitochondrial chimera possesses editing activity, while in bacterial and cytosolic PheRSs this type of activity was detected for the (αβ)2 architecture only. The fusion protein catalyzes aminoacylation of tRNAPhe with cognate phenylalanine and effectively hydrolyzes the noncognate aminoacyl‐tRNAs: Tyr‐tRNAPhe and m‐Tyr‐tRNAPhe.
Keywords: chimera, aminoacyl‐tRNA synthetases, ROS‐damaged amino acid, fusion protein, aminoacylation, editing
Introduction
The aminoacyl‐tRNA synthetases (aaRSs) play a fundamental role in the translation of the genetic code, catalyzing the attachment of the correct amino acid (aa) to its cognate tRNA in a two‐step reaction.1 At the first step, the activation of the aa by ATP gives rise to the formation of an enzyme‐bound aminoacyl adenylate. At the second step, the aa moiety is transferred to the 3′‐terminal ribose of the cognate tRNA, forming the aminoacyl‐tRNA. Prior to activation, at the aa recognition step, some aaRSs face a challenge of discrimination among aa with closely similar chemical structure. To ensure a high level of fidelity in protein biosynthesis, aaRSs developed an editing activity associated with cleavage of the ester bond between the carbonyl carbon of aminoacyl moiety and the oxygen atom of terminal ribose.2, 3, 4 In majority of aaRSs hydrolysis occurs at a site located 30–40 Å away from the aminoacylation active site and it is associated with a structural domain or with interdomain interface.5
In eukaryotes, protein synthesis proceeds concurrently both in the cytoplasm and in the organelles, such as mitochondria and chloroplasts.6 Mitochondrial aaRSs (mit‐aaRSs) are encoded in the nucleus and posttranslationally transported into the organelle. In individual cases, mit‐aaRSs are essentially identical to their cytosolic counterparts, as with the yeast mit‐ValRS,7 mit‐HisRS,8 human mit‐GlyRS,9 and plant mit‐ThrRS.10 However, it is more common situation when eukaryotic mit‐aaRSs exhibit higher homology to the corresponding bacterial enzymes, other than cytosolic analogs from the same organism. Human mit‐MetRS for one exhibits homology to the bacterial MetRS rather than to the cytosolic MetRS although it lacks a 100‐aa‐long dimerization fragment found in the former. Thus, in mitochondria it functions as a monomer, and not as a homodimer, as it does in bacteria.11 Two independent coding sequences have been revealed for mit‐GluRS and mit‐ProRS, as distinct from a single gene with both activities in the human cytosol associated with appearance of bifunctional GluProRS.12
Notable attention to the mitochondrial systems is stimulated by progressive accumulation of reports that organelles are considered as the primary source of reactive oxygen species (ROS) in nearly all cells during respiration. Thus, it should not be all that surprising that mitochondrial‐produced ROS‐damaged aa and nucleotides are conventionally regarded as having pathological potentials.13 ROS‐mediated oxidation can lead to hydroxylation of aromatic groups and aliphatic aa side chains, nitration of aromatic aa residues, chlorination of aromatic groups and primary amino groups, and to conversion of some aa residues to carbonyl derivatives.14 Among other metabolites, phenylalanine exposure to ROS‐generating systems produces multiple isomers of tyrosine‐m‐Tyr, o‐Tyr, l‐Dopa, and so on—which have been widely used as indexes of oxidative damage in tissue proteins. Recently we provided evidence that eukaryotic mitochondrial and cytoplasmic phenylalanyl‐tRNA synthetases (PheRS) catalyze direct attachment of m‐Tyr and l‐Dopa to tRNAPhe, thereby opening up the way for delivery of misacylated tRNAs to the ribosome and incorporation of nonprotein aa into eukaryotic proteins.15, 16 Such possibility is related to unique structure‐function characteristics of the eukaryotic mitochondrial and cytoplasmic PheRSs.17, 18, 19 Human mitochondrial PheRS (HsmtPheRS) is the smallest known nuclear‐encoded synthetase (415 aa) exhibiting higher homology to bacterial PheRSs than to the corresponding cytosolic counterpart. Similar to other PheRSs, HsmtPheRS has specific sequence motifs of a class II aaRS. However, while the bacterial enzyme is an (αβ)2 heterodimer,20, 21 the HsmtPheRS homolog is a single‐chain enzyme, composed of the α‐subunit and the anticodon binding domain (ABD) from the β‐subunit of the bacterial PheRS.22
Mitochondrial protein biosynthesis machinery lacks an editing activity against the misaminoacylated Tyr‐tRNAPhe. Biochemical and structural data evidenced that editing module (EMD) has been lost during the evolution of HsmtPheRS.23 Thus, HsmtPheRS does not secure against incorporation of m‐Tyr, l‐Dopa, and apparently other nonprotein aa into polypeptide chain, as cytosolic homolog does. However, in some cases, mitochondrial aaRSs can efficiently discriminate at the active site the cognate amino acid from the noncognate one and thus, do not need extra mechanism to edit misacylated tRNA. For example, mitochondrial PheRS efficiently discriminates Tyr from Phe at the aminoacylation active site: the catalytic efficiency (k cat/K m) of Tyr attachment is ∼103 times lower than that of the correct amino acid, primarily because of higher K m value.16 At the same time human cytosolic PheRS aminoacylates tRNAPhe with nonprotein aa m‐Tyr, but its EMD is unable to hydrolyze m‐Tyr‐tRNAPhe, thereby not securing polypeptide chain against m‐Tyr incorporation in it (ibid). Thus, different cellular compartments have distinct tolerances for a translation inaccuracy. Despite evidence that even mild mis‐incorporation may have a severe impact on cellular viability proofreading activities are not universally conserved in aaRSs. Moreover, for each aaRS it depends on the stereochemical characteristics of individual amino acids, and should be analyzed and investigated on a one‐by‐one basis. In order to enhance the protective characteristics in eukaryotic cells, we generated a chimera protein composed of the catalytic module (CAM) and the anticodon binding domain (ABD) from HsmtPheRS, and EMD adopted from Escherichia coli PheRS (EcPheRS).24 The new fusion protein acylates tRNAPhe with Phe, and at the same time demonstrates specific editing activity against noncognate aminoacyl‐tRNAs: Tyr‐tRNAPhe and m‐Tyr‐tRNAPhe.
