

Abstract
Cytological studies have shown many newly formed allopolyploids (neoallopolyploids) exhibit chromosomal variation as a result of meiotic irregularities, but few naturally occurring neoallopolyploids have been examined. Little is known about how long chromosomal variation may persist and how it might influence the establishment and evolution of allopolyploids in nature. In this study we assess chromosomal composition in a natural neoallotetraploid, Tragopogon mirus, and compare it with T. miscellus, which is an allotetraploid of similar age (~40 generations old). We also assess whether parental gene losses in T. mirus correlate with entire or partial chromosome losses. Of 37 T. mirus individuals that were karyotyped, 23 (62%) were chromosomally additive of the parents, whereas the remaining 14 individuals (38%) had aneuploid compositions. The proportion of additive versus aneuploid individuals differed from that found previously in T. miscellus, in which aneuploidy was more common (69% Fisher's exact test, P=0.0033). Deviations from parental chromosome additivity within T. mirus individuals also did not reach the levels observed in T. miscellus, but similar compensated changes were observed. The loss of T. dubius-derived genes in two T. mirus individuals did not correlate with any chromosomal changes, indicating a role for smaller-scale genetic alterations. Overall, these data for T. mirus provide a second example of prolonged chromosomal instability in natural neoallopolyploid populations.
Introduction
Allotetraploids are formed through interspecific hybridization together with genome duplication; thus, each parental chromosome is expected to be present in two copies (disomy). However, almost all newly formed allopolyploids (neoallopolyploids) produce some chromosomally variable progeny, due to pairing or segregation errors at meiosis (Gottschalk, 1978; Ramsey and Schemske, 2002). Few allopolyploids have been studied using modern cytogenetic tools that allow the complete karyotype to Florida Museum of Natural History be discerned unequivocally. As a result, little is known about what types of chromosomal variation arise in neoallopolyploids and to what extent they may persist. To begin to address these questions in natural neoallotetraploids, we previously surveyed the neoallotetraploid Tragopogon miscellus and observed a surprisingly high amount of chromosomal variation in these ~40-generation-old populations (Chester et al., 2012). Most individuals of T. miscellus showed either rearrangements (translocations) or aneuploidy, often both, but variation typically followed a compensated pattern, similar to that observed in resynthesized allotetraploid Brassica napus. In B. napus, changes often resulted in substitutions of homeologous chromosomes or homeologous chromosome segments (Xiong et al., 2011).
Similarities between the two systems (Tragopogon and Brassica) and classical cytological studies (Poole, 1932; Clausen et al., 1940; Longwell and Sears, 1963) support gene dosage as an important constraint on chromosomal variation in allotetraploids (Xiong et al., 2011; Birchler, 2012). More natural and synthetic polyploid systems need to be studied to determine how widely these putative ‘rules' apply. For example, in contrast to B. napus and T. miscellus, chromosomal variation in synthetic lines of allohexaploid wheat did not adhere to a compensated pattern, and no chromosomal rearrangements were detected (Zhang et al., 2013b).
Here we survey chromosomal variation in a natural neoallotetraploid, T. mirus. Like T. miscellus, this biennial formed repeatedly in North America, ~80 years ago. The two neoallotetraploids also share a similar genetic background; T. mirus is derived from the diploids T. dubius and T. porrifolius and T. miscellus is derived from T. dubius and T. pratensis (Ownbey, 1950). T. porrifolius and T. pratensis are closely related, and both are more distantly related to T. dubius (Mavrodiev et al., 2005, 2007). Plants from six populations in Washington State, USA, were analyzed (Figure 1), five of which (Rosalia, Tekoa, Oakesdale, Palouse and Pullman-1) were previously confirmed as having formed independently based on nuclear microsatellites (Symonds et al., 2010). The origin and genetic structure of the sixth collection site, Pullman-3, has not been analyzed previously, but may represent an additional origin.
Figure 1.
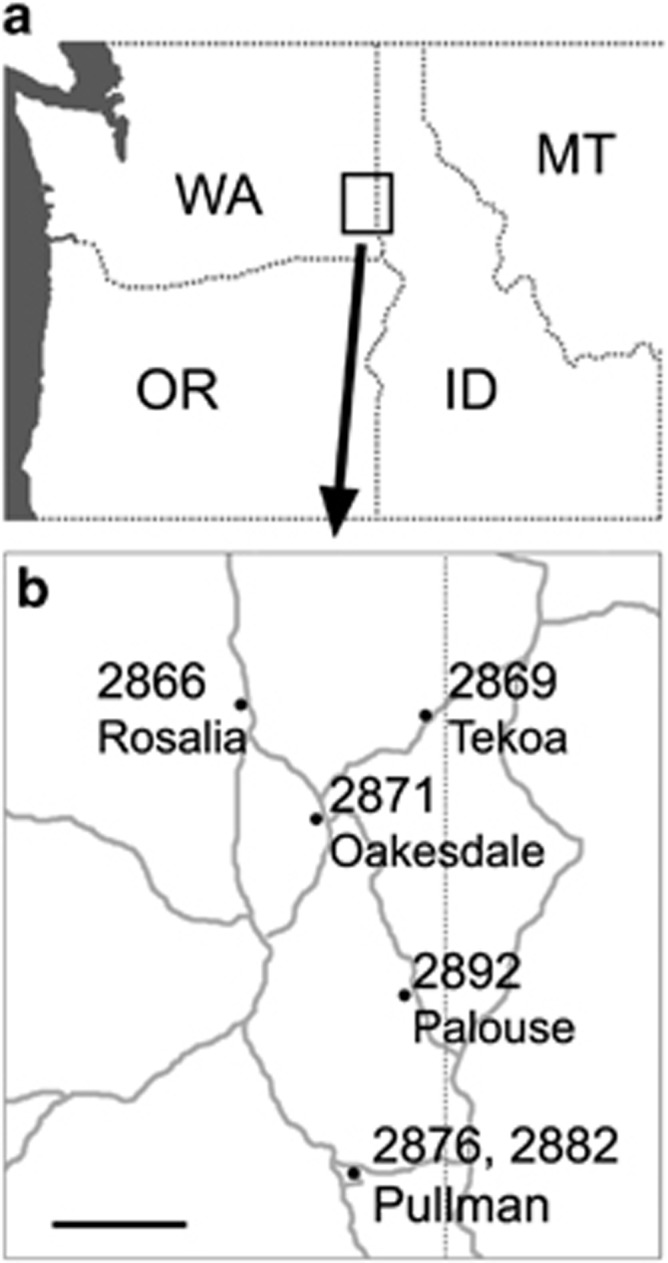
Map showing collection sites of Tragopogon mirus. (a) Pacific Northwest of USA, showing the states of Washington (WA), Oregon (OR), Idaho (ID) and Montana (MT). (b) Populations from where seeds of T. mirus were collected. Major roads are indicated by gray lines. Scale bar, 10 mi.
