

Abstract
Vaccination is the most effective means to prevent influenza and its serious complications. Influenza viral strains undergo rapid mutations of the surface proteins hemagglutinin (HA) and neuraminidase (NA) requiring vaccines to be frequently updated to include current circulating strains. It is nearly impossible to predict which strains will be circulating in the next influenza season. It is, therefore, imperative that the process of producing a vaccine be streamlined and as swift as possible. We have developed an isotope dilution mass spectrometry (IDMS) method to quantify HA and NA in H7N7, H7N2, and H7N9 influenza. The IDMS method involves enzymatic digestion of viral proteins and the specific detection of evolutionarily conserved target peptides. The four target peptides that were initially chosen for analysis of the HA protein of H7N2 and H7N7 subtypes were conserved and available for analysis of the H7N9 subtype that circulated in China in the spring of 2013. Thus, rapid response to the potential pandemic was realized. Multiple peptides are used to quantify a protein to ensure that the digestion of the protein is complete in the region of the target peptides, verify the accuracy of the measurement, and provide flexibility in the case of amino acid changes among newly emerging strains. The IDMS method is an accurate, sensitive, and selective method to quantify the amount of HA and NA antigens in primary liquid standards, crude allantoic fluid, purified virus samples, and final vaccine presentations.
Keywords: Hemagglutinin, Neuraminidase, Mass spectrometry, Influenza, Proteins, Quantification
Graphical Abstract
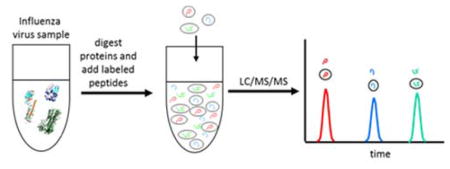
Introduction
Vaccination is the key strategy to prevent severe illness and death from influenza. Vaccine manufacturers, government regulatory agencies, and public health agencies face many challenges in the effort to develop human vaccines against avian influenzas.1 Influenza viruses contain two major surface proteins, hemagglutinin (HA) and neuraminidase (NA). HA is the primary antigen in influenza vaccines. The HA and NA are subject to either subtle or radical mutations that occur continually. Because of these changes, the influenza vaccines are usually updated each year.2
There are two groups of avian influenza virus. One group is highly pathogenic avian influenza (HPAI) and has a mortality that can be as high as 100% while the other group is low pathogenic avian influenza (LPAI). LPAI viruses cause a much milder, primarily respiratory disease, which may be aggravated by other infections or environmental conditions.3 HPAI subtypes have been limited to H5 and H7, but not all H5 and H7 subtypes are HPAI. The H7N9 subtype is unique as it is of low pathogenicity to birds while being highly pathogenic to humans making it very difficult to track and detect outbreaks.4
Recently, there have been several outbreaks of avian influenza. The first human cases of avian influenza A virus (H5N1) were reported in 1997 in Hong Kong after exposure to infected poultry.5,6 A highly pathogenic avian influenza virus caused disease in poultry in at least eight East Asian countries between 2003 and 2004. Fatalities among humans have been attributed to H5N1 in Cambodia, China, Indonesia, Thailand, and Vietnam.7 Thailand alone reported a total of 17 cases of H5N1 influenza which coincided with the disease observed in poultry.8 Human cases continued to be reported in Egypt, Iraq, Turkey, Djibouti, and Azerbaijan.9
The production of the vaccine for the H5N1 virus faced several challenges. The first batch of inactivated human influenza vaccine against the1997 H5N1 virus was not ready for clinical trial until 7 months after the second case of human infection arose. The success of the vaccine against this virus is still unclear.10 A main cause for this delay in the production of an H5N1-specific vaccine was the nature of the virus itself. The H5N1 virus is highly pathogenic in both humans and poultry. The agent must be handled, at a minimum, under biosafety level 3 (BSL3) conditions. It can also be detrimental to fertilized chicken eggs, which is the standard medium for the reassortment and spread of influenza virus before it is inactivated and formulated for use in vaccines.11 Furthermore, the emergence of antigenically distinct groups of influenza viruses poses a significant challenge for the design of vaccines against H5N1 viruses because of the possible need for group-specific vaccines.12
The H7 subtype viruses, which include the North American lineages (H7N2 and H7N3) and Eurasian lineages (H7N7, H7N3 and H7N9), have caused human disease.13,14 These avian flu viruses can infect humans, birds, pigs, seals and horses in the wild and have infected mice in laboratory studies. Between February and May 2003, a serious outbreak occurred in the Netherlands.15 A highly pathogenic avian influenza A virus of subtype H7N7, closely related to low pathogenic virus isolates obtained from ducks was also isolated in chickens. This virus was later detected in 86 humans who handled affected poultry and in three of their family members.16 Influenza-like illnesses were generally mild, but a fatal case of pneumonia in combination with acute respiratory distress syndrome did occur.16 Because humans are rarely exposed to H7N7, there is little or no antibody protection against these viruses in the general population. If a person were to become infected with H7N7 and the virus gain the capability to spread efficiently from person to person, an influenza pandemic could begin.