Results
Activity of the EMD and its fragments
Prior to assembly of chimera protein, we examined whether the standalone editing module EMD composed of 5 structural domains (B1–B5) or its various fragments (B1–B4 сomposed of 4 domains and the smallest one composed of B3/B4 module) retain the ability to hydrolyze misacylated tRNAPhe. The individual fragments of EcPheRS β‐subunit24 were expressed in E. coli. To investigate their possible editing activity, hydrolysis of presynthesized radiolabeled Tyr‐tRNAPhe was followed (Figure 1A,B). Specific deacylation of Tyr‐tRNAPhe exceeding the spontaneous degradation was detected only in the presence of high concentrations of EMD or its fragments. The apparent rate constants estimated from the kinetic data are very similar for EMD and its fragment B1–B4 (0.026 and 0.023 min−1, respectively). For comparison, hydrolysis of Tyr‐tRNAPhe in the presence of EcPheRS or TtPheRS was investigated in identical reaction conditions (data not shown). These experiments revealed that the editing activity of both wild‐type enzymes is much more (∼three orders of magnitude) higher than that of the individual EMD or its fragment.
Figure 1.
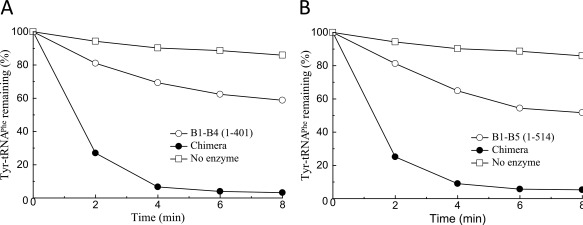
Editing activity of EMD fragments of EcPheRS against Tyr‐tRNAPhe: (A) B1–B5 and (B) B1–B4. Reactions were performed with 1.2 µM Tyr‐tRNAPhe prepared from in vitro transcribed E. coli tRNAPhe, with the addition of 4 µM B1–B5, 4 µM B1–B4, and 1.2 µM chimera or in the absence of enzyme.
It is apparent that the very low editing activity of EMD and its fragment is accounted by the lack of multiple stabilizing contacts with tRNAPhe formed in the complex with wild‐type enzyme. Previously, editing activities of various fragments of EMD isolated from EcPheRS toward Tyr‐tRNATyr have been explored.25 The only fragment which possessed no editing activity against Tyr‐tRNATyr was the B1–B5 module. Availability of the hydrolytic activity demonstrated by the B3/B4 fragment toward Tyr‐tRNATyr in the absence of B2 domain looks unique, considering its contribution to stabilizing mischarged tRNA on the pathway to the editing site of PheRS.26
Design of the chimeric protein
Design of monomeric mitochondrial chimera keeping the dual functions of aminoacylation and proofreading requires an in‐depth analysis of heterotetrameric bacterial and monomeric mitochondrial PheRSs. The crystal structure of TtPheRS complexed with cognate tRNA revealed that one tRNAPhe molecule binds across all four PheRS subunits.21 In particular, the CCA end of tRNA is located within the CAM of α‐subunit, while the anticodon stem is recognized by the C‐terminal domain of the β‐subunit (ABD). Binding and hydrolysis of misacylated tRNA species, on both cis‐ and trans‐editing pathways, are related to the β‐subunit. HsmtPheRS aminoacylates CCA‐end of tRNA and provides recognition of the anticodon stem within the same monomer.18 The editing site of the tetrameric enzyme is localized at the B3/B4 interface, 35 Å from the aminoacylation active site.27 The major architectural problem upon construction of monomeric mitochondrial PheRS chimera having desired hydrolytic activity lies in the placement of the editing site at the correct distance from the aminoacylation site, i.e., at the distance analogous to that observed in the tetrameric enzyme. The crystal structure of HsmtPheRS‐tRNAPhe complex, cross‐linking the catalytic domain with ABD and data on small angle scattering experiments give proof to the idea that formation of the complex with tRNA requires a significant rearrangement of the ABD from the “closed” (Figure 2A) to the productive “open” conformation (Figure 2B).17, 18, 28 Thus, flexibility of the anticodon binding domain in monomeric HsmtPheRS is an additional complication in the chimera design.
Figure 2.