The existence of chromosomal variation in T. mirus was revealed in an earlier study, with six out of seven plants showing deviations from the expected parentally additive karyotype (Lim et al., 2008). By applying an updated methodology that allows all individual chromosomes to be identified (Chester et al., 2012, 2013), we can now identify cytogenetic similarities and differences on a per-chromosome basis. We use this approach in the present study to conduct a survey of chromosomal variation in T. mirus and compare the results with those obtained earlier for T. miscellus (Chester et al., 2012). We also make use of the karyotyped T. mirus individuals to examine the phenomenon of homeologous gene loss, where allotetraploids show non-additivity of homeologous progenitor genes (Tate et al., 2006; Buggs et al., 2009; Tate et al., 2009; Koh et al., 2010; Buggs et al., 2012). To assess whether parental gene non-additivity correlates with chromosome loss or non-reciprocal translocations, three pairs of homeologous genes are assayed via PCR for the T. mirus individuals that were karyotyped. In summary, we assess how much chromosomal variation is present in T. mirus and if this variation can provide an explanation for parental gene non-additivity.
Materials and methods
Plant materials
For mitotic chromosome preparations, T. mirus plants were grown from seed collected from the following locations in Washington State, USA: Rosalia (Soltis & Soltis collection number: 2866), Tekoa (2869), Oakesdale (2871), Pullman (site 1; SW Elm Street; 2876), Pullman (site 3; North Grand Avenue near the intersection with Stadium Way; 2882) and Palouse (2892) (Figure 1 and Supplementary Table S1). Specimen vouchers for the above populations are deposited at the University of Florida Herbarium (FLAS). Seeds from five to seven maternal individuals from each population were germinated and grown in a temperature-controlled greenhouse at the University of Florida. After 3 months, chromosome preparations were carried out on one plant per maternal individual. Parental species were also grown from seed to provide leaf material as a source of genomic DNA for GISH: T. dubius (2674-4) was collected from Oakesdale, and T. porrifolius (2677-8) was collected from Pullman.
Chromosome preparation
The final 2 cm of growing roots were harvested and pretreated in an aqueous solution of 2 mM 8-hydroxyquinoline (Sigma-Aldrich Co., St Louis, MO, USA) overnight for 16 h at 4 °C. Pretreated roots were then fixed in ice-cold 90% acetic acid for 10 min and transferred to 70% ethanol for −20 °C storage, as described by Kato et al. (2011). Roots were digested and chromosome spreads were prepared following Kato et al. (2011). Following washing in 1 × citric buffer (10 mM sodium citrate, 10 mM EDTA; pH 5.5), roots were digested individually for 41–48 min (depending on size) at 37 °C in 20-μl aliquots of enzyme mixture containing 1% pectolyase Y-23 (MP Biochemicals, LLC, Solon, OH, USA) and 2% cellulase Onozuka R-10 (Research Products International Corp., Mt. Prospect, IL, USA) in 1 × citric buffer. Cold 70% ethanol was used to wash digested roots, after which ethanol was removed except for the final 2 μl, and 26–30 μl of glacial acetic acid was added. After maceration with a blunt dissecting needle, 3.5-μl aliquots of the cell suspension were pipetted onto slides in a humid chamber. After 10 min, slides were inspected with a phase contrast microscope; those containing metaphase spreads with non-overlapping chromosomes were stored at −20 °C for a maximum of 1 week prior to in situ hybridization.
Fluorescence and genomic in situ hybridization
We followed the methods of Kato et al. (2011) for the labeling and hybridization of DNA probes. Tandem repeat and GISH probes were made from 5 μg of double-stranded DNA (dsDNA); the β-fructosidase probe was made using 2 μg of dsDNA substrate. Probes were directly labeled by incorporating one of the following by nick translation: fluorescein-12-dUTP, Cyanine-3-dUTP or Cyanine-5-dUTP (PerkinElmer, Inc., Waltham, MA, USA). Nick translation products were purified using a QIAquick Nucleotide Removal Kit (Qiagen, Inc., Valencia, CA, USA).
For the GISH probes, total genomic DNA (gDNA) of the parental species was isolated using a cetyltrimethylammonium bromide method (Doyle and Doyle, 1987), RNase was treated and purified using a DNeasy Extraction Kit (Qiagen, Inc.). For fluorescence in situ hybridization (FISH), 18S ribosomal DNA (rDNA) was amplified by PCR from a cloned 1.3-kbp fragment from T. dubius; for 5S rDNA, centromeric TPRMBO (Pires et al., 2004), subtelomeric TGP7 (Pires et al., 2004) and interstitial TTR3 (Chester et al., 2013), dsDNA was made by annealing complementary oligonucleotides of ~60 bp in length as previously described (Chester et al., 2013). Three different in situ hybridization probe mixtures were used for karyotyping: (1) FISH with 18S rDNA, TPRMBO, TGP7 and TTR3 repetitive DNA sequences, and/or (2) genomic in situ hybridization (GISH) with gDNA of T. porrifolius and T. dubius, or (3) a modified GISH that in addition to the gDNA of the diploid progenitors also included a 5S rDNA probe.