The first identified cases of human infection with a novel influenza A (H7N9) virus occurred in eastern China during March and April 2013. The H7N9 virus causes rapidly progressive pneumonia, respiratory failure, acute respiratory distress syndrome (ARDS), and fatal outcomes.17,18 The H7N9 virus subtype have infected 128 and killed 26 people in China as of May 1, 2013.4 Fortunately, there have not been any confirmed cases of human-to-human transmission, but if there had been, we could have had a pandemic with no vaccine available. HA and NA genes in the H7N9 strain probably originated from Eurasian avian influenza viruses; the remaining genes are closely associated to avian H9N2 influenza viruses.19 H7 subtype vaccine production will face the same potential problems and similar challenges to those of the H5 subtype because they are antigenically distinct. After the unprecedented geographic spread of H5 subtype viruses since 2003, the continued occurrence of random cases of H5N1 infections in humans, and the recent outbreaks of H7N9 in China, major importance has been given to the pandemic threat posed by the H5 and H7 avian influenza viruses.
Regulatory standards require that seasonal influenza vaccines contain 30 μg/mL of HA for each strain of influenza. A trivalent vaccine containing strains of H1N1, H3N2, and B should therefore contain 90 μg/mL of total HA. The United States does not require a commercial vaccine to contain NA. However, vaccines sold in European nations are required to exhibit proof that the vaccine includes NA, even though no specified amount is required. HA is currently quantified using the single radial immuno-diffusion (SRID) assay. This method involves strain-specific reagents including a purified whole virus HA preparation and anti-HA sheep serum.20, 21 Barring any unforeseen complications, production and characterization of SRID reagents can take approximately 2–3 months to complete. Manufacturers are unable to formulate, fill, or release the pandemic vaccine doses until successful quantification of HA by SRID which necessarily requires waiting on the strain-specific SRID reagents. Adding the requirement of quantification of NA using SRID would increase the vaccine production time schedule since production of strain-specific NA reagents would also be required. Since the limit of quantification of SRID is approximately 4 μg/ mL, SRID is not sufficiently sensitive for the quantification of NA. HA is the most abundant protein on the surface of the virus with NA being at least 3 or more times less abundant. Therefore, SRID would not be an appropriate method for NA quantification in final presentations of most seasonal vaccines as NA would likely be at concentrations less than 4 μg/mL.
We have previously described an isotopic dilution mass spectrometry or IDMS method to quantify the viral proteins of H5, H3, and H1 subtypes as well as influenza B.22,23 We have expanded this method to quantify HA and NA in H7N7, H7N2, and H7N9 influenza samples in a single run. The IDMS method involves enzymatic digestion of viral proteins and the specific detection of evolutionarily conserved target peptides. Four HA peptides were used for the quantification of HA in all H7 samples while peptides specific to N2, N7, and N9 were used for each type of neuraminidase. The use of multiple peptides in the analysis ensures complete digestion of the protein in the vicinity of the target peptides, which is crucial for accuracy of the measurement and provides redundancy in the case of amino acid changes among newly emerging strains. Choosing target peptides that can cover the majority of strains of interest or of future potential interest is ideal so that fewer peptide standards have to be generated or updated annually. However, should the strain of interest be a novel one not covered by the selected target peptides, it is a simple matter to detect the difference and quickly synthesize and prepare a new peptide standard. This process takes two weeks.23 The IDMS method is an accurate, sensitive, and selective method to quantify the amount of HA and NA antigens in a variety of matrices including crude allantoic fluid, purified virus samples, and final vaccine presentations.
Materials and Methods
Virus and Samples
An inactivated whole virus preparation from the A/Netherlands/219/2003 (H7N7) was provided by the U.S. Food and Drug Administration’s Center for Biologics Evaluation and Research (CBER) and was used without further purification. Virus samples in allantoic fluid and purified virus of the A/Shanghai/2/2013 (H7N9) were obtained from the National Center for Immunization and Respiratory Diseases (NCIRD), Influenza Division, Centers for Disease Control and Prevention (CDC) and used without further purification.
Synthesis of Native and Labeled Peptides
The HA and NA peptides were custom synthesized at a 1–5 mg scale by MidWest Bio-Tech, Inc. (Fishers, IN). The targeted peptides are listed in Table 1. The 5 mg vials of the peptides were reconstituted by adding 8 mL of 10% formic acid. Then, they were diluted by adding 48 mL of 0.1% formic acid to 2 mL of the peptide. Each peptide was separated into four aliquots. These peptides were aliquoted in 200 μL volumes into 1.5 mL vials using an automated liquid handler (Beckman Coulter, Fullerton, CA). They were later lyophilized and stored at −70°C until used. Each vial should contain 3–6 nmol of peptide. An isobaric-tagged isotope dilution mass spectrometry (IT-IDMS) method for amino acid analysis (AAA) was used to quantify the peptides. The IT-IDMS method incorporates NIST certified isotopically labeled amino acids as internal standards to ensure accurate results.24
Table 1.