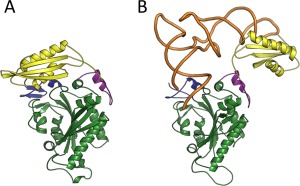
(A) “Closed” and (B) “Open” conformations of HmstPheRS. The “Open” conformation is depicted in complex with tRNAPhe (PDB codes: 3MCQ and 3TUP accordingly). CAM colored green, ABD colored yellow, while tRNA complexed with “Open” state of HmstPheRS colored brown.
Our initial experimental trial consisted in transplantation of B3/B4 fragment into the monomeric enzyme. The insertion point within the aa sequence of HsmtPheRS varied over a wide range of residues and structural elements. However, aminoacylation and requested editing activities were not working in tandem. After a few attempts we transplanted the whole structural module engaging B1–B5 domains into HsmtPheRS. Complementary point for a given solution was supported by the data indicative of the B2 domain involvement in the tRNA pathway from the aminoacylation site to the editing site in EcPheRS.26 The “prototype” model of chimera presents the combination of two structural blocks of HsmtPheRS as a whole and B1–B5 module of the EcPheRS β‐subunit (Figure 3).
Figure 3.
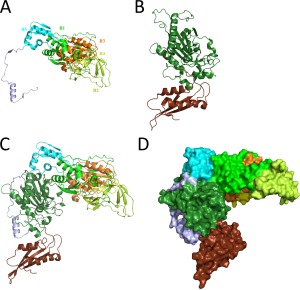
Overview of assembly process for human mitochondrial chimeric PheRS. (A) Structural module EMD from EcPheRS. Components of the module–structural domains B1–B5 depicted in different colors and marked accordingly. (B) Overall structure of the HsmtPheRS enzyme in “closed” inactive conformation. (C) The 3D‐model of human mitochondrial chimeric PheRS ribbon representation. (D) The 3D‐model space‐filling representation.
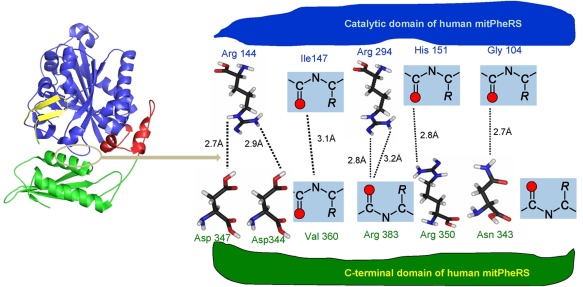
Chimera protein was modeled in both close and open conformations using apo form of HsmtPheRS and its complex with cognate tRNAPhe. The trial structures were further adjusted by remodeling external loops with Modeller29 and refined using molecular dynamics protocols (see Materials and Methods). Transition between inactive closed and active open conformational states is related to formation and disruption of the hydrogen bonding and salt bridges network at the interface between ABD and CAM. There is a remarkable arginine‐rich interface between the ABD and CAM in closed state17 (Figure 5). The arginine residues located on both sides of the domains forming HB and salt bridges with negatively charged carbonyl oxygens, and carboxyl groups of Asp347 and Asp 344. The Arg144, Arg294, Arg350, and Asn343 making hydrogen bond network at the interface are strictly conserved in all known aa sequences of eukaryotic mitochondrial PheRSs.
Figure 5.
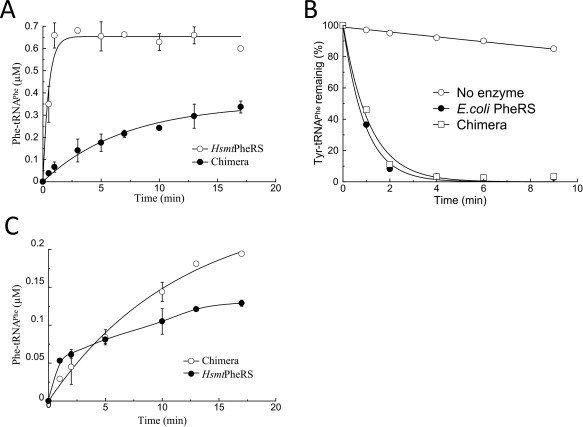
Aminoacylation and editing activities of chimera protein in comparison with various wild‐type PheRSs: (A) Charging of E. coli tRNAPhe transcript (0.8 μM) with Phe by chimera (0.4 µM) or HsmtPheRS (0.4 µM). Reactions were performed at 37°C in the presence of 5 mM ATP and 4 μM [3H]Phe; (B) Editing activities of chimera (1 µM) and EcPheRS (20 nM) toward exogenous Tyr‐tRNAPhe (1.2 μM); (C) Reaminoacylation of m‐Tyr‐tRNAPhe with Phe by chimera or HsmtPheRS. The E. coli tRNAPhe transcript was preaminoacylated with m‐Tyr and purified, then incubated (at 1.2 µM concentration) in the presence of ATP, [3H]Phe, and chimera (0.4 μM) or HsmtPheRS (0.4 µM).
