

Abstract
The natural history of Fanconi anemia remains hard to establish because of its rarity and its heterogeneous clinical presentation; since 1994, the Italian Fanconi Anemia Registry has collected clinical, epidemiological and genetic data of Italian Fanconi Anemia patients. This registry includes 180 patients with a confirmed diagnosis of Fanconi anemia who have either been enrolled prospectively, at diagnosis, or later on. After enrollment, follow-up data were periodically collected to assess the clinical course, possible complications and long-term survival; the median follow up was 15.6 years. The main goal of the study was to describe the natural history of Fanconi anemia, focusing on the following variables: family history, disease presentation, development of hematological manifestations, development of malignancies, occurrence of hematopoietic stem cell transplantation and survival. Typical morphological and/or hematological abnormalities and/or growth retardation were the most common manifestations at diagnosis; the majority of patients (77%) exhibited hematological abnormalities at the initial presentation, and almost all (96%) eventually developed hematological manifestations. More than half of the patients (57%) underwent a bone-marrow transplant. The occurrence of cancer was quite rare at diagnosis, whereas the cumulative incidence of malignancies at 10, 20 and 30 years was 5%, 8% and 22%, respectively, for hematological cancers and 1%, 15% and 32%, respectively, for solid tumors. Overall survival at 10, 20 and 30 years were 88%, 56% and 37%, respectively; the main causes of death were cancer, complications of the hematological presentation and complications of transplantation. These data clearly confirm the detrimental outcome of Fanconi anemia, with no major improvement in the past decades.
Introduction
Fanconi anemia (FA)1 is a rare inherited hematological disorder biologically characterized by hypersensitivity to DNA cross-linking agents. FA is mainly an autosomal recessive disease (except the rare X-linked FANC-B form), which was first reported in 1927 by the Swiss pediatrician Guido Fanconi as familial, infantile anemia.2 FA is now defined as a chromosomal instability (CI) syndrome, and it shows a wide clinical and genetic heterogeneity. Indeed, genetically FA can be caused by mutations in at least 18 different genes, mostly cooperating in a pathway which has not yet been fully elucidated. These gene products somehow interact with proteins encoded by genes which, when mutated, cause other CI syndromes, such as Ataxia Teleangiectasia, Bloom syndrome, and Nijmegen Breakage Syndrome.3 All these proteins cooperate in a pathway which appears to be involved in DNA and oxidative stress damage repair.4 The FA cellular phenotype is characterized by a G2 cell cycle phase delay5 and by CI, typically appearing as characteristic rearrangement figures (triradial and quadriradial figures made by nonhomologous chromosomes). The CI is both spontaneous6 and/or induced by alkylating DNA cross-linking agents, such as mytomicin C (MMC)7 or the more specific diepoxybutane (DEB);8 the cytogenetic DEB-test makes the FA diagnosis possible in those patients not showing typical malformations or still asymptomatic, and also in patients not showing spontaneous CI at standard cytogenetic evaluation.
The phenotype of FA patients is largely heterogeneous, since the natural history of the disease entails different clinical manifestations which may either be present at birth, or develop later during the course of the disease. Clinically, FA patients present bone marrow failure at various times in life, typically beginning in childhood as platelet deficiency, and then progressing to pancytopenia,1 eventually leading to life-threatening complications. FA patients can show variable congenital malformations9 and are prone to hematologic and solid neoplasias, which are ultimately the leading cause of death.1,4,10–12 At present, hematopoietic stem cell transplantation (HSCT) represents the only effective treatment for FA,13 although unfortunately it cannot improve the patient’s growth rate or reduce the propensity to develop non-hematological cancers.
FA is a rare disease, with an incidence rate of 0.1–0.5 new cases for every 100000 newborn children;1,14 thus, large multicenter studies are needed to better describe the natural history of the disease. Ideally, to prevent any bias, such studies should include all patients diagnosed with FA in a broad but well-defined geographic area, possibly with a prospective collection of data and an adequate long-term follow-up. Until now, the most reliable data on FA has come from the International Fanconi Anemia Registry (IFAR)15,16 and some national Registries, mostly the North American Survey of Fanconi Anemia (NAS)12,17 and the German Fanconi Anemia Registry (GEFA).18 In a rare and highly heterogeneous disease such as FA, it is very difficult to establish the natural history of the disease, and even more difficult to organize research projects which require the collection of samples from patients showing common features. Moreover, some significant differences are possible among different populations, due to the existence of geographic isolates or genetic derives. A specific national registry, collecting patients’ clinical, epidemiological, genetic and familial data, becomes a powerful tool, which creates for physicians and the scientific community the possibility of better knowing the disease, hence preventing misdiagnosis and delayed diagnosis. A national registry also creates a network that facilitates the participation of patients in research projects and clinical trials.
To fulfill the need for a National Database including most (if not all) FA patients diagnosed in Italy, in 1994 some of us started “Il Registro Italiano Anemia di Fanconi” (RIAF), which translates as ‘The Italian Fanconi Anemia Registry’.19 This project was established within the Italian Public National Health System (NHS), at the Genetic Unit of the local health unit “ASL Napoli 1”, taking advantage of their established expertise in diagnostics and genetic counseling for CI syndromes. The aims of the project were: i. to create a National database, recording all Italian FA cases; ii. to collect information about the epidemiology of FA in Italy, as well as about its natural history and therapeutic interventions; iii. to promote a robust scientific network among health workers (physicians, but also scientists) dealing with FA in Italy, eventually increasing awareness about the disease and promoting the strongest possible collaboration among Italian physicians and scientists; iv. to provide patients and their families with an established national network for the diagnosis, treatment and follow-up of FA patients; v. to share with the Italian authorities all the information above, aiming to assess the real impact of FA on the Italian NHS, eventually promoting further health policy strategies. Herein we report on the 20-year experience of this Registry, focusing on the natural history of the disease, frequent therapeutic modalities and long-term outcomes of FA patients. The major aim of this work was to identify possible factors affecting the survival of FA patients, as well as to identify possible changes in outcome emerging over the two decades of this study.