For each hybridization mixture, we used the following probe amounts, probe labels and blocking DNA amounts. The FISH mixture comprised 350 ng Cy5-labeled TPRMBO probe, 180 ng Cy3-labeled TGP7 probe, 300 ng fluorescein-labeled TTR3 probe, 20 ng Cy3-labeled 18S rDNA probe, 20 ng fluorescein-labeled 18S rDNA probe and 700 ng of unlabeled sheared salmon sperm DNA in 0.7 × SSC (300 mM NaCl, 30 mM sodium citrate; pH 7.0). The GISH mixture comprised 400 ng fluorescein-labeled T. dubius gDNA, 400 ng Cy3-labeled T. pratensis gDNA and 560 ng sheared salmon sperm DNA in 0.7 × SSC. The modified GISH mixture comprised 40 ng fluorescein-labeled T. dubius gDNA, 40 ng Cy3-labeled T. porrifolius gDNA, 300 ng Cy5-labeled 5S rDNA and 560 ng sheared salmon sperm DNA in 0.5 × SSC.
Prior to in situ hybridization, slides were ultraviolet cross-linked (120 mJ cm−2), the hybridization mixture was added to the slide and a plastic coverslip was then placed on top. The slides containing the probe mixtures were denatured at 82–83 °C for 2 min 30 s and transferred to a sealed humid box for incubation at 55 °C for 16 h in the case of FISH or 36 h in the cases of GISH (including or excluding 5S rDNA probe). Following hybridization, slides were washed briefly in 2 × SSC to remove the coverslip. Glass coverslips (Corning, Inc., Corning, NY, USA) were then mounted using Vectashield containing 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Vector Laboratories, Inc., Burlingame, CA, USA). Hybridized chromosome spreads were observed and imaged using a Zeiss Axio Imager.M2 fluorescence microscope (Carl Zeiss MicroImaging, Inc., Thornwood, NY, USA) with an X-Cite Series 120 Q Lamp (Lumen Dynamics Group, Inc., Mississauga, ON, Canada). The brightness and contrast of captured images was adjusted in AxioVision (version 4.8 Special Edition 64 bit, Carl Zeiss MicroImaging, Inc.) by moving the upper and lower cutoffs in the histogram of signal intensity. Images captured on individual channels were pseudocolored in the following way: DAPI: gray, Cy3: red, Cy5: lilac and fluorescein: green. For chromosome preparations that were GISH-reprobed, after FISH imaging, the glass coverslip was removed in 55° 2 × SSC, and GISH was carried out as described above.
Karyotype construction and analysis
Karyotypes based on FISH and/or GISH were assembled in Photoshop CS3 (Adobe Systems Inc., San Jose, CA, USA) using merged and pseudocolored TIFF images exported from AxioVision. Subgenome designations were as follows: Du (T. dubius origin) and Po (T. porrifolius origin). Chromosome designations A to F for chromosomes in each subgenome (Du and Po) were based on Chester et al. (2013).
A Fisher's exact test was conducted using R (R Core Team, 2012) to assess whether there was a significant difference in the proportions of euploids and aneuploids between T. mirus and T. miscellus. To test if aneuploidy deviated from a random distribution across the six chromosome groups in T. mirus, a multinomial test was conducted on the total aneuploidy counts per group (A–F) in R using the EMT package (Menzel, 2013). Binomial tests were conducted in R to assess whether T. mirus aneuploidy counts for the individual chromosome groups deviated from the expected 1/6th frequency using a Bonferroni-corrected significance level of 0.0083.
The numbers of intergenomic translocation homozygotes, non-recombined homozygotes and heterozygotes expected under Hardy–Weinberg equillibrium (HWE) were calculated in Excel (Microsoft Corp., Redmond, WA, USA). Calculations were only conducted on translocations that occurred on chromosomes that were consistently disomic and if the breakpoint position was unique to the arm in the population (that is, arms with multiple translocations were excluded). To test for a significant departure from HWE, a Hardy–Weinberg exact test (Haldane, 1954) was conducted on each set of translocations using Genepop (version 4.2; Rousset, 2008).
Gene homeolog analysis
gDNA was extracted from leaves for all 37 individuals of T. mirus that were karyotyped, and also the parent species, T. dubius and T. porrifolius, using a cetyltrimethylammonium bromide method (Doyle and Doyle, 1987). Three genes that were previously found to be fixed in the parents, but show genetic non-additivity in some T. mirus, were PCR amplified: TDF36.3, a gene putatively encoding thioredoxin M-type I (Tate et al., 2006; Koh et al., 2010); TDF85, a gene putatively encoding β-fructosidase (Tate et al., 2006; Koh et al., 2010); and MHC, a gene putatively encoding myosin heavy chain (Koh et al., 2010). PCR primer sequences were THIOR_F1: 5′-AATCAGAAGCATCCCGACTG-3′ and THIOR_R1 5′-CACAATCTTTTTGTGAAATGCAA-3′ BFRUCT4_F1: 5′-GGAAGACCTTGATTGATCGG-3′ and BFRUCT4_R1: 5′-AAGGATGTTGTGGTGGAAGC-3′; MHC_F1: 5′-CGACACGGAATATAGCATCC-3′ and MHC_R1: 5′-GGATAAAGTGATGCTCATATGG-3′. The PCR profile was 2 min at 95 °C, 34 cycles of 30 s at 95 °C, 30 s at 55 °C and 45 s at 72 °C and a final extension of 5 min at 72 °C. The size difference between homeologs of β-fructosidase and myosin heavy chain allowed them to be identified by separation of PCR products on 2% agarose gels. For thioredoxin, PCR products were digested with DdeI to identify parental homeologs, with a single cut site present in the T. dubius homeolog. For the three genes examined, we confirmed that the strongest amplification products matched the size that was documented previously (Tate et al., 2006; Koh et al., 2010).