Target peptides employed for the quantification of hemagglutinin (HA) and neuraminidase (NA). Underlined amino acids correspond to 13C- and 15N-labeled amino acids. N9, neuraminidase of H7N9 strains; H7, hemagglutinin of H7N7 strains; N7, neuraminidase of H7N7 strains.
| Target Peptide | influenza protein | precursor ion m/z | fragment ion (quantification) | fragment ion (confirmation) | fragment ion (confirmation) | collision energy (eV) |
|---|---|---|---|---|---|---|
| IGESSDVLVTR | N9 | 588.3 (+2) | 702.4 (y6) | 789.4 (y7) | 876.5 (y8) | 23 |
| IGESSDVLVTR | N9 | 591.3 (+2) | 708.4 (y6) | 795.5 (y7) | 882.5 (y8) | 23 |
| FYALSQGTTIR | N9 | 628.8 (+2) | 762.4 (y7) | 875.5 (y8) | 946.5 (y9) | 24 |
| FYALSQGTTIR | N9 | 632.3 (+2) | 769.4 (y7) | 882.5 (y8) | 953.5 (y9) | 24 |
| VPNALTDDR | N9 | 500.8 (+2) | 619.3 (y5) | 690.3 (y6) | 804.4 (y7) | 20 |
| VPNALTDDR | N9 | 504.3 (+2) | 626.3 (y5) | 697.3 (y6) | 811.4 (y7) | 20 |
| FGESEQIIVTR | N7 | 639.8(+2) | 729.4 (y6) | 858.5 (y7) | 945.5 (y8) | 23 |
| FGESEQIIVTR | N7 | 642.8 (+2) | 735.5 (y6) | 864.5 (y7) | 951.6 (y8) | 23 |
| IPNAGTDPNSR | N7 | 571.3 (+2) | 473.2 (y4) | 689.3 (y6) | 931.4 (y9) | 25 |
| IPNAGTDPNSR | N7 | 574.3 (+2) | 479.3 (y4) | 695.3 (y6) | 937.4 (y9) | 25 |
| SGYETFR | N2 | 430.2(+2) | 715.3 (y5) | 772.4 (y6) | 552.3 (y4) | 18 |
| SGYETFR | N2 | 435.2 (+2) | 725.4 (y5) | 782.4 (y6) | 562.3(y4) | 18 |
| VNTLTER | H7 | 416.7(+2) | 518.3 (y4) | 619.3 (y5) | 733.4 (y6) | 17 |
| VNTLTER | H7 | 420.2 (+2) | 525.3 (y4) | 626.4 (y5) | 740.4 (y6) | 17 |
| FVNEEALR | H7 | 489.3 (+2) | 617.3 (y5) | 731.4 (y6) | 830.4 (y7) | 20 |
| FVNEEALR | H7 | 492.8 (+2) | 624.3 (y5) | 738.4 (y6) | 837.5 (y7) | 20 |
| IQIDPVK | H7 | 406.7(+2) | 343.2 (y3) | 458.3 (y4) | 571.3 (y5) | 17 |
| IQIDPVK | H7 | 409.7 (+2) | 349.2 (y3) | 464.3 (y4) | 577.4 (y5) | 17 |
| STQSAIDQITGK | H7 | 624.8(+2) | 661.3 (y6) | 774.4 (y7) | 845.5 (y8) | 25 |
| STQSAIDQITGK | H7 | 628.3 (+2) | 668.4 (y6) | 781.5 (y7) | 852.5 (y8) | 25 |
A labeled analog of the target peptides STQSAIDQITGK and FYALSQGTTIR were made by incorporating the isoleucine closest to the carboxy terminus with 13C and 15N to give a peptide that is 7 Da heavier than the native peptide. Leucine was 13C and 15N labeled to give a peptide that is 7 Da heavier than the native peptide for VNTLTER, VPNALTDDR and FVNEEALR. For IQIDPVK, IGESSDVLVTR and FGESEQIIVTR, valine was 13C and 15N labeled resulting in a peptide that is 6 Da heavier than the native peptide. The carboxy-terminal proline was similarly labeled for the peptide IPNAGTDPNSR producing a peptide that is 8 Da heavier than the native peptide. A labeled analogue of the target peptide SGYETFR was synthesized by incorporating 13C and 15N labeled phenylalanine to give a peptide that is 10 Da heavier than the native peptide.
Preparation of Working Stock, Calibration, and Labeled Solutions
The native and labeled peptide standards were reconstituted by adding 100 μL of 10% (v/v) formic acid and diluted with 0.1% (v/v) formic acid to yield a 5 pmol/μL stock solution. In order to facilitate the sample preparation of unknowns, a 0.5 pmol/μL cocktail of the ten labeled H7,N7,N2 and N9 peptides was made by adding 100 μL of each appropriately reconstituted peptide. These cocktails were dried via lyophilization, stored at −70° C until needed, and then reconstituted in 0.1% formic acid (v/v) in water. A similar cocktail was made with the ten native H7, N7, N2 and N9 peptides. Two vials were used to prepare the calibration standards for all peptides used in the analysis in order to minimize the chances for error. Seven-point calibration curves were prepared using the 0.5 pmol/μL solutions of the native and labeled cocktails at average levels of 10, 30, 50, 70, 90, 180, and 250 fmol/μL. The 0.5 pmol/μL spiked solutions of the labeled peptides were used for the internal standards. Mean area ratios (unlabeled/labeled) were plotted against concentrations for each standard. Linear regression without weighting was applied to the data sets, and calibration curves were generated for each peptide. We found the corresponding R2 values of 0.9975, 0.9968, 0.9981, 0.9949, 0.9955 and 0.9925 for VNTLTER, IPNAGTDPNSR, FVNEEALR, IQIDPVK, STQSAIDQITGK and FGESEQIIVTR, respectively.