Methods
Study design and data handling
The RIAF was officially established in 1994 as a prospective, non-interventional study, approved by the local Government (serving as the institutional review board, IRB) and operated according to National laws within the NHS. The program was approved and funded by the Regional Government (Regione Campania), and supported by the Italian Association for Research on Fanconi Anemia (AIRFA). All patient data were collected through dedicated case report forms (CRFs), designed by the geneticists working at the ASL Napoli 1, and further developed thanks to the contributions of collaborating physicians (listed in the Appendix). In accordance with the Declaration of Helsinki, before enrollment all patients or their parents/guardians gave written, informed consent, after discussing the RIAF aims and policy and their own rights with an authorized delegate (geneticist or physician), as listed in a written information sheet. Follow-up CRFs were periodically filed by the geneticists or the physicians, through a continuous sharing of critical information with both treating physicians and patients or their families. The CRFs and informed consent were approved by the local Government/IRB. The data were stored both on paper and in digital records, strictly protected, accessible only to the authorized staff and always made anonymous for publications or sharing with other researchers.
Inclusion criteria, enrollment and data collection
Patients were enrolled only after a positive DEB or MMC test, for the most part carried out or confirmed in our laboratory. The chromosome breakage assay was always performed on peripheral blood cells. Patients were enrolled either at the time of initial diagnosis, or later, during the course of the disease, according to patients’ and/or parents’ decisions. Following informed consent, family history and medical information were recorded by the treating physicians, according to the specific CRFs. Given the epidemiologic purpose of the RIAF, the data of patients belonging to non-Italian ethnic populations were collected separately and are not considered here. Indeed, the patients geographic designation was established on the basis of their parents’ and grandparents’ birthplaces (Caucasian ethnicity and proven Italian descent covering at least two generations); the same criteria were used to assign a patient to a specific Italian Region.
Data analysis and statistics
Statistical analysis was performed on the population of 180 patients (fetal losses and miscarriages were excluded), focusing on the following categories: family history, disease presentation, hematologic manifestations, HSCT, treatment impact on survival, malignancies, overall and cancer-free survival, and causes of death. Standard descriptive statistical tests were applied as appropriate, using SPSS software (PSP, Bologna, Italy). Student’s t-test, Mann-Whitney test and Fisher’s exact test were used for most descriptive analysis. The time to developing specific disease presentation (i.e., hematological presentation, hematological malignancies, solid tumors) was presented as cumulative incidence, using a competing risk approach, with birth treated as the FA onset date; death and HSCT (the latter only for hematological presentation and hematological malignancies) were considered as competing risks. The Kaplan-Meier curve was used to estimate overall survival; again birth was considered as the FA onset date. The following variables were tested for a possible impact on survival: gender, date of birth, age at diagnosis, all congenital abnormalities (presence, total number, type; with and without skin abnormalities), hematological presentation (at diagnosis, or at any time), hematological malignancies, solid tumors (all together and head/neck) and HSCT. Univariate and multivariate analyses were performed using a Cox regression model on all patients, as well as separately on transplanted and non-transplanted patients.
Results
Diagnosis and genetics of FA
A preliminary diagnosis of FA was made by treating physicians, based on clinical presentation at birth, or later on; in some patients their family history was the main reason to hypothesize the presence of FA. The diagnosis of FA was based on a standard chromosome breakage test by exposure to DEB or MMC, performed on peripheral blood samples. Given the possible challenges in the diagnosis of CI, all tests were confirmed at the Genetic Unit of the ASL Napoli 1, or other laboratories with specific expertise for the diagnosis of FA, eventually limiting subjective interpretations and inter-laboratory technical variability. Lymphoblastoid cell lines were established for research aims and as diagnostic positive controls. A single DEB test was sufficient for the diagnosis in the majority of cases; however, the protocol adopted at the Genetic Unit of the ASL Napoli 1 was used to confirm the diagnosis on two different samples, allowing a robust consistency of data. Between 1989 and 2014, out of a total of 1340 DEB tests performed on 1185 subjects, the number of positive tests was 206 (for 135 patients). Notably, a prior misdiagnosis was proven in 11 patients: in 7 cases a previous diagnosis of FA was not confirmed, whereas in 4 patients the diagnosis of FA was missing. Mosaicism was suspected in 9% of patients.20 They showed chromosome breaks in <40% of their cells, but typical DEB-induced rearrangements were demonstrated; CI testing performed on different tissues, together with clinical, family and/or molecular data, confirmed the FA diagnosis. Patients lacking a confirmatory positive chromosome breakage test were not enrolled in the RIAF, irrespective of their clinical presentation; thus, possible revertant phenotypes may be underrepresented in this cohort. Complementation groups were available for 55 patients; the most common complementation group was A (91%), followed by G (5%) and D2 (4%) (Online Supplementary Table S1).
Subject characteristics
Between 1994 and 2014, a total of 180 patients were included in the RIAF, belonging to 151 distinct families (median number of affected subjects per family was 1, range 1–4; Table 1); a few (n=3) cases of miscarriage diagnosed as FA (by DEB and/or molecular tests on amniocytes or chorionic villi) were also recorded, but were not included in this study. The geographical distribution was spread throughout the country, even if a significant number of patients were from the North-East or the South of Italy;21 however, we were unable to identify any founder effect. The characteristics of enrolled patients are described in Table 2. There were 94 (52%) male and 86 (48%) female patients, with no statistical difference in gender. The median age at diagnosis was 7.48 years; when patients were divided according to the date of birth, by quartiles (≤1980, 1981–1987, 1988–1995, ≥1996), the age at diagnosis was significantly lower in patients born in more recent periods (Mann-Whitney test, P<0.001; Figure 1A).
Table 1.