To generate a FISH probe for the β-fructosidase gene, we first searched assembled transcriptome sequence data from T. dubius (Buggs et al., 2010) using tBLASTx (Altschul et al., 1997) and querying with a partial β-fructosidase coding sequence (GenBank accession number: DQ267230; Tate et al., 2006). Primers were then designed as close as possible to the beginning and end of a tentative transcript that showed 99% similarity to the query (BFRUCT_1F: 5′-GAGGTTTCCGGTACACATCA-3′ and BFRUCT_2043R: 5′-AACGACAATCATCATGCCACG-3′). A PCR amplification was conducted on gDNA of T. dubius, and a FISH probe was made from the purified PCR product (QIAquick PCR Purification Kit, Qiagen, Inc.) following the ‘small chromosomal targets' protocol in the study by Kato et al. (2011). The probe was tested on mitotic chromosomes of T. mirus individual 2876–1–1.
Results
Karyotyping of T. mirus
GISH to metaphase chromosome preparations of T. mirus allowed the diploid parental chromosome sets (subgenomes) to be differentiated (for example, see Figure 2). Identification of individual chromosomes was also possible when taking into account chromosome size, arm length and the heterogeneous GISH signal along the chromosomes. We found that the E and F chromosomes of T. porrifolius-origin (EPo and FPo) were difficult to differentiate in cells where chromosomes were more condensed. To address this problem, a 5S rDNA probe was also included in subsequent GISH cocktails to help identify EPo, as this has a 5S rDNA site on the long arm (Chester et al., 2013; Supplementary Figures S1–S6). To validate GISH karyotyping, a FISH cocktail of repeated sequences was applied to five individuals showing varying amounts of parental chromosome non-additivity (Figure 3). Although there was no conflicting information on chromosome origin provided by FISH and GISH, some small subterminal (or distal) reductions or losses of arrays were only resolvable with FISH probes specific to the TGP7 or TTR3 repeats (Figure 3). Subtelomeric TGP7 signals, for example, were missing on the long arm of CPo (Figures 3e and f), but there was no indication of an intergenomic translocation based on the GISH signals at this position.
Figure 2.

Mitotic karyotypes of seven individuals from Rosalia, WA, USA (a–g). Chromosomes are shown following GISH using total genomic DNA of the parents, T. dubius (green) and T. porrifolius (red). Diamond symbols are placed below aneuploid chromosomes. Arrows indicate the position of translocation breakpoints. (a) 2866-8-1, (b) 2866-9-1, (c) 2866-10-1, (d) 2866-11-1, (e) 2866-12-1, (f) 2866-13-1 and (g) 2866-15-1. Scale bar, 5 μm.
Figure 3.

Karyotyping using FISH and GISH reprobing. (a, c, e, g and i) FISH probes were as follows: centromeric TPRMBO (lilac), subtelomeric TGP7 (red), interstitial TTR3 (green) and 18 S rDNA (yellow/orange). (b, d, f, h and j) GISH reprobing of the same chromosome preparations using total genomic DNA of the parents, T. dubius (green) and T. porrifolius (red). Diamond symbols are placed below aneuploid chromosomes. Arrows indicate the position of translocation breakpoints revealed by GISH. Arrowheads indicate missing FISH markers that did coincide with a translocation detected with GISH. (a and b) 2866-11-1, (c and d) 2866-15-1, (e and f) 2869-9-1, (g and h) 2869-10-1 and (i and j) 2876-10-1. Scale bar, 5 μm.
Chromosome copy number variation
Of 37 individuals, 23 (62%) were euploid, where each parental chromosome was disomic. The remaining 14 individuals (38%) had one or more chromosomes that were aneuploid, that is, in more or fewer than two copies. Individuals that had aneuploid chromosomes were classified as either numerical aneuploids or compensated aneuploids (Table 1; Ising, 1966). Five plants were numerical aneuploids with counts of 2n=23 (N=3) or 2n=25 (N=2); the remaining nine were compensated aneuploids, as they had the expected count of 2n=24, but not all chromosomes were disomic. In some cases there were three or four copies of the T. dubius chromosome and one or none, respectively, of the corresponding chromosome from T. porrifolius; in other cases there were three copies of the T. porrifolius chromosome and one corresponding chromosome from T. dubius (see Table 1; for a detailed summary see Supplementary Table S2). The incidence of aneuploidy across the six chromosome groups (A: 0, B: 6, C: 4, D: 4, E: 7, F: 0) deviated from a random distribution (exact multinomial test, P=0.0279). However, post-hoc testing of aneuploidy frequency for each individual chromosome group did not reveal any significant deviations from the expected 1/6th frequency (in all cases, exact binomial test with Bonferroni-corrected P>0.05).
Table 1. Summary of T. mirus karyotypes.
| Population | Euploid | Compensated aneuploid | Numerical aneuploid |
|---|---|---|---|
| Rosalia | 6 | 1 | 0 |
| Tekoa | 2 | 4 | 1 |
| Oakesdale | 3 | 0 | 2 |
| Pullman-1 | 6 | 0 | 0 |
| Pullman-3 | 4 | 1 | 1 |
| Palouse | 2 | 3 | 1 |
Chromosomal variation in eight of the nine compensated aneuploids followed a pattern where aneuploidy resulted in the swapping of diploid parental chromosomes of the same group, for example, substituting EDu with EPo (Figure 2b). The one exception to this pattern was individual 2882-2-1, which was monosomic for DDu and monosomic for a chromosome appearing to comprise the short arm of EPo and the long arm of FPo (Supplementary Figure S5a). For the other eight compensated aneuploids (where intragenomic recombination was not suspected), either mono-trisomy (1:3 copies) or nulli-tetrasomy (0:4 copies) was observed. Mono-trisomy was more frequent, with one or two occurrences involving chromosome groups B, D and E in a total of six individuals (Supplementary Table S2). Nulli-tetrasomy was only observed in two individuals (2869-10-1 and 2892-2-1), for groups B and C, respectively. Chromosome substitutions involving multiple chromosome groups per individual were not observed.