Sample Preparation and LC-MS/MS Quantification
A volume of 20 μL of the A/Netherlands/219/2003 (H7N7) sample was used in the digestion protocol. A purified virus sample of the A/Shanghai/2/2013 (H7N9) was separately diluted to a 1:5 ratio with 50 mM ammonium bicarbonate solution containing 0.1% RapiGest SF (Waters Corporation, Milford, MA). A volume of 10 μL of the resulting diluted H7N9 purified virus was used for sample digestion. A volume of 10 μL of 0.2% Rapigest was added to the sample and it was boiled for 5 min at 100°C. After to room temperature, 5 μL (~86 pmol) of sequencing grade modified trypsin (Promega, Madison, WI) were added to each sample and incubated at 37°C for 2 hr. The digested samples were cooled to room temperature, and 10 μL of 0.475 M HCl was added to reduce the pH to 2.0 to cleave the acid labile surfactant.25 To the resulting mixture, 10 μL each of the 0.5 pmol/μL H7, N7, N2 and N9 labeled cocktail solution was added. A 0.1% formic acid solution was used to dilute the final sample volume to 100 μL. The digested H7N7 and H7N9 samples were prepared separately, centrifuged for 10 sec (3,000 x g), and transferred to autosampler vials where they ran on the same LC-MS/MS analysis.
LC/MS/MS Instrumentation Parameters
Peptides were separated using an Agilent Technologies 1200 Series Quaternary pump and a High-Performance Autosampler G-1367B HiP-ALS. The analytical column was a 150 mm × 1 mm i.d. Symmetry300 reverse phase C18 (3.5 μm particle size, Waters Corporation, Milford, MA). The aqueous mobile phase (A) consisted of HPLC-grade water with 0.1% formic acid, while the organic phase (B) was acetonitrile (ACN) with 0.1% formic acid. A 5-μL volume injection was utilized. The gradient profile utilized a 150 μL/min flow rate. Initially, the mobile phase, consisting of 98% A and 2% B, was held constant for 5 min. A 1.2% change per min was then utilized over the next 15 min, where the mobile phases were 80% A and 20% B, respectively, followed by a 1.0% change per min over the next 5 min, where the mobile phases were 75% and 25%, respectively. After 37 min run time, the gradient was increased to 98% A and 2% B for the next 20 min to equilibrate the column to its initial conditions. The total run time was 57 min.
The eluent was introduced into a Thermo Scientific Vantage TSQ triple quadrupole tandem mass spectrometer with an electrospray interface (Thermo Scientific, Waltham, MA). The TSQ mass spectrometer was operated in positive ion mode. Table 1 shows the multiple reaction monitoring (MRM) 26m/z with quantifying ion pair transitions. Two additional ion transition pairs utilizing the same conditions were monitored for HA peptide confirmation and are provided in Table 1. Two peptide transitional pairs were monitored for each NA protein: one for detection purposes and two for confirmation. The TSQ parameters were as follows: spray voltage 3400 V, sheath gas 20, auxiliary gas 0, capillary tube temperature 300°C, and a collision gas of 1.5 mTorr. Collision energies and tube lens were optimized for each peptide. Data processing and instrument control were performed with the Thermo Scientific Xcalibur software.
QC Preparation and LC-MS/MS Quantification
A QC material was prepared using an expired commercial monovalent H1N1 vaccine. To this vaccine matrix, the native peptides of the H7N7, H7N2, and H7N9 subtypes were added. A 5 μL spike was used for the low QC while a 10 μL spike was used for the high QC. The digestion efficiency of the HA and NA of the H1N1 strain was monitored. This QC material was prepared and analyzed using the same protocol and LC/MS/MS instrument method as unknown samples. Characterization of the QC material was completed by preparing 20 individual samples on 20 different days. Modified Westgard rules26 were followed to determine QC limits and to report the data.
Results and Discussion
The ability to quantify the viral proteins of the H7N7, H7N2, and H7N9 influenza strains with improved accuracy, precision, and strain specificity of HA and NA may reduce the time needed to develop vaccines against newly emerging influenza viruses. Isotope dilution mass spectrometry (IDMS) for accurate quantification of proteins in complex mixtures was demonstrated in our laboratory in 199627 and has been expanded to include quantification of viral proteins in seasonal vaccine strains. Tryptic peptides conserved among the H7N7, H7N2, and H7N9 proteins were identified, selected as stoichiometric representatives of the HA and NA proteins from which they were cleaved, and quantified against a spiked internal standard to yield a measure of protein concentration. The standard procedure in our method uses enzymatic digestion to generate unique viral peptide fragments, separation of these peptides by liquid chromatography and quantification by multiple-reaction monitoring (MRM).22,23 The steps taken to assure complete digestion of the viral proteins have been previously discussed. 28
In order to assure complete digestion of HA and NA in the region of the target peptides of the virus strain of interest, we quantified four unique peptides from different regions of HA protein, two unique peptides from the NA of N7 strains, one peptide from the NA of N2 strains, and three peptides from the NA of N9 strains. The amino acid sequences for HA and NA proteins are shown in Figure 1. The target peptides used for quantitation are underlined. Obtaining similar results using at least two peptides that are quantified independently demonstrate that the protein is completely digested in the region of the target peptides.
