

Abstract
Downregulation of the unfolded protein response mediates proteasome inhibitor resistance in multiple myeloma. The Human Immunodeficieny Virus protease inhibitor nelfinavir activates the unfolded protein response in vitro. We determined dose-limiting toxicity and recommended dose for phase II of nelfinavir in combination with the proteasome inhibitor bortezomib. Twelve patients with advanced hematologic malignancies were treated with nelfinavir (2500–5000 mg/day p.o., days 1–14, 3+3 dose escalation) and bortezomib (1.3 mg/m2, days 1, 4, 8, 11; 21-day cycles). A run in phase with nelfinavir monotherapy allowed pharmakokinetic/pharmakodynamic assessment of nelfinavir in the presence or absence of concomittant bortezomib. End points included dose-limiting toxicity, activation of the unfolded protein response, proteasome activity, toxicity and response to trial treatment. Nelfinavir 2×2500 mg was the recommended phase II dose identified. Nelfinavir alone significantly up-regulated expression of proteins related to the unfolded protein response in peripheral blood mononuclear cells and inhibited proteasome activity. Of 10 evaluable patients in the dose escalation cohort, 3 achieved a partial response, 4 stable disease for 2 cycles or more, while 3 had progressive disease as best response. In an exploratory extension cohort with 6 relapsed, bortezomib-refractory, lenalidomide-resistant myeloma patients treated at the recommended phase II dose, 3 reached a partial response, 2 a minor response, and one progressive disease. The combination of nelfinavir with bortezomib is safe and shows promising activity in advanced, bortezomib-refractory multiple myeloma. Induction of the unfolded protein response by nelfinavir may overcome the biological features of proteasome inhibitor resistance.
Introduction
Proteasome inhibitors are the backbone of multiple myeloma (MM) therapy in Europe.1 However, the majority of MM patients ultimately develop proteasome inhibitor resistance, and proteasome inhibitor therapy yielded disappointing results in other hematologic malignancies. Response of bortezomib-refractory MM to next generation drugs (carfilzomib, pomalidomide) is in the 20%–30% range,2;3 leaving the majority of bortezomib-resistant patients currently without active therapy. Proteasome inhibitor sensitivity of MM cells is modulated by the unfolded protein response (UPR),4–6 a conserved pathway7 that prevents accumulation of misfolded and dysfunctional protein in the endoplasmic reticulum (ER) by acting on mRNA translation, protein folding and destruction. The latter is orchestrated by the ER-associated degradation machinery (ERAD), with the proteasome as its rate-limiting terminal protease.8 Excessive activation of the UPR (terminal UPR) results in apoptosis and is a major mechanism of cytotoxicity of proteasome inhibitors in MM.6
The level of UPR pre-activation modulates both maturation stage and proteasome inhibitor-sensitivity of MM, so that pharmacological activation of the UPR may overcome proteasome inhibitor resistance.9 Activation of the UPR is initiated via three ER-resident transmembrane proteins, including inositol-requiring kinase 1 (IRE1). IRE1 drives activation of Xbox-binding protein (XBP1), a major regulator of chaperones and ERAD, while a pro-apoptotic pathway is triggered via CCAAT/-enhancer-binding protein homologous protein (CHOP) upon excessive UPR activation. Silencing of IRE1 or XBP1 in MM results in proteasome inhibitor resistance,4 and the response of MM to bortezomib correlates with high XBP1 expression.10 The status of UPR activation links proteasome inhibitor sensitivity of MM to the differentiation pathway from pre-plasmablasts to mature plasma cells. Full plasma cell maturation requires UPR activation via the IRE1/XBP1 axis11 and results in a mature, proteasome inhibitor-sensitive MM cell type.4 In contrast, IRE1-/XBP1- MM cells are immature, proteasome inhibitor-resistant, lack a fully developed ER,12 and accumulate in proteasome inhibitor-resistant MM patients.4
While IRE1-targeting drugs are in early development13 the HIV protease inhibitor nelfinavir has UPR- and IRE1/XBP1-inducing pre-clinical activity,14–16 allowing proof-of-concept clinical trials to test the effect of UPR induction on proteasome inhibitor-sensitivity of MM. The UPR-inducing activity of nelfinavir on mammalian cells may involve interference with UPR-activating proteases,17 the pAKT pathway18–20 and/or the proteasome.21–23 Nelfinavir has single agent pre-clinical activity against MM, leukemia and solid tumors in vitro and in vivo,15;24–28 and re-sensitizes proteasome inhibitor-resistant tumor cells, including MM, at low micromolar concentrations.21;22;29 Nelfinavir is registered at 2×1250 mg/day. A dose of 2×3125 mg is safe in patients with advanced solid tumors,30 and nelfinavir is under investigation as a sensitizer for chemotherapy or radiation.31
The primary aim of this trial was to evaluate the safety and establish the recommended dose for a phase II trial (RP2D) of nelfinavir in combination with standard-dose bortezomib in patients with hematologic malignancies, including MM. Molecular studies assessed the effect of nelfinavir on UPR and proteasome activity. Early signs of activity were explored in patients with bortezomib-refractory MM.