The number of aneuploidy observations was low, but the trend was towards an overall gain of T. dubius-origin chromosomes and a loss of T. porrifolius-origin chromosomes. Excluding the two individuals where intragenomic recombination was suspected (2882-1-1 and 2882-2-1; Supplementary Figures S5a and d), the number of chromosome losses (copy number was less than two) was 3:9 (Du: Po), and the number of chromosome gains (copy number was more than two) was 7:2 (Du: Po).
Genome rearrangements
Aside from individual 2882-2-1 (see above for details), the only other case of putative intragenomic recombination was found for another individual from Pullman-3, 2882-1-1 (2n=25), where a monosome appeared to comprise the short arm of EDu and long arm of FDu (Supplementary Figure S5a). Although 33 of the 37 individuals showed intergenomic translocations, the number per individual was low (N=4 had 0; N=19 had 1; N=10 had 2; N=2 had 3; N=2 had 4). Furthermore, due to some translocations being shared within a population, most individuals had either zero (N=22) or one (N=11) unique intergenomic translocation (Supplementary Tables S2–S7). Individuals 2869-14-1, 2869-15-1 and 2882-3-1 showed neither rearrangements nor aneuploidy and so appeared to be fully additive of the parents, in terms of both chromosome content and structure (Supplementary Figures S2j, k and S5e).
Almost all intergenomic translocations involved chromosomes of the same group, but the origin of the smallest distal translocated segments was difficult to ascertain due to a lack of diagnostic signals, so these may represent exceptions. The one clear exception was where part of ADu and part of BPo seem to have been exchanged in individual 2882-6-1 (Supplementary Figure S5j). Apart from group E, intergenomic translocations within groups were also consistently between the same arm of each parental chromosome, for example, short and short or long and long. For group E chromosomes, the 5S rDNA array on the EPo long arm was transferred to the short arm of EDu; this was observed in three individuals from three different populations (2882-1-1, 2876-15-1 and 2869-12-1) (Supplementary Figures S5c, S4j, k and S2f, g).
In some cases, plants from different populations showed intergenomic translocations at similar positions in the genome. The most abundant translocations were positioned in (i) the distal region of the group A long arms, observed in at least one individual from Rosalia, Pullman-3 and Palouse; (ii) the distal region of the group C short arms, observed in at least one individual from Rosalia, Tekoa, Pullman-1 and Palouse; (iii) the distal region of the group C long arms, observed in at least one individual from Rosalia, Tekoa, Pullman-1, Pullman-3 and Palouse.
The numbers of individuals that were homozygous and heterozygous for intergenomic translocations were compared with those expected under HWE. For 16 translocation breakpoints where a comparison could be made, two showed a significant departure from HWE, namely, the translocation on the CDu short arm in the Tekoa population and on the BDu long arm in the Pullman-3 population (Supplementary Tables S3–S8). In both cases, no translocation heterozygotes were recovered, but both recombined and non-recombined homozygotes were observed.
Only in one population (Oakesdale) was a homozygous reciprocal intergenomic translocation, on the long arm of chromosome ADu and APo, present in all individuals analyzed (Supplementary Figures S3a and f). One individual, 2871-10-1, showed an additional translocation on an APo chromosome, with the breakpoint appearing to be close to the 5S rDNA array (Supplementary Figure S3c).
Gene loss
PCR amplification of three genes encoding proteins with similarity to β-fructosidase, myosin heavy chain and thioredoxin was conducted on T. mirus individuals that had been karyotyped, as well as on a representative of each diploid parental species. In most cases the amplicons generated from T. mirus were addititive of the parents. A novel 500-bp fragment, not amplified from either diploid parent, was generated in the T. mirus β-fructosidase amplifications. The PCR assays revealed homeolog loss in two individuals from Palouse (2892-6-1 and 2892-7-1) (Figures 4a–c). For both individuals, the T. dubius homeolog amplification product of all three genes was missing. Visual inspection of the GISH karyotypes from these two individuals did not reveal shared chromosomal changes that would explain the loss of genes from the Du subgenome (Figures 5d and e). We attempted to localize the β-fructosidase gene in the Du genome using FISH with the largest gene fragment that could be amplified. With a 2-kbp β-fructosidase gene fragment, no clear hybridization signal was produced, most likely because the probe length was too short.
Figure 4.

Stained agarose gels showing genomic amplification products for the following three genes: (a) β-fructosidase, (b) myosin heavy chain and (c) thioredoxin M-type 1. Arrows indicate missing amplification products, which in all cases were of T. dubius–origin for the same two individuals (2892-5-1 and 2892-6-1). Diploid parental amplification products are shown on the left (T. dubius) and right (T. porrifolius) of each gel. The smallest and largest fragments of the size standard are annotated.
Figure 5.

Mitotic karyotypes of seven individuals from Palouse, WA, USA. Chromosomes are shown following GISH using total genomic DNA of the parents, T. dubius (green) and T. porrifolius (red). Diamond symbols are placed below aneuploid chromosomes. Arrows indicate the position of translocation breakpoints. (a) 2892-1-1, (b) 2892-2-1, (c) 2892-5-1, (d) 2892-6-1, (e) 2892-7-1 and (f) 2892-8-1. Scale bar, 5 μm.
Discussion
T. mirus plants were analyzed with a GISH method that allowed all parental chromosomes to be identified. The types of chromosomal variation detected were similar to those observed in T. miscellus (Chester et al., 2012), namely the predominance of partial or entire chromosome substitutions. In terms of the frequency of aneuploidy, the most variable T. mirus populations, Tekoa and Palouse, were comparable to the least variable T. miscellus populations analyzed previously (Chester et al., 2012). The number of euploid and aneuploid individuals differed between T. mirus (23:14) and T. miscellus (18:40) (Fisher's exact test, P=0.0033). Furthermore, in T. mirus, substitutions among multiple chromosome groups were not observed, compared with a 17% incidence (10 of 58 individuals) in T. miscellus (Fisher's exact test, P=0.0134).