Figure 1.

The amino acid sequences of the HA and NA proteins of the H7N7 strains and the NA of the H7N2 and the H7N9 proteins are shown with target peptides in bold and underlined. To ensure complete protein digestions, four unique H7 peptides were chosen from different regions of the HA protein. Obtaining similar results using at least two peptides located in different regions demonstrates that the protein is completely digested in the region of the peptides used for protein quantitation.
There are several key steps to follow when choosing peptides to use for targeted protein quantification. Others have described the general rules on choosing the best peptides for quantification.29 Briefly, IDMS is a targeted quantification method and requires knowledge of the accurate mass of the peptides to be used for quantification. Amino acids that can be readily modified must be avoided in the chosen target peptide. Also, sample preparation protocols that induce changes in the molecular weight of an amino acid are not desired unless conversion is 100% efficient. For this reason, reduction and alkylation of the sample is not part of our sample digestion procedure. Peptides that contain oxidizable amino acids (methionine or tryptophan) or cysteine which might be involved in a disulfide bond with another cysteine in a distal region of the protein are not chosen as quantification targets.30 Peptides containing an N-linked glycosylation consensus sequence are avoided as glycosylation is a very heterogeneous post-translational modification in which the mass of the glycan cannot be accurately predicted.31
To avoid the need to make new standards annually, we aim for target peptides that are conserved throughout the subtype. We chose four peptides for the quantification of the HA of the influenza H7. Two of the peptides are on the HA1 portion of the protein while the other two are on HA2 The amino acid sequences of several strains of H7 (H7N2, H7N3, H7N7, and H7N9) is shown in Figure 2. Even though the subtypes are different and the strains were collected in various regions of the world including Mexico, China, United States, and the Netherlands, the same four regions of the protein sequence could be targeted by IDMS. The peptides and the extent to which they are conserved throughout the subtype are shown in Table 2. STQSAIDQITGK is conserved in 1178 of the 1463 strains (81%) that have been sequenced in this region of the protein and for all available sequences deposited in the NCBI Influenza Virus Resource Database as of July 24, 2013.32 The VNTLTER is conserved in 1116 of the 1463 strains (76%) while the IQIDPVK is conserved in 1120 of the 1463 strains (77%). FVNEEALR is conserved in 434 strains while a peptide with two amino acid differences (FTNEESLR) was present in 800 of the strains in the database. Both sequences combined are conserved in 1234 of the 1463 strains (84%). To increase the chances of quantifying any H7 strain, we incorporated both FVNEEALR and FTNEESLR in our method.
Figure 2.

Amino acid sequence of strains of H7N7, H7N2, H7N3, and H7N9 showing the conserved peptides used for IDMS quantification of HA. The peptides chosen can be used to quantify strains collected from various areas of the world including China, Mexico, United States, and the Netherlands.
Table 2.
We identified the target peptides among the H7, N7 and N2 subtypes of concern. The N* and H* means from any NA or HA strain. The peptides that are shown in gray were not analyzed in this method. All full-length HA and NA sequences for the H7N7 and H7N2 strains were analyzed through July 24, 2013. Peptides that are conserved among the strains are most valuable as the need to generate new peptide standards upon annual strain changes is less likely.
| Target Peptide Sequence | # strains of this subtype in NCBI database as of July 24, 2013 | # of strains containing target peptide | Extent of conservation of target peptide | |
|---|---|---|---|---|
| H7N* | VNTLTER | 1463 | 1116 | 76% |
|
|
|
|||
| FVNEEALR | 434 | 84% (combined) | ||
| FTNEESLR | 800 | |||
|
|
|
|||
| IQIDPVK | 1120 | 77% | ||
|
|
|
|||
| STQSAIDQITGK | 1178 | 81% | ||
|
| ||||
| H*N7 | IPNAGTDPNSR | 688 | 400 | 83% (combined) |
| IPNAETDPNSK | 172 | |||
|
|
|
|||
| FGESEQIIVTR | 542 | 79% | ||
|
| ||||
| H9N2, H7N2 | SGYETFR | 1693 | 1249 | 74% |
|
| ||||
| H*N9 | IGESSDVLVTR | 538 | 67 | 89% (combined) |
| IGENSDVLVTR | 163 | |||
| IGEDSDVLVTR | 247 | |||
|
|
|
|||
| FYALSQGTTIR | 502 | 93% | ||
|
|
|
|||
| VPNALTDDR | 499 | 93% | ||
Our efforts were focused on identifying conserved peptides among HA subtypes of concern, (either seasonal influenza viruses or emerging avian viruses with pandemic potential (i.e., H7 from H7N9)). Therefore, peptides had been prepared in anticipation of H7N7 or H7N2 strains of influenza. These peptides were conserved and applicable for quantitation of HA in the H7N9 strains that were circulating in China in the spring of 2013. However, we had not prepared standards suitable for the analysis of NA of N9 strains. We were able to rapidly identify three target peptides for NA of N9 sequences and new isotopically labeled peptides were synthesized and accurately characterized in a matter of two weeks. By anticipating potential circulating influenza subtypes and choosing peptides that are conserved within the subtype allows for a quantitative method to be developed for a broader range of influenza strains. The flexibility of the IDMS method and the straight-forward manner in which peptide standards are produced allows rapid method development, if necessary, and quantification of HA in newly emerging strains of influenza. The implementation of IDMS, therefore, could shorten development time for new vaccines.