Methods
Eligibility
Patients with advanced MM, acute leukemia or malignant lymphoma lacking active standard treatment options were eligible for the trial. Eligibility criteria included less than 5 prior chemotherapy lines, ECOG performance score of 2 or under, adequate hematologic, hepatic and renal function. Major exclusion criteria included uncontrolled, clinically relevant medical conditions, CTC grade over 1 peripheral polyneuropathy, and use of strong CYP3A4 modulators. In the extension cohort of the trial, only bortezomib-resistant MM patients were eligible after at least 2 lines of systemic treatment. As according to the protocol developed before 2011, “bortezomib-resistant” disease was defined as “nonresponsiveness or progression to bortezomib-containing therapy, or progression < 6 months after completing such therapy”. In agreement with IMWG criteria, we here refer to “bortezomib-refractory myeloma” as myeloma “nonresponsive while on bortezomib-containing therapy, or progressive within 60 days of last bortezomib-containing therapy”.32
Trial design
The primary end point was dose-limiting toxicity (DLT). Secondary end points included adverse events (AE), pharmacodynamic/pharmacokinetic parameters and response to trial treatment. Dose escalation was performed in a classical 3+3 design. AE were graded according to CTCAE 4.0. DLT was assessed during cycle 1 in all patients who had received at least one dose of bortezomib in combination with nelfinavir, and defined as hematologic toxicity grade 3–4 persisting for more than two weeks, or any grade 3 or over non-hematologic AE judged to be possibly related to nelfinavir or bortezomib, excluding self-limiting grade 3 ALT, bilirubin or metabolic changes (cholesterol, blood glucose, triglycerides), grade 3 nausea/vomiting without adequate symptomatic therapy, or grade 3 neurotoxicity without dose reduction of bortezomib.
After establishing the recommended dose, the protocol was amended to treat an exploratory extension cohort of 6 additional patients with bortezomib-resistant myeloma at the recommended dose to assess toxicity and detect early signs of activity.
All patients gave written informed consent prior to trial inclusion, the trial was approved by the independent canton research ethics committees, performed in accordance with national Swiss law, ICH-GCP, and the Declaration of Helsinki, and registered at clinicaltrials.gov identifier:01164709.
Drug administration
Bortezomib was given at 1.3 mg/m2 days 1, 4, 8, 11 i.v. for three cycles of 21 days. No dexamethasone was added during the dose escalation part. Nelfinavir [dose levels 1250 mg (DL0), 1875 mg (DL1) and 2500 mg (DL2)] was taken on days 1–14 as 625 mg capsules q 12 h p.o. together with a full meal. In cycle 1 only, combination treatment with bortezomib+nelfinavir was preceded by nelfinavir monotherapy on days −7 to −1 (run in phase for PK/PD assessment). Patients without disease progression after cycle 3 could continue trial therapy until completion of 7 cycles.
Dose-limiting toxicity and response assessment
Adverse events were recorded throughout the trial for all patients. Response assessment was peformed centrally by the SAKK.
Pharmacokinetic and pharmacodynamic assessment
For details regarding PK/PD samples collection and measurements, please refer to the Online Supplementary Methods.
Further details of the methods used are available in the Online Supplementary Methods.