To gain a deeper understanding of nature of the changes observed in editing and aminoacylation reactions, we carried out computer simulations of the chimera model. Molecular dynamics simulations revealed that in‐effect ABD generates two clusters of interdomain contacts as distinct from WT HsmtPheRS. First cluster represents basically the same set of contacts observed in closed “inactive” conformation of HsmtPheRS (Figure 5). The new cluster of the HBs is arisen between ABD and transplanted EMD: His850 (ABD) forms HB with Glu320 (EMD) (2.9 Å), Glu816 (ABD) with Thr293 (EMD) (3.1 Å), and Glu816 (ABD) with His 350 (EMD, 2.9 Å).
Diffusional association of tRNA and aaRS, including monomeric HsmtPheRS, is governed by long‐range electrostatic interactions.30 These are the driving forces behind their primary stickiness. Bending of the long stretch of aa linking ABD to CAM (Figure 5) enables large‐scale rearrangement of ABD, and formation of open state in complex with tRNA. It is evident that appearance of three extra HB between ABD and EMD raises the energy barrier on ABD rotation, slowing transition from closed inactive to open active state.
To confirm the importance of extra HBs in chimera, we mutated residues within the interface between ABD and EMD. The mutation Thr293Val, reducing just the number of HBs in the interface, did not exert an influence on the kinetic parameters of aminoacylation. The aminoacylation activity of the mutant was virtually the same as that of chimera. A double mutation (His850Asp and Glu816Gly) might presumably destabilize the interface in chimera, generating repulsion between the ABD and EMD. Indeed, this double mutation leads to a complete loss of the aminoacylation activity. Double mutant considerably changes the dynamic equilibrium between the closed and open states, preventing formation of the complex with tRNA.
Arrangement of the structural domains in chimera substantially differs from that in the bacterial PheRS. In bacterial PheRSs, aminoacylation and editing activities are associated with different subunits, while in chimera, both the synthetic and editing sites are located at the same monomer. Moreover, ABD in the heterodimer is immovable, while in HsmtPheRS, it undergoes hinge‐type rotation through 160° upon tRNA binding. Chimera protein has a molecular weight of 100.5 kDa, encompasses 893 aa and consists of the following building blocks (numbering as in the crystal structures of EcPheRS and HmstPheRS): block‐I [EcoliPheRS (residues from 1 to 514)] + block‐II [HsmtPheRS (residues from 37 to 415)].
Kinetic characterization of the mitochondrial chimera PheRS
Kinetic experiments on aminoacylation and editing activities have been carried out for EcPheRS, HsmtPheRS, and chimera comparatively. The production of Phe‐tRNAPhe by chimera demonstrates that insertion of the B1–B5 module does not interfere with the aa activation, tRNA binding, and ultimately with formation of aminoacylated tRNAPhe (Figure 4A). We have determined the kinetic parameters of aminoacylation for E. coli tRNAPhe‐transcript by chimera protein and compared them with respective constants of the wild‐type HsmtPheRS (Table 1).
Figure 4.
Interface area in the “closed” inactive configuration of HsmtPheRS. CAM is colored blue, while ABD is colored green. Long stretch of aa (11 aa) connecting CAM and ABD colored red. Interface area is outlined by oval colored grey.
Table 1.
Kinetic Parameters of E. coli tRNAPhe‐Transcript Aminoacylation
| PheRS | K m (µM) | k cat (min−1) | k cat/K m |
|---|---|---|---|
| Wild‐type HsmtPheRS | 0.84 | 3.58 | 4.3 |
| Mitochondrial chimera | 0.57 | 0.32 | 0.56 |
Chimera aminoacylates tRNAPhe with an eightfold lower catalytic efficiency (k cat/K m), as compared to the wild‐type HsmtPheRS. The lower aminoacylation efficiency of tRNAPhe by chimera is conditioned by decrease in the turnover number.
To analyze the editing activity of chimera, the enzyme‐assisted hydrolysis of misacylated tRNA was investigated. Radiolabeled Tyr‐tRNAPhe presynthesized in the presence of HsmtPheRS was further incubated with chimera or EcPheRS. The enzyme possessing editing activity should hydrolyze the exogenous Tyr‐tRNAPhe. Hydrolysis reactions were performed at varied concentrations of the enzymes to find conditions at which the rates of hydrolysis by the two enzymes are comparable (Figure 4B). The kinetic data testify that the specific editing activity of chimera is lower than that of the wild‐type EcPheRS. At the same time, chimera displays a substantially higher editing activity than the individual EMD. The distinctions between chimera and wild‐type HsmtPheRS and EcPheRS revealed in both aminoacylation and editing activities might be due to variations in dynamic characteristics of structural domains in the chimera. This is not unexpected since CAM, ABD, and EMD in wild‐type EcPheRS or TtPheRS passed through the mutual adjustment and optimization of their activities for a long evolutionary history.
For verification of trans‐editing activity of chimera toward other noncognate aa‐tRNA, deacylation of m‐Tyr‐tRNAPhe was additionally investigated, using the reaminoacylatiuon assay. The nonradiolabeled m‐Tyr‐tRNAPhe pre‐synthesized in the presence of HsmtPheRS was isolated and further incubated under the hydrolysis conditions in the presence of HsmtPheRS or chimera, [3H]phenylalanine and ATP, and incorporation of the radioactive amino acid into tRNA was measured (Figure 4C). The charging level after 1–2 min of incubation corresponds to the extent of spontaneous deacylation of presynthesized aa‐tRNA determined independently.16 The further slow increase of the charging level in the presence of HsmtPheRS is caused by phenylalanylation of the aa‐tRNA spontaneously degraded during the incubation. A higher charging level in the presence of chimera is indicative of enzyme‐assisted hydrolysis of m‐Tyr‐tRNAPhe and its reaminoacylation.