Parental divergence
A lower incidence and severity of aneuploidy in T. mirus relative to T. miscellus may be a result of (i) meiosis being more regular and/or (ii) aneuploidy being more deleterious in the former species. One likely cause underlying both (i) and (ii) would be a lower genetic similarity between individual Du and Po chromosomes (in T. mirus) compared with Du and Pr chromosomes (in T. miscellus). Phylogenetic analyses so far indicate that T. porrifolius and T. pratensis are more closely related to each other than either is to T. dubius (Mavrodiev et al., 2005, 2007). Despite the close relationship between T. porrifolius and T. pratensis, chromosomal differences are apparent. Comparing the distribution of high-copy tandem repeats localized by FISH between T. porrifolius and T. pratensis revealed differences for chromosomes B through F (Chester et al., 2013). Therefore, greater chromosomal divergence between T. dubius and T. porrifolius remains a possible factor contributing to the reduced variation observed in T. mirus. The chromosome painting techniques employed here are unlikely to have revealed intragenomic rearrangements such as paracentric inversions (for example, Mandáková et al., 2014). To address the question of progenitor divergence, comparative genomic analyses will be needed to assess differences in gene content and organization.
Genetic dosage and genome structure
Eight of the nine T. mirus individuals that showed substitutions resulted in putatively homeologous chromosomes being in 3:1 or 4:0 ratios, rather than the expected 2:2 ratio. Rearrangements also largely followed a pattern where putatively homeologous segments were exchanged (only one out of the 53 translocations was an exception). These types of compensated changes were also found in synthetic neoallotetraploid B. napus (Xiong et al., 2011) and synthetic allotetraploid wheats (Zhang et al., 2013a). Thus, T. mirus provides another example in which alterations in parental gene balance appear to be tolerated more than alterations in total gene copy number, as expected based on the gene balance hypothesis (Birchler, 2012). For rearrangements that were rare within a population but found across multiple populations (for example, translocations in the long arm of chromosome A), the compensated pattern may perhaps reflect an elevated propensity for homeologous recombination as much as compensated changes being less deleterious.
In T. mirus, no aneuploidy was uncovered for group A chromosomes when using GISH signals at and around the centromere to determine the parental origin of the chromosome. No clear-cut cases of aneuploidy involving entire F chromosomes were observed either (that is, where intragenomic recombination was not involved). Similarly, a trend of reduced aneuploidy for A and F chromosomes was also observed in T. miscellus. This suggests that for A and F chromosomes in particular, within both T. mirus and T. miscellus (i) aneuploidy may be more deleterious and/or (ii) meiosis may be more regular in the former species. Both of these factors could be explained by increased chromosomal divergence between homeologs. For group A chromosomes, hypothesis (ii) seems unlikely given that multiple independent intergenomic translocations were found in both T. mirus and T. miscellus, indicating that strict diploid-like meiotic pairing is not operating. Therefore, there may be loci residing on the A group that are highly sensitive to dosage alterations—perhaps in the haploid stage, as copy number alterations above or below four were not seen in T. mirus or T. miscellus. To test this hypothesis, dosage-sensitive genes would need to be identified and localized by combining gene expression, gene dosage and mapping data from neoallopolyploid populations.
No individuals of T. mirus showed substitutions between entire non-homeologous chromosomes, yet there were plants that were numerical aneuploids (that is, 2n=23 or 25). The same was previously observed in T. miscellus (Chester et al., 2012) and in a spontaneous Crepis neoallotetraploid (Poole, 1932). Such a pattern indicates that alterations in dosage can in some cases be tolerated at the haploid stage (that is, +1 or −1 for a single chromosome) to produce numerical aneuploids. In contrast, based on their absence, there was no evidence for non-homeologous substitutions persisting in the sporophyte stage.
In T. mirus, several changes in chromosome structure paralleled what was seen in T. miscellus. Repeated independent intergenomic translocations occurred at three distal regions in the genome of T. mirus (A long arm, C short arm and C long arm). Multiple T. miscellus populations also had intergenomic translocations in similar regions (Chester et al., 2012). Application of FISH to T. mirus, as for T. miscellus (Chester et al., 2013), showed that repetitive DNA was also lost from these regions in some individuals.
Although translocations were common in T. mirus, most individuals typically showed only one or two translocations differentiating them from a non-rearranged karyotype. This observation also applies to the T. miscellus populations that were analyzed previously (Chester et al., 2012). Only one reciprocal intergenomic translocation in the Oakesdale population of T. mirus (Supplementary Figure S3) and one reciprocal intergenomic translocation in the Spokane-2 population of T. miscellus (Chester et al., 2012) appeared close to fixation. Interestingly, these translocations both occurred in similar regions on the group A chromosomes. The limited number of rearrangements per individual suggests that more highly rearranged karyotypes are not persisting in natural populations. The trend towards underrepresentation of (intergenomic) translocation heterozygotes in T. mirus suggests some segregating translocations may be disadvantageous (Supplementary Tables S3–S8). In resynthesized allotetraploid B. napus and several allotetraploid wheats, lineages displaying the highest fertility were those that had the lowest number of alterations (Xiong et al., 2011; Zhang et al., 2013a).
The two individuals of T. mirus that showed all three tested genes to be absent (from the Du subgenome) did not show shared non-reciprocal translocations or nullisomy. Therefore, smaller-scale changes, not detected with GISH, must have taken place to explain the genetic losses uncovered here. Smaller-scale genetic changes may be the result of gene-conversion events (Salmon et al., 2010; Flagel et al., 2012) or deletions (Chantret et al., 2005; Lukens et al., 2006). The PCR assay employed here could only have revealed homeolog loss if all copies of a gene from one parent were missing. In cases where one gene copy was lost but a second copy was still present, no changes would have been detected. Therefore, absolute genetic dosage will need to be ascertained to distinguish between processes such as gene conversion and deletion.