Characteristics of the 151 families.*
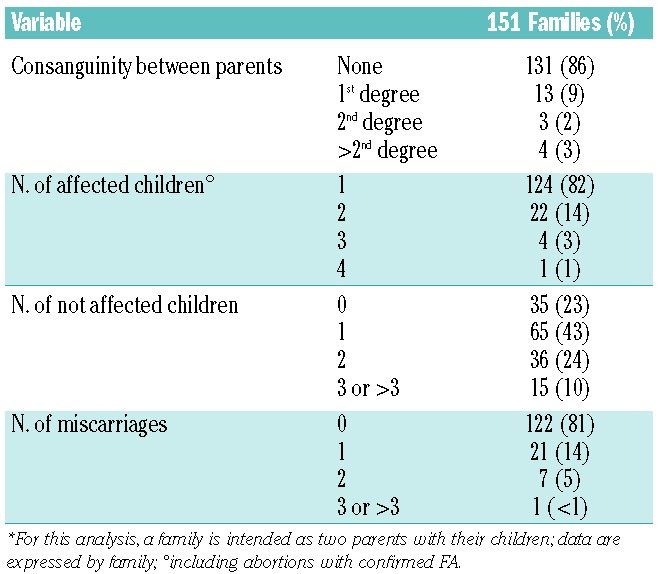
Table 2.
Patient characteristics at diagnosis.
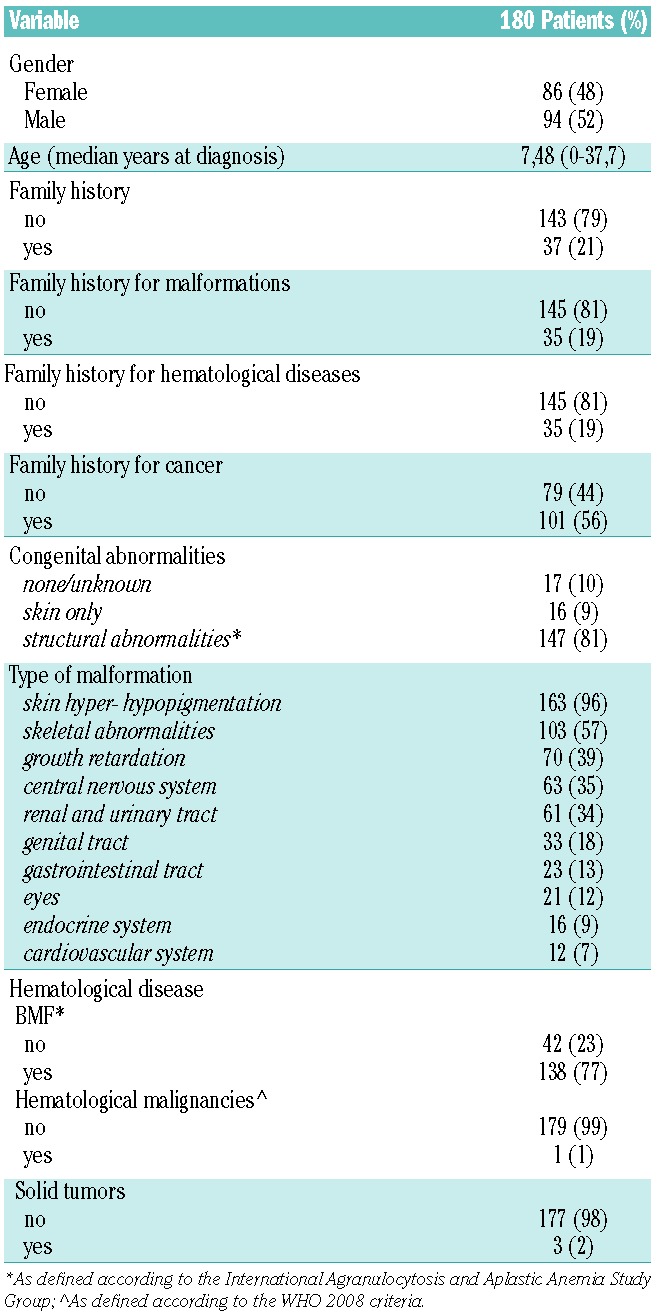
Figure 1.

Time to most common FA complications. (A) Age at diagnosis, according to quartiles of date of birth (DOB); (B) Cumulative incidence of bone marrow failure (BMF), hematological malignancies (MDS and AML; HEM TUM), solid tumors (SOL TUM) and HSCT; (C) Age at HSCT, according to DOB quartiles; (D) Cumulative incidence of solid tumor and of head/neck tumors: HSCT vs. no HSCT.
Family history
Family history was carefully collected for all patients included in the registry; parents consanguinity was recorded in 20 of the 151 families (14%; Table 1). For each family (considered as two parents), the median number of affected children was 1 (range 1–4), the median of unaffected siblings (all confirmed by DEB test: all alive siblings were tested even as potential HSCT donors) was 1 (range 0–10); the median of miscarriages was 0 (range 0–3). Globally, in the 151 families enrolled we recorded 310 babies, of whom 183 (60%) were FA cases (180 enrolled in the RIAF and 3 miscarriages), and 127 were unaffected siblings. Thirty-seven of the patients included in the registry reported a family history of FA. We also looked for the occurrence of hematological disorders and malignancies in the relatives of enrolled FA patients (up to the second degree of kinship); family history for cancer (taking into account up to the second degree) was 56% (85 out of 151). Family history for hematological disorders was demonstrated in 19% of patients. Morphological abnormalities in some relatives were recorded in 19% of patients.
Disease presentation
In the majority of patients the diagnosis was suspected based on typical morphological and/or hematological abnormalities and/or growth retardation. As detailed in Table 2, congenital abnormalities were demonstrated in 90% of patients at the time of diagnosis; the most common were the typical abnormalities of skin pigmentation, which affected 96% of RIAF patients (Table 2); skeletal abnormalities were also very frequent (57%). Other common congenital abnormalities involved growth retardation (39%), the central nervous system (35%), the urinary system (34%), the genital tract (18%), the gastrointestinal tract (13%), the eyes (12%), the endocrine system (9%) or the cardiovascular system (7%). Hematological manifestations were defined according to the definition of aplastic anemia22 and in accordance with the WHO 2008 classification of myeloid malignancies.23 The majority of patients (77%) exhibited some hematological abnormalities at diagnosis, which in most cases was a mild-to-moderate cytopenia eventually associated with some degree of bone marrow failure (BMF), whereas hematological malignancies (e.g., myelodysplastic syndromes, MDS) and solid tumors were very rarely observed at diagnosis (Table 2). Thanks to the long-term follow-up of the enrolled patients, we were able to assess the further course of the disease with the development of the most common complications of FA, as well as the impact of different factors on survival.