Results
Patients and dose escalation
The dose escalation cohort of the trial was treated between July 2010 and April 2012 at 4 academic hospitals in Switzerland. Twelve patients were included in the dose escalation cohort (Table 1). Treatment was performed as outlined (Figure 1). In cycle 1, more than 93% of the planned nelfinavir and bortezomib dose was administered per protocol. The only DLT was a G4 ALT elevation at DL2 that resolved after trial drug discontinuation. Nine patients discontinued treatment before completion of cycle 3: 5 progressive disease (PD), one due to toxicity, one death related to the underlying disease, 2 premature discontinuations for putative disease progression (see below). An average of 2.6 treatment cycles was administered in the dose escalation cohort.
Table 1.
Characteristics of the patients, diseases, treatment durations and responses in the phase I dose escalation cohort of the trial.
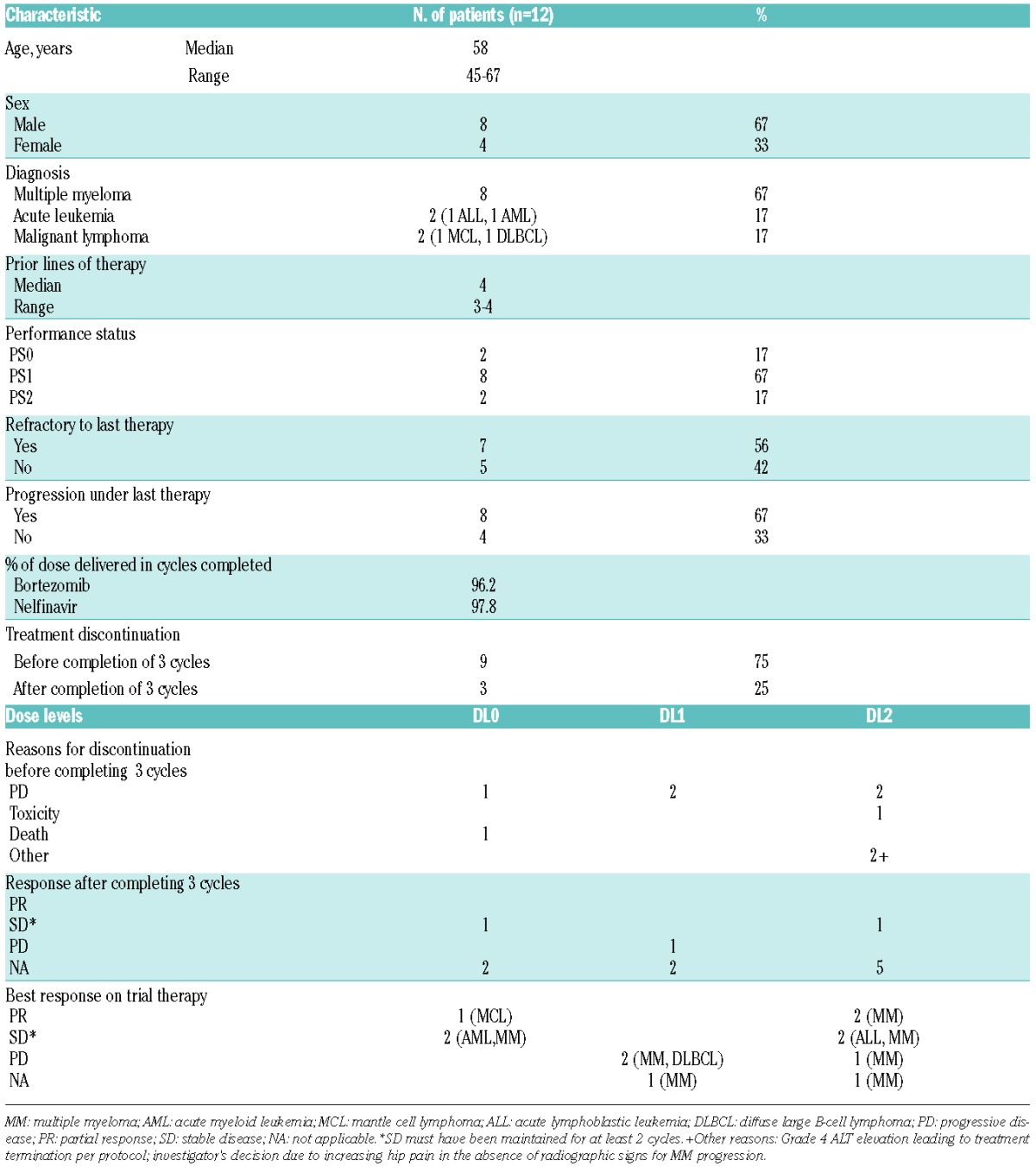
Figure 1.