The N9 peptide IGESSDVLVTR is only present in 67 of the 538 N9 strains that had been deposited in the NCBI database. IGENSDVLVTR in and IGEDSDVLVTR are the peptides that are present in other strains of H9. Combined, these three peptides would cover 89% of the strains that had been deposited in the NCBI database. IGENSDVLVTR would be a necessary peptide to quantify H2N9 while H11N9 would be quantified using IGEDSDVLVTR. Since we are currently focused only on the analysis H7N9, only IGESSDVLVTR was prepared as a standard. The FYALSQGTTIR peptide is present in 502 of the 538 N9 strains (93%) in the NCBI database. The sequence VPNALTDDR is present in 499 of the 538 strains (93%). These two peptides would be useful for the quantification of all N9 strains, even those of H2N9 and H11N9.
Two peptides, IPNAGTDPNSR and FGESEQIIVTR, are incorporated into this method for the quantification of N7. When all strains of N7 are considered, there are two closely related peptides that cover 83% of the strains. IPNAGTDPNSR and IPNAETDPNSK, when combined, cover 572 of the 688 strains. However, IPNAETDPNSK is present predominantly in H10N7 strains, not H7N7 strains. That peptide, therefore, was not prepared as a standard and is shown in Figure 2 in grey font since it is not useful for the analysis of the H7N7 or the H7N9 strains. However, it would need to be prepared if H10N7 strains ever became of interest. The second N7 peptide, FGESEQIIVTR, is present in 542 of the 688 (79%) N7 strains in the NCBI database which suggests that this peptide would work for both H7N7 and H10N7 strains. The peptide used to quantify N2 strains, SGYETFR, is present in 1249 of the 1693 N2 sequences (74%) in the NCBI database.
Quantification of N2 neuraminidase was integrated into the method, but currently only one peptide is used. If regulatory agencies become interested in quantifying NA, then the time and cost required to add additional peptides to the method may be justified. However, since NA is digested and quantified at the same time as HA, we assume that if digestion is complete for H7, N7, and N9, which all include multiple peptide targets for each protein that it is likely complete for similarly sized N2. One peptide is sufficient for protein quantification purposes related to research or characterization.
Chromatograms of the peptide pairs corresponding to HA and NA of the H7N7 subtype are shown in Figure 3. All six peptides are analyzed in one analytical run. The area ratio between the native peptide and the isotopically labeled peptide is calculated and compared to that of the known standard amounts used to generate the calibration curve. The IDMS method, which is a targeted method, provides a chromatographic reference for peak selection and adds another degree of selectivity to the method. The clarity of this chromatogram is typical of any sample regardless of matrix. Thus IDMS is able to quantify viral proteins from purified samples, split and subunit vaccine preparations, and even crude allantoic fluid.
Figure 3.

Liquid chromatography-tandem mass spectrometry (LC-MS/MS) extracted ion chromatograms of target H7 and N7 peptides for quantifying a standard in which 6 peptide pairs were simultaneously monitored. Amino Acids shown in red are 13C and 15N stable isotope labeled. Data were acquired on a Thermo Scientific TSQ Vantage.
Typical quantitative results are presented in Table 3. Since the assumption is that one mole of peptide equals one mole of protein, each peptide is an independent quantitative measurement of the amount of protein in the sample. The method requires complete digestion in the region of the peptide for an accurate measurement. Similar quantitative results using multiple peptides provide assurance that the digestion of the protein in the region of the target peptides is complete and that the method produces an accurate result. The inactivated whole virus sample of the A/Netherlands/219/2003 (H7N7) and the purified virus A/Shanghai/2/2013 (H7N9) were analyzed using the same comprehensive method. In one analytical run, all four HA peptides from the two H7 strains were analyzed as were the NA peptides. The values obtained using the various peptides were then averaged. The average amount of HA in the inactivated whole virus sample A/Netherlands/219/2003 (H7N7) using all four H7 peptides was calculated to be 81.8 fmol/μL. The average value obtained using the two N7 peptides was 35.8 fmol/μL. A 10 μL aliquot of the purified virus A/Shanghai/2/2013 (H7N9) contained 150.0 fmol/μL of HA and 12.7 fmol/μL of NA determined by three N9 peptides. A final vaccine preparation contains 30 μg/mL (40 fmol/μL) of HA. The sensitivity of the IDMS method is such that HA levels as low as 1.5 μg/mL can be quantified—an amount much lower than required by regulatory agencies.
Table 3.