Discussion
Along with their key activity of specific tRNA aminoacylation, aaRSs are known to perform various other biological functions. The molecular basis of these alternative functions of aaRSs lies in their modular composition, and PheRS is a particularly significant example. In cytoplasmic PheRSs, the aminoacylation and editing sites are located in different structural modules, and well away from each other. Such an arrangement of the functional modules in PheRSs, where the CCA‐end of the substrate tRNA migrates from the aminoacylation site to the editing site upon cis‐editing, underlies the conformational ability for the EMD to be isolated and transplanted into the HsmtPheRS.20, 24, 27
The fully active fusion protein consisting of human mitochondrial PheRS and EMD of EcPheRS was constructed. Chimera protein retains significant rearrangement of the ABD from the “closed” to the productive “open” state despite the doubling of its molecular weight as compared to the mitochondrial PheRS. Chimera possesses both the aminoacylation and editing activities toward the noncognate aa‐tRNAPhes (misacylated with tyrosine, and nonprotein aa), thereby retaining fidelity of protein biosynthesis.
Editing activity is of prime importance in eukaryotic cells, where ROS‐damaged aa are incorporated into proteins by the cell biosynthetic pathways rather than via chemical reactions.31, 32, 33 It was reported that m‐Tyr and o‐Tyr are misincorporated instead of Phe in recombinantly produced monoclonal antibodies considered as the effective drugs for treating various diseases.31 Editing activity of the chimera in this case may have considerable utility when supporting the fidelity of biomedical production of these monoclonal antibodies.
Many of nonprotein aa formed as a by‐product in plants demonstrate phytotoxic effect.33 Exposure to m‐Tyr and l‐Dopa results in growth inhibition of a wide range of plant species including commercially important monocots and dicots that is reflected in the inhibition of root growth.34 It has been especially important that phytotoxicity of m‐Tyr is caused by its incorporation into proteins in place of phenylalanine during protein synthesis.33 Chimera can be efficiently implemented in the production of transgenic plants resistant to natural bioherbicides, i.e., to m‐Tyr and l‐Dopa and other nonprotein aa possessing herbicidal activity. Resistance of this type, using ability of E. coli PheRS to hydrolyze mischarged m‐Tyr‐tRNAPhe, recently was generated on small flowering genetically modified plant Arabidopsis thaliana. It was manifested in the presence of exogenous m‐Tyr at concentrations that have a deleterious effect on unmodified plant (Patent: US 2014/0237686 A1). Chimera, whose aa composition is more appropriate to mitochondria and chloroplasts, can easily substitute E. coli PheRS in transgenic plant A. thaliana.
Materials and Methods
Chemicals, proteins, and tRNAs
l‐[3H]Phenylalanine and l‐[3H]tyrosine were purchased from PerkinElmer Inc. dl‐m‐tyrosine was from Sigma‐Aldrich. The E. coli tRNAPhe was synthesized by using runoff transcription of synthetic genes with T7 RNA polymerase followed by electrophoretic isolation of the correct‐length transcripts as described.15 Phenylalanyl‐tRNA synthetases were purified from natural (TtPheRS) or overexpressed sources (HsmtPheRS and EcPheRS) as previously described.15, 24
tRNA aminoacylation assay
The activity of HsmtPheRS and chimera enzymes was tested at 37° in reaction mixtures containing 50 mM Tris–HCl, pH 8.5, 30 mM MgCl2, 5 mM ATP, 10 mM 2‐mercaptoethanol, 4 μM L‐[3H]Phe, ∼1 μM E. coli tRNAPhe transcript [a heterologous substrate of HsmtPheRS18], and 0.3–0.4 μM HsmtPheRS or chimera protein. Kinetic parameters (k cat and K M) for tRNAPhe were determined using a range of 0.3–2.2 μM of E. coli tRNAPhe transcript. At the appropriate times, aliquots of 6 µL were spotted onto Whatman filter paper impregnated with 5% trichloroacetic acid (TCA). Then the filters were extensively washed with ice‐cold 5% TCA, and TCA‐insoluble radioactivity was measured by liquid scintillation counting. The kinetic parameters were calculated by a nonlinear regression fit of the data to a Michaelis–Menten equation. The reported k cat and K m values represent the average of at least two determinations with experimental errors within 15% of the indicated values.