Time to hematological manifestations: bone marrow failure and hematological malignancies
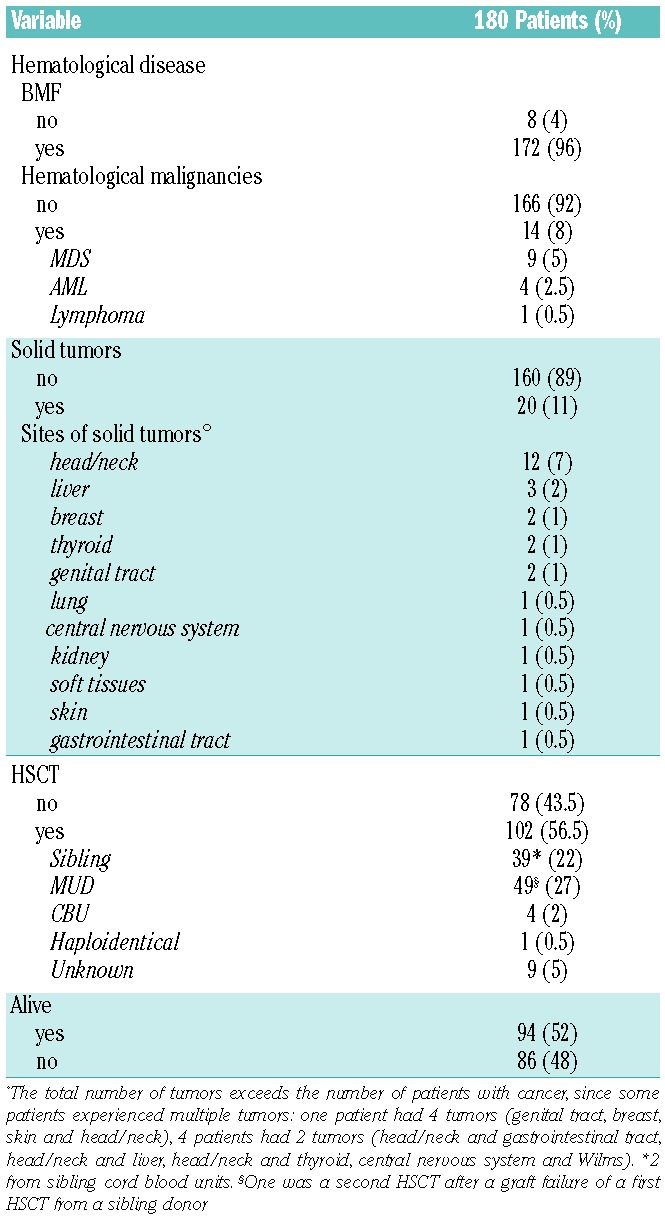
Even if hematological abnormalities were present at diagnosis in only 77% of cases, a total of 172 (96%) of FA patients enrolled in the RIAF had some hematological manifestations during their disease course; in almost all cases (172, 96%) this included cytopenia due to BMF, whereas a hematological malignancy (mostly MDS or acute leukemia, n=9 and n=4, respectively) was recorded in 8% of patients (see Table 3). As expected, in many cases an initial BMF progressed to either MDS or more aggressive hematological cancers; 1 MDS, 1 leukemia and 1 lymphoma patient did not evolve from a previous BMF. Considering death and HSCT as competing events, the cumulative incidence of any hematological disorder was 62%, 88% and 94% at 10, 20 and 30 years respectively, whereas the incidence of hematological malignancies was 5%, 8% and 22% at 10, 20 and 30 years, respectively. ‘The cumulative incidence of the first hematological presentation and of the first hematological malignancy is depicted in Figure 1B.
Table 3.
Disease manifestations during the whole disease course.
Time to hematopoietic stem cell transplantation
The development of a hematological presentation is the main indication for HSCT in FA patients; indeed, more than half of the patients enrolled in the RIAF (102 out of 180, 57%) had received a HSCT from either a non-affected sibling or matched unrelated donor (Table 3). The first HSCT was performed from a non-affected sibling in 38% of cases, from a matched unrelated donor in 48%, and quite rarely from cord blood (4%) or a mismatched related donor (0.9%). The cumulative incidence of HSCT in our patient cohort was 33%, 64% and 72% at 10, 20 and 30 years, respectively, as depicted in Figure 1B. The age at transplant was significantly different according to the date of birth cohorts, since patients born in more recent years were transplanted earlier (Mann-Whitney test, P<0.001; Figure 1C). Since National and European Registries collecting transplant-specific information exist,15,16 in the RIAF we decided not to duplicate this information. A formal analysis of HSCT outcome in these patients is beyond the scope of this study. However, follow-up data on survival and the possible development of malignancies were also collected for those RIAF patients who received a HSCT.
Cumulative incidence of solid tumors
A total of 27 solid cancers were diagnosed in 20 of the 180 RIAF patients (11%); a few patients experienced multiple cancers. The most common sites of cancer were the head and neck (n=12, 44% of all solid tumors), liver (n=3, 11%), breast, thyroid and genital tract (n=2 for each, 7%) (see Table 3 for details). The cumulative incidence of solid tumors was 1%, 15% and 32% at 10, 20 and 30 years respectively, as depicted in Figure 1B. The incidence of all solid cancers and of head and neck tumors was not statistically different between patients who had received a HSCT and those who had not (P=0.43 and P=0.50, respectively; Figure 1D), even if the analysis is limited by the small number of events. In transplanted patients, all but one tumor occurred after HSCT.