The amount (in fmol/μL) of H7, N7 and N9 determined using individual peptides from three replicate sample preparations is shown. The average and standard deviation between the peptides are calculated using each peptide and each replicate run as independent measurements (n=12, 6, and 9 respectively).
| H7N7 HA (fmol/uL) | H7N7 NA (fmol/uL) | H7N9 NA (fmol/uL) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| VNTLTER | FVNEEALR | IQIDPVK | STQSAIDQITGK | IPNAGTDPNSR | FGESEQIIVTR | IGESSDVLVTR | FYALSQGTTIR | VPNALTDDR | |
| A/Netherlands/219/2003 (H7N7) | 79.2 | 76.2 | 93.0 | 78.8 | 31.9 | 39.7 | |||
| 81.8 ± 8.6 | 35.8 ± 5.5 | ||||||||
| A/Shanghai/2/2013 (H7N9) | 143.4 | 160.9 | 137.6 | 157.9 | 13.3 | 10.9 | 14.0 | ||
| 150.0 ± 11.2 | 12.7 ± 1.6 | ||||||||
Conclusions
We have developed an accurate and precise isotope-dilution mass spectrometry (IDMS) method using a purified virus preparation of A/Netherlands/219/2003 (H7N7) to quantify the HA and the NA proteins in one analytical run. This IDMS method was also adapted for N2 and N9 neuraminidases and is also successfully used for quantifying the HA and NA content in the purified virus sample of A/Shanghai/2/2013 (H7N9). IDMS is an accurate, precise, and sensitive method that can be used to quantify multiple influenza viral proteins. The mass spectrometer offers a high level of selectivity based on the mass (and therefore, amino acid sequence of the peptide) so that multiple proteins from different viral subtypes can be simultaneously quantified. The IDMS method utilizes peptides that are conserved among H7, N7, N2 and N9 strains to provide insurance that the protein can be quantified regardless of strain. However, should a strain emerge that has a mutation in the selected peptide, the sequence of a new peptide target can easily be identified and new peptide standards can be synthesized and accurately characterized in a matter of a few weeks. The flexibility and sensitivity of the IDMS method would enable rapid quantification of HA in newly emerging strains of influenza, and consequently, it would shorten development time for new vaccines.
Acknowledgments
We thank Dr. Adrian Woolfitt and Ms. Maria Solano for AAA analyses of all peptides used as standards in this research. We also thank Dr. Ruben O. Donis and Dr. Li-Mei Chen of the Influenza Division, OID, NCIRD, CDC, Atlanta, GA, for manuscript suggestions and providing inactivated H7N9 virus samples.
We acknowledge the Center for Biologics Evaluation and Research (CBER) of the U.S. Food and Drug Administration for providing H7N2 and H7N7 samples. Reference in this article to any specific commercial products, process service, manufacturer, or company does not constitute an endorsement or a recommendation by the U.S. government or the Centers for Disease Control and Prevention. The findings and conclusions reported in this article are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
References
- 1.Subbarao K, Joseph T. Nature reviews Immunology. 2007;7:267–78. doi: 10.1038/nri2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerdil C. Vaccine. 2003;21:1776–9. doi: 10.1016/s0264-410x(03)00071-9. [DOI] [PubMed] [Google Scholar]
- 3.Alexander DJ. Veterinary microbiology. 2000;74:3–13. doi: 10.1016/s0378-1135(00)00160-7. [DOI] [PubMed] [Google Scholar]
- 4.Liu W, Zhu Y, Qi X, Xu K, Ge A, Ji H, Ai J, Bao C, Tang F, Zhou M. Journal of biomedical research. 2013;27:163–6. doi: 10.7555/JBR.27.20130071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bridges CB, Lim W, Hu-Primmer J, Sims L, Fukuda K, Mak KH, Rowe T, Thompson WW, Conn L, Lu X, Cox NJ, Katz JM. The Journal of infectious diseases. 2002;185:1005–10. doi: 10.1086/340044. [DOI] [PubMed] [Google Scholar]
- 6.Mounts AW, Kwong H, Izurieta HS, Ho Y, Au T, Lee M, Buxton Bridges C, Williams SW, Mak KH, Katz JM, Thompson WW, Cox NJ, Fukuda K. The Journal of infectious diseases. 1999;180:505–8. doi: 10.1086/314903. [DOI] [PubMed] [Google Scholar]
- 7.Webster RG, Peiris M, Chen H, Guan Y. Emerging infectious diseases. 2006;12:3–8. doi: 10.3201/eid1201.051024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Areechokchai D, Jiraphongsa C, Laosiritaworn Y, Hanshaoworakul W, O'Reilly M Centers for Disease C and Prevention. MMWR Morbidity and mortality weekly report. 2006;55(Suppl 1):3–6. [PubMed] [Google Scholar]