Preparation of aa‐tRNAPhe and post‐transfer editing assay
Aminoacylation of 1–3 μM E. coli tRNAPhe‐transcript with noncognate amino acids was performed in the presence of 30 μM l‐[3H]Tyr (5.4 mCi/mmol) or 200 µM m‐Tyr and definite concentration of HsmtPheRS (0.4 µM for charging with m‐Tyr; 2 µM for tyrosylation). After 30 min of incubation at 37° in the mixtures containing all the other components described above for tRNA aminoacylation, the reaction was stopped by addition of four volumes of cold solution of 300 mM sodium acetate (pH 4.9). The remaining ATP and amino acid were separated by dialysis at 4° against 300 mM sodium acetate (pH 4.9), using Vivaspin microconcentrator. The aa‐tRNA was extracted with phenol (buffered with 100 mM sodium acetate, pH 4.9), followed by chloroform extraction, ethanol precipitation and stepwise washing with 70% and absolute ethanol. The aa‐tRNA pellet was dried, stored at −20°, and dissolved in water directly before hydrolysis experiments. The aa‐tRNA concentration in the [3H]Tyr‐tRNA preparations was determined by precipitation with 5% TCA on filter discs, followed by washing in 5% TCA, drying and scintillation counting. The yields of tyrosylation did not exceed 25% of the total charging level with Phe (due to a higher K m value for Tyr as compared to that for Phe). The reaction conditions for m‐Tyr‐tRNA synthesis were optimized previously to obtain the yield of charged tRNA about 80%.16
For post‐transfer editing reaction, dry pellet of charged l‐[3H]Tyr‐tRNAPhe was dissolved in the appropriate buffer containing 50 mM Tris–HCl, pH 8.0, 20 mM MgCl2. The editing reaction was initiated by adding chimera (whose concentration varied from 0.3 to 1.5 μM) or B1/2–B3/4–B5 (Δ1–514), B1/2–B3/4 (Δ1–395), and B3/4 (Δ214–396) fragments from EcPheRS β‐subunit (0.8–22 μM) or intact EcPheRS or TtPheRS (2–20 nM). At the appropriate times aliquots of 5 µL were spotted onto Whatman filter paper impregnated with 5% TCA. Then the filters were extensively washed with ice‐cold 5% TCA, and TCA‐insoluble radioactivity was measured by liquid scintillation counting.
Post‐transfer editing was also assayed by reaminoacylation of m‐Tyr‐tRNAPhe with l‐[3H]Phe. The nonradiolabeled aa‐tRNA (0.8–1 µM) synthesized and purified as described above was incubated at 37° in the hydrolysis reaction mixture in the presence of 5 mM ATP, 4 µM l‐[3H]Phe, and 5–20 nM EcPheRS or 0.1–4 μM chimera. The yield of radiolabeleded aa‐tRNA was determined by measuring TCA‐insoluble radioactivity as described above.
Preparation of Isolated E. coli PheRS structural modules and chimeric protein
The fragments from EcPheRS β‐subunit were cloned in a pET‐21c (+) vector and expressed and purified without His‐tag. The vector was transformed into E. coli Rosetta DE3 strain cells (Novagen). Fresh colonies were inoculated in the 2 L of LB medium in presence of 100 µg/mL ampicillin and grown at 37°C. The cells were then induced with 1 mM IPTG and cultured overnight at 298°K. Cells were harvested by centrifugation at 6000 rpm for 30 min and stored at 193°K. Fragments were purified by chromatography on a 5 ml HiTrap heparin‐affinity column (25 × 16 mm, GE Healthcare) followed by a 600 × 16 mm size‐exclusion HiLoad Superdex 200 column (GE Healthcare). Purified sample was concentrated and dialyzed against buffer (20 mM Tris–HCl pH 8, 100 mM NaCl, 7 mM MgCl2, 5 mM 2‐mercaptoethanol, and 1 mM EDTA). The protein was stored in small aliquots and flash‐frozen at 193°K.
Modeling
A model comprising EMD from E. coli PheRS (residues B1–B514 from β‐subunit) and catalytic and ABD modules from mitochondrial PheRS (residues 37–415) was assembled into “chimera” using a set of special restraints implemented in MODELLER.29 The initial structure was further adjusted by remodeling of the protein loops using Modeller protocols, and further refined by using minimization/molecular dynamics protocol.35 Restraints include (a) homology‐derived restraints on the distances and dihedral angles in the target sequence, extracted from its alignment with the template structures; (b) stereochemical restraints such as bond length and bond angle preferences, obtained from the CHARMM‐22 molecular mechanics force field36; (c) statistical preferences for dihedral angles and nonbonded interatomic distances, obtained from a representative set of known protein structures.
Molecular dynamics
Molecular dynamics simulations presented in this work were performed using the GROMACS software package.35 For chimera enzyme, the GROMOS96 force field was used.37 For water, the rigid simple point charge (SPC) force field was applied. The Berendsen thermostat with a reference temperature of 300°K was applied with a time constant of 0.1 ps.38 The potential energy of the protein was relaxed, before addition of water and subsequent equilibration for 100 ps. Eleven sodium ions were added randomly to compensate for negative charges of the chimera protein. The “open” and the “closed” conformation systems were solvated by 14,658 and 10,795 water molecules, respectively. After addition of ions and water molecules, energy minimization was performed followed by a 50 ps equilibration in the NVT (constant number of particles, constant volume, and constant temperature) ensemble.
Author Contribution
E.K. performed biochemical and kinetic experiments, wrote the article; M.P. carried out molecular biology and kinetic experiments, wrote the article; D.T. did molecular modeling and MM calculations; N.M. performed kinetic experiments, wrote the article; M.G.S. formulated problem, supervised the work, and wrote the article.
Acknowledgments
Authors are grateful to Drs. L. Klipcan and I. Finarov for helpful discussions and advices.