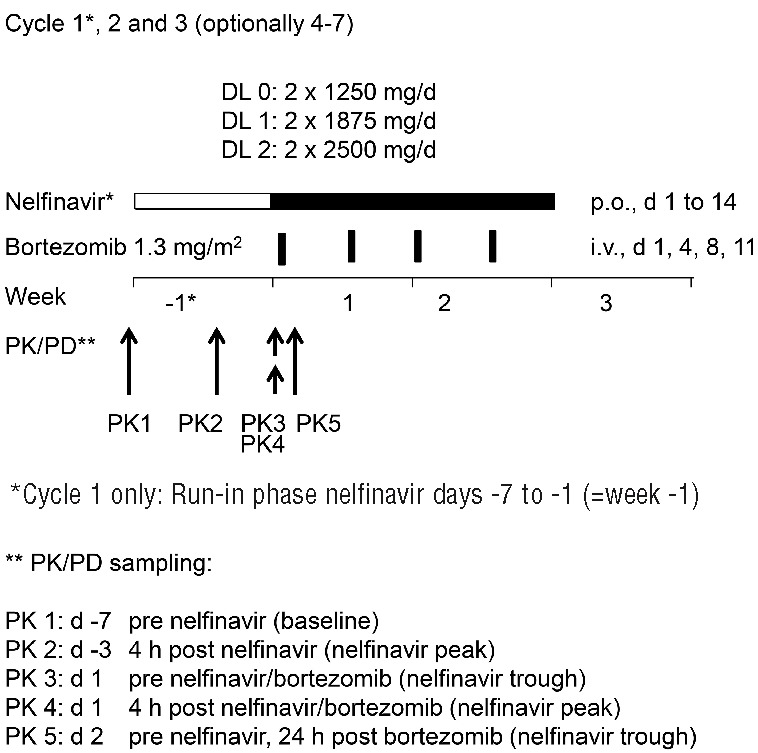
Graphical overview of dosing of nelfinavir (dose levels DL0, DL1, DL2) and bortezomib together with the time points at which samples for pharmakokinetic/pharmakodynamic (PK/PD) analysis were collected (PK1-5). Nelfinavir dosing in week -1 was only performed in cycle 1.
Toxicity and anti-tumor activity
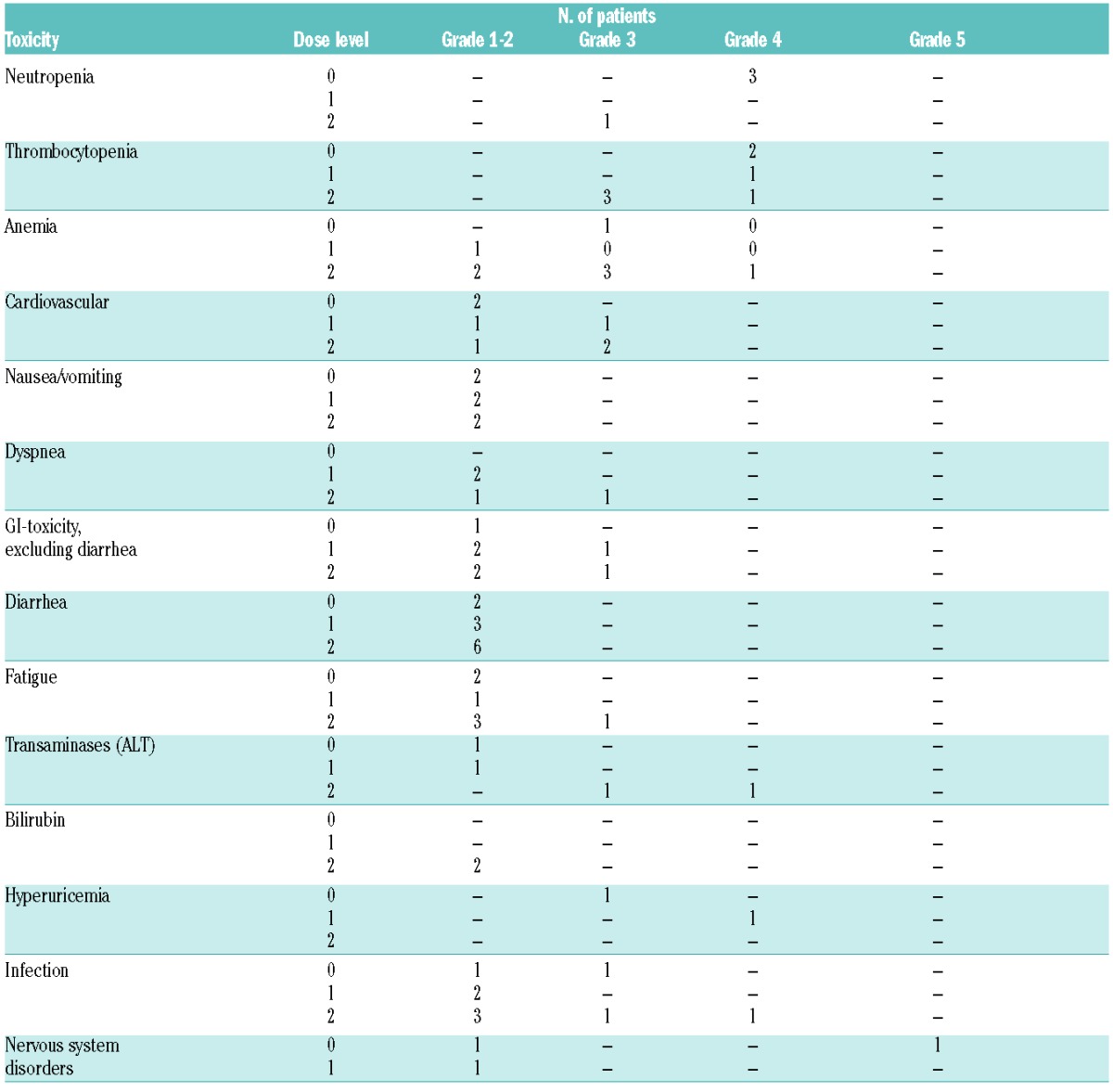
Seven serious adverse events were observed in 5 patients (Table 2): one fatal thrombotic cerebral ischemia (DL0) occurring after withdrawl of the trial drug in a patient with leukemic mantle cell lymphoma who was hospitalized with pneumonia, 2 cases of pneumonia, one neutropenic fever, one gastrointestinal hemorrhage and one hyperviscosity syndrome (all grade 3), and one pulmonary embolism that resolved without sequelae. Except for one infection and the hyperviscosity syndrome, all SAE were judged to be possibly related to trial medication. Because the thrombotic ischemia occurred after trial drug withdrawl, it was not considered a DLT. The other SAEs occurred after therapy cycle 1 and did not meet the criteria for DLT.
Table 2.
Overview of the number of adverse events graded according to Common Toxicity Criteria 4.0 per dose level in the phase I dose escalation cohort of the trial.
Ten patients received at least one full cycle of trial therapy and were evaluable for best treatment response during trial therapy (Table 1). Of these, 3 achieved a partial response (PR), 4 remained in stable disease (SD) for at least 2 cycles, while 3 progressed within 2 therapy cycles. Therapy was prematurely stopped in 2 MM patients, based on putative progression of MM lesions on radiological images assessed by the local investigators; however, independent analysis of imaging results later concluded that there had been no objective evidence for MM progression. These patients were, therfore, considered non-evaluable for anti-tumor activity. Of the patients with malignancies other than MM, one patient with MCL achieved a PR, 2 patients with relapsed, refractory acute leukemia [1 acute myeloid leukemia (AML), 1 acute lymphoblastic leukemia (ALL)] remained stable for two cycles, while one patient with relapsed diffuse large B-cell lymphoma (DLBCL) experienced progressive disease (PD). Of the 3 patients from the dose escalation cohort that did complete three therapy cycles, 2 patients with MM experienced SD and one patient with DLBCL had PD.