Overall survival and prognostic factors
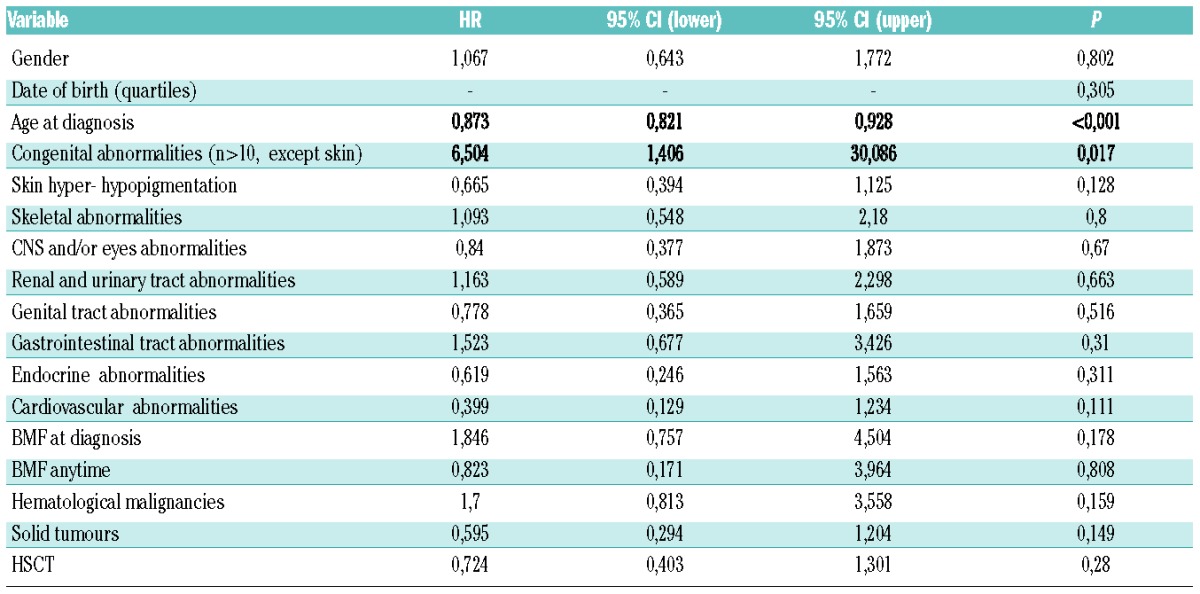
Ninety-four of the 180 patients were still alive at the time of the last follow-up. For all patients enrolled in the RIAF, overall survival was calculated starting from the day of birth. With a median follow up of 15.6 years, median survival was 22.5 years (Figure 2A); probabilities of survival at 10, 20 and 30 years were 88%, 56% and 37%, respectively (without censoring HSCT patients). Looking for the natural history of the disease, when patients who had received an allogeneic HSCT were censored at the time of transplant, the probabilities of survival at 10, 20 and 30 years were 85%, 39% and 24%, respectively (Figure 2B). In univariate analysis, no patients feature affected overall survival, except age at diagnosis and the number of structural abnormalities (excluding skin anomalies; Online Supplementary Table S2). In multivariate analysis, an older age at diagnosis (HR=0.873, P<0.001) and the presence of more than 10 structural abnormalities (excluding skin; HR=6.504, P=0.017) were associated with a better or worse survival rate, respectively. This distinction resulted in statistically significant differences in overall survival based on age at diagnosis (Figure 2C; P<0.001) and on higher numbers of structural abnormalities (Figure 2D; P<0.005). Notably, BMF at initial presentation, date of birth cohorts and HSCT seemed not to affect survival (Table 4). Indeed, looking at overall survival by quartiles of date of birth there was no improvement in survival over time (Figure 2E; P=n.s.). Similarly, grouping patients by HSCT, the 10, 20 and 30 year survival rate of non-transplanted patients (n=78, median follow-up 15.8 years) were 84%, 49% and 34%, respectively, while those of transplanted patients (n=102, median follow-up 16 years) were 90%, 62% and 40%, respectively (Figure 2F; P=0.17). Multivariate analysis was also performed separately on non-HSCT and HSCT patients; in this context, age at diagnosis remained associated with a better survival rate in both groups (Online Supplementary Table S3).
Figure 2.

Overall survival. (A) Overall survival, HSCT not censored (filled area represents 95% confidence interval); (B) Overall survival, HSCT censored (filled area represents 95% confidence interval); (C) Overall survival, according to age at diagnosis (patients were grouped based on the median age at diagnosis of 7,48 years); (D) Overall survival, according to number of structural abnormalities; (E) Overall survival, according to DOB quartiles (HSCT not censored); (F) Overall survival: HSCT vs. no HSCT.
Table 4.
Multivariate analysis.
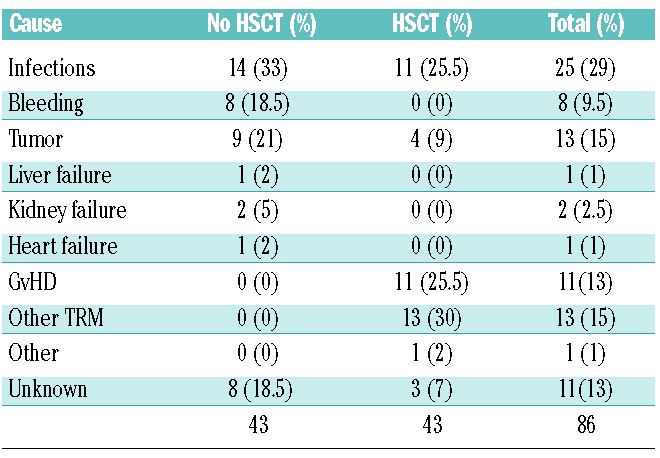
Cause of death
Eighty-six of the 180 FA patients enrolled in the RIAF died during their follow-up; the main causes of death are listed in Table 5. As expected, the causes of death were different in patients who had not received an HSCT as compared with those of transplanted patients (P<0.001; Chi-square test). Indeed, in non-HSCT patients the main causes of death were related to the underlying disease (i.e., for the most part the hematological abnormalities), such as infections (n=14, 33% of total deaths in non-HSCT patients), bleeding (n=8, 18.5%) and solid tumors (n=9, 21%). In contrast, in HSCT patients the majority of deaths were somehow related to treatment complications, such as infections (n=11, 25.5% of total deaths in HSCT patients), graft versus host disease (GvHD) (n=11, 25.5%) and other transplant related mortality (TRM) (n=13, 30%). Solid tumors accounted for 9% of deaths (n=4).