Highlights: Chimeric human mitochondrial PheRS charges tRNAPhe with cognate Phe, and hydrolyzes the noncognate Tyr‐tRNAPhe. Thus, the editing activity described previously for the heterodimeric PheRSs only is exhibited by the monomeric PheRS as well. The novel chimeric protein acts as a sieve rejecting nonprotein amino acids harmful to proteins and as such may be applied in agriculture and medicine.
References
- 1. Ibba M, Soll D (2000) Aminoacyl‐tRNA synthesis. Annu Rev Biochem 69:617–650. [DOI] [PubMed] [Google Scholar]
- 2. Eldred EW, Schimmel PR (1972) Rapid deacylation by isoleucyl transfer ribonucleic acid synthetase of isoleucine‐specific transfer ribonucleic acid aminoacylated with valine. J Biol Chem 247:2961–2964. [PubMed] [Google Scholar]
- 3. Fersht AR (1977) Editing mechanisms in protein synthesis. Rejection of valine by the isoleucyl‐tRNA synthetase. Biochemistry 16:1025–1030. [DOI] [PubMed] [Google Scholar]
- 4. Jakubowski H. Accuracy of aminoacyl‐tRNA synthetases: proofreading of amino acids In: Ibba M, Francklyn C, Cusack S, Eds. (2005) Aminoacyl‐tRNA synthetases. LANDES Bioscience, Georgetown, Texas, USA pp. 384–396. [Google Scholar]
- 5. Perona JJ, Gruic‐Sovulj I Synthetic and editing mechanisms of aminoacyl‐tRNA synthetases In Topics in current chemistry. (2014) Springer‐Verlag: Berlin‐Heidelberg, pp. 1–41. [DOI] [PubMed] [Google Scholar]
- 6. Christian BE, Spremulli LL (2012) Mechanism of protein biosynthesis in mammalian mitochondria. Biochim Biophys Acta 1819:1035–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chatton B, Walter P, Ebel JP, Lacroute F, Fasiolo F (1988) The yeast VAS1 gene encodes both mitochondrial and cytoplasmic valyl‐tRNA synthetases. J Biol Chem 263:52–57. [PubMed] [Google Scholar]
- 8. Natsoulis G, Hilger F, Fink GR (1986) The HTS1 gene encodes both the cytoplasmic and mitochondrial histidine tRNA synthetases of S. cerevisiae. Cell 46:235–243. [DOI] [PubMed] [Google Scholar]
- 9. Sissler M, Putz J, Fasiolo F, Florentz C. Mitochondrial aminoacyl‐tRNA synthetases In: Ibba M, Francklyn C, Cusack S, Eds. (2005) The Aminoacyl‐tRNA synthetases. Georgetown: LANDES Bioscience, pp. 271–284. [Google Scholar]
- 10. Souciet G, Menand B, Ovesna J, Cosset A, Dietrich A, Wintz H (1999) Characterization of two bifunctional Arabdopsis thaliana genes coding for mitochondrial and cytosolic forms of valyl‐tRNA synthetase and threonyl‐tRNA synthetase by alternative use of two in‐frame AUGs. FEBS Lett 266:848–854. [DOI] [PubMed] [Google Scholar]
- 11. Spencer AC, Heck A, Takeuchi N, Watanabe K, Spremulli LL (2004) Characterization of the human mitochondrial methionyl‐tRNA synthetase. Biochemistry 43:9743–9754. [DOI] [PubMed] [Google Scholar]
- 12. Kaiser E, Hu B, Becher S, Eberhard D, Schray B, Baack M, Hameister H, Knippers R (1994) The human EPRS locus (formerly the QARS locus): a gene encoding a class I and a class II aminoacyl‐tRNA synthetase. Genomics 19:280–290. [DOI] [PubMed] [Google Scholar]
- 13. Schwenzer H, Zoll J, Florentz C, Sissler M (2014) Pathogenic implications of human mitochondrial aminoacyl‐tRNA synthetases. Top Curr Chem 344:247–292. [DOI] [PubMed] [Google Scholar]
- 14. Stadtman ER, Levine RL (2003) Free radical‐mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 25:207–218. [DOI] [PubMed] [Google Scholar]
- 15. Klipcan L, Moor N, Kessler N, Safro MG (2009) Eukaryotic cytosolic and mitochondrial phenylalanyl‐tRNA synthetases catalyze the charging of tRNA with the meta‐tyrosine. Proc Natl Acad Sci USA 106:11045–11048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moor N, Klipcan L, Safro MG (2011) Bacterial and eukaryotic phenylalanyl‐tRNA synthetases catalyze misaminoacylation of tRNA(Phe) with 3,4‐dihydroxy‐L‐phenylalanine. Chem Biol 18:1221–1229. [DOI] [PubMed] [Google Scholar]
- 17. Klipcan L, Levin I, Kessler N, Moor N, Finarov I, Safro M (2008) The tRNA‐induced conformational activation of human mitochondrial phenylalanyl‐tRNA synthetase. Structure 16:1095–1104. [DOI] [PubMed] [Google Scholar]
- 18. Klipcan L, Moor N, Finarov I, Kessler N, Sukhanova M, Safro MG (2012) Crystal structure of human mitochondrial PheRS complexed with tRNA(Phe) in the active “open” state. J Mol Biol 415:527–537. [DOI] [PubMed] [Google Scholar]
- 19. Finarov I, Moor N, Kessler N, Klipcan L, Safro MG (2010) Structure of human cytosolic phenylalanyl‐tRNA synthetase: evidence for kingdom‐specific design of the active sites and tRNA binding patterns. Structure 18:343–353. [DOI] [PubMed] [Google Scholar]