Chromosomal variation in neoallotetraploid populations
Chromosomal instability was apparent in all analyzed populations of T. mirus, similar to what was seen previously in T. miscellus (Chester et al., 2012). This variation is a result of meiotic errors such as when homeologous chromosomes pair (forming either bivalents or multivalents) and possibly recombine, or when homologs fail to pair (univalents), leading to some form of unbalanced segregation. However, as discussed above, deviations from the expected parentally additive karyotype were less marked than in T. miscellus. To explain the karyotyping results for the two Tragopogon neoallotetraploid species, we propose a model where chromosomal variation repeatedly expands and contracts at each generation. Chromosomal variation would be at its maximum immediately following meiosis. Thereafter, selection must operate at the haploid stage to limit homeolog ratios to 1:1 (null), or 2:0 or 0:2 to explain the predominance of 2:2, 1:3 or 0:4 ratios in individuals grown from seed. Sporophytes that are genetically imbalanced are expected to be at a metabolic or developmental disadvantage; however, overwintered adult T. miscellus plants showing mono-trisomy were found in Pullman, showing that moderate aneuploids can persist in nature (Chester et al., 2012). Any individuals that persist to flowering, but do not have a fully diploid-like meiosis, may then contribute to the re-expansion of chromosomal variation in the next generation (for example, translocations or aneuploidy). Finding out at what stages T. mirus differs from T. miscellus may help to explain why different amounts of chromosomal variation were observed.
For an allopolyploid genome to be maintained, a diploid-like meiotic behavior needs to be in place, if not immediately, then soon after polyploid formation. For several crops, there is evidence that homeologous pairing suppressors could in some cases have evolved prior to formation, or were operating at formation or in a few generations after (reviewed in the study by Jenczewski and Alix, 2004). The need for rapidly restoring diploid-like meiosis is underscored by the concept of the ‘polyploid ratchet', where after a certain threshold, rearrangements become difficult to reverse and may also promote further chromosomal destabilization (Gaeta and Pires, 2010). The finding of some T. mirus (N=3) and T. miscellus (N=3) individuals showing neither translocations nor aneuploidy may be due to chance or perhaps their derivation from meiotically stable lineages. Future work could test whether a diploid-like meiotic regulation has already arisen by analyzing meiosis in these and other meiotically stable candidates from natural populations and synthetic allotetraploid lines. Interestingly, the establishment of some allotetraploid wheat species has been associated with certain diploid progenitor combinations generating chromosomally stable lineages from the first generation (Zhang et al., 2013a).
T. mirus represents another example of a natural, recently formed allotetraploid (along with T. miscellus) that is still generating considerable chromosomal variation in natural populations that are ~40 generations old. The finding that all populations show heterogeneity in chromosome composition reflects an inability of natural selection to eliminate meiotically unstable lineages over the timeframe since the allopolyploids formed. This prolonged window of instability opens up possibilities for alterations and the generation of karyotypic novelty in neoallopolyploids, for example, see Soltis et al. (2014).
Data archiving
There were no data to deposit.
Acknowledgments
We thank Srikar Chamala for assistance with the bioinformatic analysis and the reviewers for their comments and suggestions. This work was supported by funding from the National Science Foundation (DEB-1146065 and DEB-0922003).
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Heredity website (http://www.nature.com/hdy)
Supplementary Material
References
- Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler J. (2012). Genetic consequences of polyploidy in plants. In: Soltis PS, Soltis DE (eds). Polyploidy and Genome Evolution. Springer: Berlin Heidelberg, pp 21–32. [Google Scholar]
- Buggs RJA, Chamala S, Wu W, Tate JA, Schnable PS, Soltis DE et al. (2012). Rapid, repeated, and clustered loss of duplicate genes in allopolyploid plant populations of independent origin. Curr Biol 22: 248–252. [DOI] [PubMed] [Google Scholar]
- Buggs RJA, Doust AN, Tate JA, Koh J, Soltis K, Feltus FA et al. (2009). Gene loss and silencing in Tragopogon miscellus (Asteraceae): comparison of natural and synthetic allotetraploids. Heredity 103: 73–81. [DOI] [PubMed] [Google Scholar]
- Buggs RJA, Elliott NM, Zhang LJ, Koh J, Viccini LF, Soltis DE et al. (2010). Tissue-specific silencing of homoeologs in natural populations of the recent allopolyploid Tragopogon mirus. New Phytol 186: 175–183. [DOI] [PubMed] [Google Scholar]
- Chantret N, Salse J, Sabot F, Rahman S, Bellec A, Laubin B et al. (2005). Molecular basis of evolutionary events that shaped the hardness locus in diploid and polyploid wheat species (Triticum and Aegilops). Plant Cell 17: 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester M, Gallagher JP, Symonds VV, da Silva AVC, Mavrodiev EV, Leitch AR et al. (2012). Extensive chromosomal variation in a recently formed natural allopolyploid species, Tragopogon miscellus (Asteraceae). P Natl Acad Sci USA 109: 1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester M, Lipman MJ, Gallagher JP, Soltis PS, Soltis DE. (2013). An assessment of karyotype restructuring in the neoallotetraploid Tragopogon miscellus (Asteraceae). Chromosome Res 21: 75–85. [DOI] [PubMed] [Google Scholar]
- Clausen J, Keck DD, Hiesey WM. (1945). Experimental Studies on the Nature of Species. II. Plant Evolution through Amphidiploidy and Autoploidy, with Examples from the Madiinae Carnegie Institution of Washington: Washington, D.C., USA, pp 62–63.