- 9.Salzberg SL, Kingsford C, Cattoli G, Spiro DJ, Janies DA, Aly MM, Brown IH, Couacy-Hymann E, De Mia GM, Dung do H, Guercio A, Joannis T, Maken Ali AS, Osmani A, Padalino I, Saad MD, Savic V, Sengamalay NA, Yingst S, Zaborsky J, Zorman-Rojs O, Ghedin E, Capua I. Emerging infectious diseases. 2007;13:713–8. doi: 10.3201/eid1305.070013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wood JM. Philosophical transactions of the Royal Society of London Series B Biological sciences. 2001;356:1953–60. doi: 10.1098/rstb.2001.0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Webby RJ, Perez DR, Coleman JS, Guan Y, Knight JH, Govorkova EA, McClain-Moss LR, Peiris JS, Rehg JE, Tuomanen EI, Webster RG. Lancet. 2004;363:1099–103. doi: 10.1016/S0140-6736(04)15892-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayden FG, Abdel-Ghafar AN, Chotpitayasunondh T, Gao ZC, Hien ND, de Jong MD, Naghdaliyev A, Peiris JSM, Shindo N, Soeroso S, Uyeki TM Consultation, n. W. H. O. New England Journal of Medicine. 2008;358:261–273. doi: 10.1056/NEJMra0707279. [DOI] [PubMed] [Google Scholar]
- 13.Ai J, Huang Y, Xu K, Ren D, Qi X, Ji H, Ge A, Dai Q, Li J, Bao C, Tang F, Shi G, Shen T, Zhu Y, Zhou M, Wang H. Euro surveillance : bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin. 2013;18:20510. doi: 10.2807/1560-7917.es2013.18.26.20510. [DOI] [PubMed] [Google Scholar]
- 14.Min JY, Vogel L, Matsuoka Y, Lu B, Swayne D, Jin H, Kemble G, Subbarao K. Journal of virology. 2010;84:11950–60. doi: 10.1128/JVI.01305-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du Ry, van Beest Holle M, Meijer A, Koopmans M, de Jager CM. Euro surveillance : bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin. 2005;10:264–8. [PubMed] [Google Scholar]
- 16.Fouchier RA, Schneeberger PM, Rozendaal FW, Broekman JM, Kemink SA, Munster V, Kuiken T, Rimmelzwaan GF, Schutten M, Van Doornum GJ, Koch G, Bosman A, Koopmans M, Osterhaus AD. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1356–61. doi: 10.1073/pnas.0308352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicoll A, Danielsson N. Euro surveillance : bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin. 2013;18:20452. [PubMed] [Google Scholar]
- 18.Li Q, Zhou L, Zhou M, Chen Z, Li F, Wu H, Xiang N, Chen E, Tang F, Wang D, Meng L, Hong Z, Tu W, Cao Y, Li L, Ding F, Liu B, Wang M, Xie R, Gao R, Li X, Bai T, Zou S, He J, Hu J, Xu Y, Chai C, Wang S, Gao Y, Jin L, Zhang Y, Luo H, Yu H, Gao L, Pang X, Liu G, Shu Y, Yang W, Uyeki TM, Wang Y, Wu F, Feng Z. The New England journal of medicine. 2013 [Google Scholar]
- 19.Kageyama T, Fujisaki S, Takashita E, Xu H, Yamada S, Uchida Y, Neumann G, Saito T, Kawaoka Y, Tashiro M. Euro surveillance : bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin. 2013;18:20453. [PMC free article] [PubMed] [Google Scholar]
- 20.Wood JM, Schild GC, Newman RW, Seagroatt V. Developments in biological standardization. 1977;39:193–200. [PubMed] [Google Scholar]
- 21.Wood JM, Schild GC, Newman RW, Seagroatt V. Journal of biological standardization. 1977;5:237–47. doi: 10.1016/s0092-1157(77)80008-5. [DOI] [PubMed] [Google Scholar]
- 22.Williams TL, Luna L, Guo Z, Cox NJ, Pirkle JL, Donis RO, Barr JR. Vaccine. 2008;26:2510–20. doi: 10.1016/j.vaccine.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 23.Williams TL, Pirkle JL, Barr JR. Vaccine. 2012;30:2475–82. doi: 10.1016/j.vaccine.2011.12.056. [DOI] [PubMed] [Google Scholar]
- 24.Woolfitt AR, Solano MI, Williams TL, Pirkle JL, Barr JR. Analytical chemistry. 2009;81:3979–85. doi: 10.1021/ac900367q. [DOI] [PubMed] [Google Scholar]
- 25.Yu YQ, Gilar M, Lee PJ, Bouvier ES, Gebler JC. Analytical chemistry. 2003;75:6023–8. doi: 10.1021/ac0346196. [DOI] [PubMed] [Google Scholar]
- 26.Caudill SP, Schleicher RL, Pirkle JL. Statistics in medicine. 2008;27:4094–106. doi: 10.1002/sim.3222. [DOI] [PubMed] [Google Scholar]
- 27.Barr JR, Maggio VL, Patterson DG, Jr, Cooper GR, Henderson LO, Turner WE, Smith SJ, Hannon WH, Needham LL, Sampson EJ. Clinical chemistry. 1996;42:1676–82. [PubMed] [Google Scholar]
- 28.Norrgran J, Williams TL, Woolfitt AR, Solano MI, Pirkle JL, Barr JR. Analytical biochemistry. 2009;393:48–55. doi: 10.1016/j.ab.2009.05.050. [DOI] [PubMed] [Google Scholar]
- 29.Andrews P, Arnott J, Farmar J, Ivanov A, Kowalak J, Lane W, Mechtler K, Ogorzalek Loo R, Raida M. The Association of Biomolecular Resource Facilities. Salt Lake City: 2008. [Google Scholar]
- 30.Mo W, Ma Y, Takao T, Neubert TA. Rapid communications in mass spectrometry : RCM. 2000;14:2080–1. doi: 10.1002/1097-0231(20001115)14:21<2080::AID-RCM120>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 31.Blake TA, Williams TL, Pirkle JL, Barr JR. Analytical chemistry. 2009;81:3109–18. doi: 10.1021/ac900095h. [DOI] [PubMed] [Google Scholar]
- 32.Sayers EWBT, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S. Nucleic Acids Res. 2009 Jan;:D5–15. doi: 10.1093/nar/gkn741. [DOI] [PMC free article] [PubMed] [Google Scholar]