- 20. Mosyak L, Safro M (1993) Phenylalanyl‐tRNA synthetase from Thermus thermophilus has four antiparallel folds of which only two are catalytically functional. Biochimie 75:1091–1098. [DOI] [PubMed] [Google Scholar]
- 21. Goldgur Y, Mosyak L, Reshetnikova L, Ankilova V, Lavrik O, Khodyreva S, Safro M (1997) The crystal structure of phenylalanyl‐tRNA synthetase from Thermus thermophilus complexed with cognate tRNAPhe . Structure 5:59–68. [DOI] [PubMed] [Google Scholar]
- 22. Bullard JM, Cai YC, Demeler B, Spremulli LL (1999) Expression and characterization of a human mitochondrial phenylalanyl‐tRNA synthetase. J Mol Biol 288:567–577. [DOI] [PubMed] [Google Scholar]
- 23. Roy H, Ling J, Alfonzo J, Ibba M (2005) Loss of editing activity during the evolution of mitochondrial phenylalanyl‐tRNA synthetase. J Biol Chem 280:38186–38192. [DOI] [PubMed] [Google Scholar]
- 24. Mermershtain I, Finarov I, Klipcan L, Kessler N, Rozenberg H, Safro MG (2011) Idiosyncrasy and identity in the prokaryotic phe‐system: crystal structure of E. coli phenylalanyl‐tRNA synthetase complexed with phenylalanine and AMP. Protein Sci 20:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oki K, Sakamoto K, Kobayashi T, Sasaki HM, Yokoyama S (2008) Transplantation of a tyrosine editing domain into a tyrosyl‐tRNA synthetase variant enhances its specificity for a tyrosine analog. Proc Natl Acad Sci USA 105:13298–13303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roy H, Ibba M (2006) Phenylalanyl‐tRNA synthetase contains a dispensable RNA‐binding domain that contributes to the editing of noncognate aminoacyl‐tRNA. Biochemistry 45:9156–9162. [DOI] [PubMed] [Google Scholar]
- 27. Kotik‐Kogan O, Moor N, Tworowski D, Safro M (2005) Structural basis for discrimination of L‐phenylalanine from L‐tyrosine by phenylalanyl‐tRNA synthetase. Structure 13:1799–1807. [DOI] [PubMed] [Google Scholar]
- 28. Yadavalli SS, Klipcan L, Zozulya A, Banerjee R, Svergun D, Safro M, Ibba M (2009) Large‐scale movement of functional domains facilitates aminoacylation by human mitochondrial phenylalanyl‐tRNA synthetase. FEBS Lett 583:3204–3208. [DOI] [PubMed] [Google Scholar]
- 29. Eswar N, Eramian D, Webb B, Shen MY, Sali A (2008) Protein structure modeling with MODELLER. Methods Mol Biol 426:145–159. [DOI] [PubMed] [Google Scholar]
- 30. Tworowski D, Feldman AV, Safro MG (2005) Electrostatic potential of aminoacyl‐tRNA synthetase navigates tRNA on its pathway to the binding site. J Mol Biol 350:866–882. [DOI] [PubMed] [Google Scholar]
- 31. Popp O, Larraillet V, Kettenberger H, Gorr IH, Hilger M, Lipsmeier F, Zeck A, Beaucamp N (2015) Molecular polygamy: the promiscuity of l‐phenylalanyl‐tRNA‐synthetase triggers misincorporation of meta‐ and ortho‐tyrosine in monoclonal antibodies expressed by Chinese hamster ovary cells. Biotechnol Bioeng 112:1187–1199. [DOI] [PubMed] [Google Scholar]
- 32. Gurer‐Orhan H, Ercal N, Mare S, Pennathur S, Orhan H, Heinecke JW (2006) Misincorporation of free m‐tyrosine into cellular proteins: a potential cytotoxic mechanism for oxidized amino acids. Biochem J 395:277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rodgers KJ, Shiozawa N (2008) Misincorporation of amino acid analogues into proteins by biosynthesis. Int J Biochem Cell Biol 40:1452–1466. [DOI] [PubMed] [Google Scholar]
- 34. Bertin C, Weston LA, Huang T, Jander G, Owens T, Meinwald J, Schroeder FC (2007) Grass roots chemistry: meta‐tyrosine, an herbicidal nonprotein amino acid. Proc Natl Acad Sci USA 104:16964–16969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lindahl E, Hess B, van der Spoel D (2001) GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model 7:306–317. [Google Scholar]
- 36. Mackerell AD, Jr (2004) Empirical force fields for biological macromolecules: overview and issues. J Comput Chem 25:1584–1604. [DOI] [PubMed] [Google Scholar]
- 37. Scott WRP, Hunenberger PH, Tironi IG, Mark AE, Billeter SR, Fennen J, Torda AE, Huber T, Kruger P, van Gunsteren WF (1999) The GROMOS biomolecular simulation program package. J Phys Chem A 103:3596–3607. [Google Scholar]
- 38. Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR (1984) Molecular‐dynamics with coupling to an external bath. J Chem Phys 81:3684–3690. [Google Scholar]