- Doyle J, Doyle J. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19: 11–15. [Google Scholar]
- Flagel LE, Wendel JF, Udall JA. (2012). Duplicate gene evolution, homoeologous recombination, and transcriptome characterization in allopolyploid cotton. BMC Genomics 13: 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaeta RT, Pires JC. (2010). Homoeologous recombination in allopolyploids: the polyploid ratchet. New Phytol 186: 18–28. [DOI] [PubMed] [Google Scholar]
- Gottschalk W. (1978). Open problems in polyploidy research. Nucleus 21: 99–112. [Google Scholar]
- Haldane J. (1954). An exact test for randomness of mating. J Genet 52: 631–635. [Google Scholar]
- Ising G. (1966). Cytogenetic studies in Cyrtanthus I. Segregation in an allotetraploid. Hereditas 56: 27–53. [Google Scholar]
- Jenczewski E, Alix K. (2004). From diploids to allopolyploids: The emergence of efficient pairing control genes in plants. Crit Rev Plant Sci 23: 21–45. [Google Scholar]
- Kato A, Lamb JC, Albert PS, Danilova T, Han F, Gao Z et al. Chromosome painting for plant biotechnology. Plant chromosome engineering: Methods and protocols, methods in molecular biology. (2011). Vol 701. Humana Press: New York, NY, USA. [DOI] [PubMed]
- Koh J, Soltis PS, Soltis DE. (2010). Homeolog loss and expression changes in natural populations of the recently and repeatedly formed allotetraploid Tragopogon mirus (Asteraceae). BMC Genomics 11: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KY, Soltis DE, Soltis PS, Tate J, Matyasek R, Srubarova H et al. (2008). Rapid chromosome evolution in recently formed polyploids in Tragopogon (Asteraceae). Plos One 3: e3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longwell JH, Sears ER. (1963). Nullisomics in tetraploid wheat. Am Nat 97: 401–403. [Google Scholar]
- Lukens LN, Pires JC, Leon E, Vogelzang R, Oslach L, Osborn T. (2006). Patterns of sequence loss and cytosine methylation within a population of newly resynthesized Brassica napus allopolyploids. Plant Physiol 140: 336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandáková T, Marhold K, Lysak MA. (2014). The widespread crucifer species Cardamine flexuosa is an allotetraploid with a conserved subgenomic structure. New Phytol 201: 982–992. [DOI] [PubMed] [Google Scholar]
- Mavrodiev EV, Soltis PS, Gitzendanner MA, Baldini RM, Soltis DE. (2007). Polyphyly of Tragopogon porrifolius L. (Asteraceae), a European native with intercontinental disjuncts. Int J Plant Sci 168: 889–904. [Google Scholar]
- Mavrodiev EV, Tancig M, Sherwood AM, Gitzendanner MA, Rocca J, Soltis PS et al. (2005). Phylogeny of Tragopogon L. (Asteraceae) based on internal and external transcribed spacer sequence data. Int J Plant Sci 166: 117–133. [Google Scholar]
- Menzel U. (2013) Exact multinomial test: Goodness-of-fit test for discrete multivariate data, http://cranr-projectorg/web/packages/EMT/indexhtml.
- Ownbey M. (1950). Natural hybridization and amphiploidy in the genus Tragopogon. Am J Bot 37: 487–499. [Google Scholar]
- Pires JC, Lim KY, Kovarik A, Matyasek R, Boyd A, Leitch AR et al. (2004). Molecular cytogenetic analysis of recently evolved Tragopogon (Asteraceae) allopolyploids reveal a karyotype that is additive of the diploid progenitors. Am J Bot 91: 1022–1035. [DOI] [PubMed] [Google Scholar]
- Poole CF. (1932). The interspecific hybrid, Crepis rubra x C. foetida, and some of its derivatives. II. Two selfed generations from an amphidiploid hybrid. Univ Calif Publ Agr Sci 6: 231–255. [Google Scholar]
- Core Team R. (2012). R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria. [Google Scholar]
- Ramsey J, Schemske DW. (2002). Neopolyploidy in flowering plants. Annu Rev Ecol Syst 33: 589–639. [Google Scholar]
- Rousset F. (2008). GENEPOP'007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8: 103–106. [DOI] [PubMed] [Google Scholar]
- Salmon A, Flagel L, Ying B, Udall JA, Wendel JF. (2010). Homoeologous nonreciprocal recombination in polyploid cotton. New Phytol 186: 123–134. [DOI] [PubMed] [Google Scholar]
- Soltis PS, Liu X, Marchant DB, Visger CJ, Soltis DE. (2014). Polyploidy and novelty: Gottlieb's legacy. Phil Trans R Soc B 369: 20130351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symonds VV, Soltis PS, Soltis DE. (2010). Dynamics of polyploid formation in Tragopogon (Asteracaeae): recurrent formation, gene flow, and population structure. Evolution 64: 1984–2003. [DOI] [PubMed] [Google Scholar]
- Tate JA, Joshi P, Soltis KA, Soltis PS, Soltis DE. (2009). On the road to diploidization? Homoeolog loss in independently formed populations of the allopolyploid Tragopogon miscellus (Asteraceae). BMC Plant Biol 9: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JA, Ni ZF, Scheen AC, Koh J, Gilbert CA, Lefkowitz D et al. (2006). Evolution and expression of homeologous loci in Tragopogon miscellus (Asteraceae), a recent and reciprocally formed allopolyploid. Genetics 173: 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Z, Gaeta RT, Pires JC. (2011). Homoeologous shuffling and chromosome compensation maintain genome balance in resynthesized allopolyploid Brassica napus. Proc Natl Acad Sci USA 108: 7908–7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bian Y, Gou X, Dong Y, Rustgi S, Zhang B et al. (2013. a). Intrinsic karyotype stability and gene copy number variations may have laid the foundation for tetraploid wheat formation. Proc Natl Acad Sci USA 110: 19466–19471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bian Y, Gou X, Zhu B, Xu C, Qi B et al. (2013. b). Persistent whole-chromosome aneuploidy is generally associated with nascent allohexaploid wheat. Proc Natl Acad Sci USA 110: 3447–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.