Table 5.
Causes of death.
Discussion
The RIAF is the first population-based Italian database, run within the Italian NHS, focusing on FA, which is rare in the frequency of the disease, but the most common among inherited bone marrow failure syndromes. Herein we report a comprehensive analysis of all patients included in the registry over the past 20 years with their long-term follow-up, eventually providing a reliable description of the natural history of FA. In our series of 180 prospectively collected patient data, we have shown a median survival of 22 years, which unfortunately has not improved in the past two decades. Our efforts of creating a robust scientific network have increased the awareness of this disease in Italy, eventually leading to objective achievements. Indeed, the diagnosis has come to be made earlier over the past decades, and the time to the only curative treatment – namely HSCT – has decreased. Nevertheless, these improvements in the management of FA patients have not yet resulted in a better survival rate, and even the outcome of patients who have received a HSCT does not appear to be better than that of those who did not. Indeed, in our multivariate analysis, the only factors associated with a better outcome were an older age at diagnosis and a lower number of structural abnormalities, indicating that different clinical phenotypes may have a different life expectancy.
The natural history of FA has been described in previous retrospective studies,15–18 which have highlighted the heterogeneity of clinical presentation. The RIAF includes only patients with a DEB test confirmed diagnosis of FA, who are unselected for specific disease presentations and have a long-term follow-up. Thus, selection biases (e.g., toward a severe phenotype) should be limited (with the only exception being a possible underestimation of patients beginning with cancers, who might not receive the correct FA diagnosis), eventually leading to a more accurate representation of the natural history of FA. However, like all registry studies, the RIAF suffers from potential limitations, since its completeness and accuracy largely depends on the commitment and dedication of collaborating physicians. Our database confirms that FA severely impairs the survival of affected patients; the median survival observed in our series (about 22 years) was slightly lower than those reported by the International Fanconi Anemia Registry16 and by the USA National Cancer Institute.24 In our Registry, we have not systematically investigated any genotype-phenotype correlation;25 however, genetic data from a subset of patients (as well as independent data on the genetics of FA in the same geographic area)26 seem predominantly influenced by the large prevalence of patients harboring FANCA mutations.21,27 Malignancies play an important role in the natural history of FA, the risk increasing with age for a wide array of cancer types;10–12,18 moreover, some patients can develop multiple cancers, possibly also due to the increased risk associated with anti-cancer treatments (i.e., chemotherapy and radiotherapy). This report confirms the cancer propensity of FA patients and further stresses the need for frequent and careful tumor evaluations, aiming at early therapeutic interventions,24,28 the only effective strategy for improving long-term survival in FA patients.
Our Registry was not designed to formally investigate the impact of specific therapeutic interventions on the natural history of the disease. However, even if a head-to-head comparison is impossible, we have separately looked for overall survival in FA patients who have received an HSCT, without showing any difference with non-transplanted patients. HSCT may cure the hematological disease associated with FA, but it does not reverse the phenotype that results from the involvement of extra-hematological tissues and organs.29–31 This is especially true for the intrinsic risk of cancer due to the genetic instability typical of FA, which might actually be increased by the pre-transplant conditioning regimen and possible detrimental effects of GvHD.28,32 Notably, in our study the risk of solid tumors remains high even after HSCT, but apparently it is not increased over that of non-transplanted patients. However, these data should be confirmed with a longer follow-up, possibly within International studies designed to specifically investigate this endpoint. Our observation that the survival of transplanted and non-transplanted patients was not different is not surprising, because HSCT in the context of FA carries specific challenges. Beyond the fact that HSCT does not affect the extra-hematological phenotype of FA, other reasons may play a role: i. HSCT patients may be biased toward a more severe phenotype; ii. initial patients may have received a HSCT with a non-optimized conditioning regimen;33 iii. initial patients have received HSCT quite late in their disease course; and iv. longer follow-up is needed to let the positive impact of HSCT emerge. Unfortunately, even if in recent decades improvements in transplant procedures (e.g., the use of reduced intensity conditioning regimens) have significantly prolonged the overall survival rate of patients,31 HSCT for FA remains associated with a poor prognosis, with a high number of patients exposed to lethal complications. Since the RIAF was not designed to study HSCT in the context of FA, the actual impact of HSCT on the natural history of FA needs to be investigated in more specific studies that also deal with all the transplant-specific factors affecting the outcome of HSCT. Indeed, the question is whether more recent HSCT, performed according to transplant protocols which have been optimized over the past years,34 have improved the outcome of FA, as compared with natural history. One may anticipate that combining earlier therapeutic intervention with improved HSCT protocols may lead in the near future to improved long-term outcomes for FA patients,35,36 especially if a lack of an increased risk of malignancies is confirmed.
In conclusion, our registry confirms the adverse natural history of FA, eventually leading to disappointing outcomes that have not improved over time; thus, there is an urgent need for effective treatment strategies. Our findings highlight that large collaborative studies are essential to investigate the impact of available therapeutic interventions (such as transplantation), to optimize their use and to define their role in the treatment algorithm of FA. It seems obvious that it will only be through stronger collaboration among physicians and scientists, National and International Registries, and healthcare networks, that we may hope to offer better long-term outcomes to patients affected by FA and to their families.
Acknowledgements
The authors are deeply grateful to the patients and their families for providing data and tissue samples, not identified here for privacy reasons. They are grateful to list in the Appendix all the physicians providing data and moral support. The authors are also grateful to Dr. G. Pagano, founder and past president of the Italian Association for Research in Fanconi Anemia (AIRFA), who strongly sustained the RIAF project and partially funded it, to Dr. M.R. Piemontese, A. Savoia and L. Zelante, who provided mutation data, to Dr. M. Amato, O. Catapano, F. D’Amico, M. Galgani, E. Montone, M. Rossi, D. Scafato, who followed each other in maintaining the Registry, and to Dr. V. Altieri, A. Lioniello and G. Peperna for daily cytogenetic and informational work. The authors thank Prof. Sharon Schuman for editing the manuscript. The authors wish to dedicate this paper to the beloved memory of Lisa Orsini, whose relatives funded this work, and to Prof. Bruno Rotoli and Dr. Angelo Rosolen, who both first collaborated to create the Registry, and to all the patients who passed away.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/3/319
Appendix
RIAF Contributors (Institutions, Physicians, number of Patients reported):
Napoli - Oncoematologia AORN “Santobono-Pausilipon” -Poggi V., Loffredo G., Misuraca A., Menna G., Ripaldi M., Parasole R., Marchese L., Schiavulli M., Boccalatte M.F. (18); Napoli – Servizio di Genetica ASL Napoli 1 – Zatterale A., Calzone R. (16); Genova - Ematologia e Oncologia Pediatrica Ist. “G. Gaslini” - Dufour C., Svahn J. (15); Padova -Oncoematologia Pediatrica AOU - Bisogno G., Cesaro S., Rosolen A., Rossetti F., Sainati L.,Varotto S. (14); Torino - Dip. Scienze Pediatriche e dell’Adolescenza OIRM - Ramenghi U. (14); Napoli - Clinica Pediatrica AOU SUN - Nobili B., Matarrese S., Ferrara M., Perrotta S. (14); Roma - Ematologia Univ. “La Sapienza” - Mandelli F, Giona F., Arcese W., Rana I., Amendola A., Barberi W., Testi A.M. (12); Cagliari - Centro Trapianti Osp. Microcitemico - Arru L., Cossu F. (8); Monza -Oncoematologia Pediatrica Policlinico “San Gerardo” – Longoni D. (8); Napoli - Ematologia AOU “Federico II” - Rotoli B., Catalano L., Fiorillo A., Risitano A. (7); Pescara - UOC Trapianto Emopoietico PO - Di Bartolomeo P. (5); Palermo - Ematologia Osp. “Cervello” - Fabbiano F., Mirto S. (5); Palermo - Oncoematologia Pediatrica Osp.“Di Cristinaǀ - Farruggia P. (4); Roma - Pediatria Osp. “Sant’Eugenio” - Del Principe D. (4); Bologna - Oncoematologia Pediatrica “Sant’Orsola” - Rosito P., Paolucci G. (3); Catania - Ematologia ed Oncologia Policlinico di Catania - Schilirò G. (3); Parma - Pediatria e Oncoematologia Pediatrica A.O. - Izzi G., Barone A. (3); Napoli - Dip. Pediatria AOU “Federico II” - Pignata C., Sebastio G., Scarcella A. (2); Napoli - Pediatria AO “A. Cardarelli” - Saviano A. (2); Nocera Inferiore - Pediatria AO “Umberto I” - Amendola G., Di Concilio R. (2); Pesaro - Pediatria Osp. “San Salvatore” – Felici L. (2); Roma - Oncoematologia pediatrica Policlinico “A. Gemelli” - Lasorella A., Mastrangelo S. (2); Trieste - Ematooncologia Pediatrica Univ. - Rabusin M. (2); Ancona -Oncoematologia Pediatrica - Pierani P., Fabrizzi B. (2); Avellino - Ematologia AORN “San Giuseppe Moscati” - Volpe E., Cantore N. (2); Bari - Dip. Biomedicina Età Evolutiva UO Pediatrica I Policlinico - Martire B. (2); Catanzaro - Ematologia PO “Pugliese-Ciaccio” - Levato L. (1); Ferrara - Clinica Pediatrica Univ. - Borgna Pignatti C. (1); Genova - Div. Pediatria “Ospedali Galliera” - Melevendi C. (1); Perugia -Pediatria AO - Mazzarino I. (1); Pisa - Oncoematologia Pediatrica AO - Favre C., De Marco E. (1); Reggio Calabria -Genetica Medica OORR “Bianchi-Melacrino” - Laganà C. (1); Roma - UOC TCS Policlinico “Tor Vergata” - Cudillo L. (1); Siena - Italy -Clinica Pediatrica Univ. - Acquaviva A. (1); Zurich (Switzerland) - Kinderspital – Albisetti M. (1).
References
- 1.Alter BP, Kupfer G. Fanconi Anemia. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews. Seattle (WA): University of Washington, 2013; pp. 1993–2015. [Google Scholar]
- 2.Fanconi G. Familiäre infantile perniziosaartige Anämie (pernizioses Blutbild und Konstitution). Jahrb Kinderh. 1927;117: 257–280. [Google Scholar]
- 3.Schroeder TM. Genetically determined chromosome instability syndromes. Cytogenet Cell Genet. 1982;33(1–2):119–132. [DOI] [PubMed] [Google Scholar]
- 4.Zatterale A. Fanconi Anemia clinical and genetic heterogeneity. In: Pagano G, ed. Fanconi Anemia and Oxidative Stress: Mechanistic Background and Clinical Prospects. NY: Nova Science Publishers Inc.; 2015:1–15. [Google Scholar]
- 5.Dutrillaux B, Aurias A, Dutrillaux AM. The cell cycle of lymphocytes in Fanconi anemia. Hum Genet. 1982;62(4):327–332. [DOI] [PubMed] [Google Scholar]
- 6.Schroeder TM, Anschultz F, Knoff A. Spontane chromosome aberrationen bei familiärer Panmyelopathie. Humangenetik. 1964;1(2):194–196. [DOI] [PubMed] [Google Scholar]
- 7.Sasaki MS, Tonomura A. A high susceptibility of Fanconi’s anemia to chromosome breakage by DNA cross-linking agents. Cancer Res. 1973;33(8):1829–1836. [PubMed] [Google Scholar]
- 8.Auerbach AD. Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Exp Hematol. 1993;21(6):731–733. [PubMed] [Google Scholar]
- 9.Glanz A, Fraser FC. Spectrum of anomalies in Fanconi anaemia. J Med Genet. 1982;19(6): 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alter BP. Inherited bone marrow failure syndromes. In: Nathan DG, Oskin SH, Ginsburg D, Look AT, Oski FA, eds. Hematology of Infancy and Childhood. Philadelphia: PA Saunders; 2003. pp. 280–365. [Google Scholar]
- 11.Alter BP. Cancer in Fanconi’s anemia, 1927–2001. Cancer. 2003;97(2):425–440. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood. 2003;101(3):822–826. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg PS, Alter BP, Socié G, Gluckman E. Secular trends in outcomes for Fanconi anemia patients who receive transplants: implications for future studies. Biol Blood Marrow Transplant. 2005;11(9):672–679. [DOI] [PubMed] [Google Scholar]
- 14.Orphanet. http://www.orpha.net/consor/cgibin/Disease_Search.php¿lng=EN&data_id=634&MISSING%20CONTENT=Fanconi-anemia&search=Disease_Search_Simple&title=Fanconi-anemia
- 15.Auerbach AD, Schroeder TM. First announcement of the Fanconi anemia International Registry. Blood. 1982;60(4): 1054. [PubMed] [Google Scholar]
- 16.Kutler DI, Singh B, Satagopan J, et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood. 2003;15(4): 1249–1256. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg PS, Huang Y, Alter BP. Individualized risks of first adverse events in patients with Fanconi anemia. Blood. 2004;104(2):350–355. [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg PS, Alter BP, Ebell W. Cancer risks in Fanconi anemia: findings from the German Fanconi Anemia Registry. Haematologica. 2008;93(4):511–517. [DOI] [PubMed] [Google Scholar]
- 19.Zatterale A, Calzone R, Montone E, Pagano G. Il Registro Italiano Anemia di Fanconi. Ann Ist Super Sanità. 1999;35(2):233–235. [PubMed] [Google Scholar]
- 20.Fargo JH, Rochowski A, Giri N, Savage SA, Olson SB, Alter BP. Comparison of chromosome breakage in non-mosaic and mosaic patients with Fanconi anemia, relatives, and patients with other inherited bone marrow failure syndromes. Cytogenet Genome Res. 2014;144(1):15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Savoia A, Zatterale A, Del Principe D, Joenje H. Fanconi’s Anaemia in Italy: high prevalence of complementation group A in two geographic clusters. Hum Genet. 1996;97(5): 599–603. [DOI] [PubMed] [Google Scholar]
- 22.International agranulocytosis and aplastic anemia study. Incidence of aplastic anemia: The relevance of diagnostic criteria. Blood. 1987;70(6):1718–1723. [PubMed] [Google Scholar]
- 23.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. [DOI] [PubMed] [Google Scholar]
- 24.Alter BP, Giri N, Savage SA, et al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol. 2010;150(2):179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faivre L, Guardiola P, Lewis C, et al. Association of complementation group and mutation type with clinical outcome in Fanconi anemia. Blood. 2000;96(13):4064–4070. [PubMed] [Google Scholar]
- 26.De Rocco D, Bottega R, Cappelli E, et al. Molecular analysis of Fanconi anemia: the experience of the Bone Marrow Failure Study Group of the Italian Association of Pediatric Onco-Hematology. Haematologica. 2014;99(6):1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savoia A, Piemontese MR, Savino M, et al. Linkage analysis of Fanconi anaemia in Italy and mapping of the complementation group A gene. Hum Genet. 1997;99(1):93–97. [DOI] [PubMed] [Google Scholar]
- 28.Masserot C, Peffault de Latour R, Rocha V, et al. Head and neck squamous cell carcinoma in 13 patients with Fanconi anemia after hematopoietic stem cell transplantation. Cancer. 2008;113(12):3315–3322. [DOI] [PubMed] [Google Scholar]
- 29.Locatelli F, Zecca M, Pession A, et al. The outcome of children with Fanconi anemia given hematopoietic stem cell transplantation and the influence of fludarabine in the conditioning regimen: a report from the Italian pediatric group. Haematologica. 2007;92(10): 1381–1388. [DOI] [PubMed] [Google Scholar]
- 30.Dufour C, Rondelli R, Locatelli F, et al. Stem cell transplantation from HLA-matched related donor for Fanconi’s anaemia: a retrospective review of the multicentric Italian experience on behalf of AIEOP-GITMO. Br J Haematol. 2001;112(3):796–805. [DOI] [PubMed] [Google Scholar]
- 31.Peffault de Latour R, Porcher R, Dalle JH, et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood. 2013; 122(26):4279–4286. [DOI] [PubMed] [Google Scholar]
- 32.Rosenberg PS, Socié G, Alter BP, Gluckman E. Risk of head and neck squamous cell cancer and death in patients with Fanconi anemia who did and did not receive transplants. Blood. 2005;105(1):67–73. [DOI] [PubMed] [Google Scholar]
- 33.Pasquini R, Carreras J, Pasquini MC, et al. HLA-matched sibling hematopoietic stem cell transplantation for fanconi anemia: comparison of irradiation and nonirradiation containing conditioning regimens. Biol Blood Marrow Transplant. 2008;14(10): 1141–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benajiba L, Salvado C, Dalle JH, et al. HLA-matched related-donor HSCT in Fanconi anemia patients conditioned with cyclophosphamide and fludarabine. Blood. 2015;125(2):417–418. [DOI] [PubMed] [Google Scholar]
- 35.Hutson SP, Han PK, Hamilton JG, et al. The use of haematopoietic stem cell transplantation in Fanconi anaemia patients: a survey of decision making among families in the US and Canada. Health Expect. 2015;18(5):929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khan NE, Rosenberg PS, Lehmann HP, Alter BP. Preemptive Bone Marrow Transplantation for FANCD1/BRCA2. Biol Blood Marrow Transplant. 2015;21(10): 1796–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]