Anti-tumor activity in relapsed, bortezomib-refractory myeloma
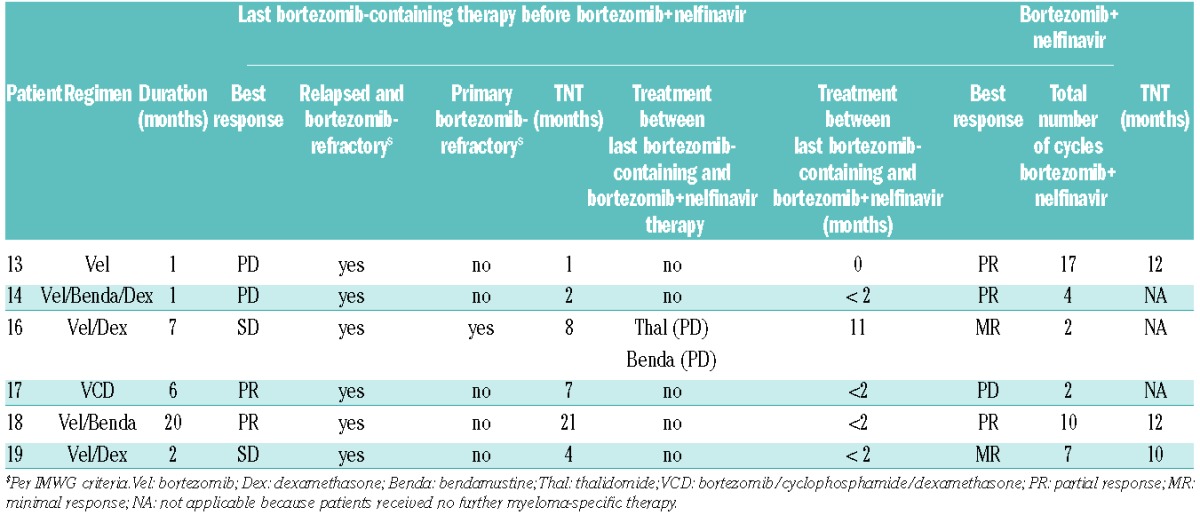
Six additional patients with bortezomib-refractory myeloma were treated with the tentative recommended dose of nelfinavir 2×2500 mg p.o. in combination with bortezomib. The disease characteristics and prior therapies and responses of the included patients are summarized in Tables 3 and 4. Importantly, all patients were bortezomib-refractory according to IMWG criteria, and all in addition had lenalidomide-resistant disease. Five out of 6 of these patients received bortezomib+nelfinavir treatment within less than two months after having experienced PD under other bortezomib-based combination regimens, inlcuding double and triple combinations with dexamethasone, bendamustine and cyclophosphamide, with no additional interposed lines of treatment. Only one patient received interposed treatment lines between the last regimen that defined bortezomib-refractory disease and the experimental bortezomib+nelfinavir therapy, and this patient had failed under both interposed therapies (thalidomide and bendamustine, respectively). Three of these patients with bortezomib-refractory MM achieved a PR with bortezomib+nelfinavir tretament. They all had been treated with bortezomib+nelfinavir immediately after (<2 months) progressing under therapies with bortezomib/bendamustine/dexamethasone (UPN 14) and bortezomib/bendamustine (UPN18) or bortezomib monotherapy (UPN 13), respectively. Another 2 patients achieved a minimal response (MR) with bortezomib+nelfinavir after having progressed under prior bortezomib/dexamethasone therapy. One of these patients, a 77-year old woman, refused trial continuation after suffering a myocardial infarction secondary to a bacteriemia from a portacath infection in cycle 2, whereas the other patient received 5 cycles per protocol. One patient did not respond to bortezomib+nelfinavir treatment. His MM had been refractory to lenalidomide/dexamethasone first-line and refractory to bortezomib/cyclophosphamide/dexamethasone second-line therapy. An average number of 4.5 therapy cycles was administered in this cohort within the trial. There was no change in the toxicity profile. One patient with rapidly progressive MM relapse after allogeneic transplant (UPN 15) developed graft-versus-host disease (GvHD) and generalized Epstein-Barr virus (EBV) infection during the first week of trial treatment. She was excluded from the trial during the first week of treatment due to a rapidly worsening clinical condition, although her paraprotein decreased. The patient died of GvHD within two weeks, was replaced in the trial, and was not evaluated for efficacy. A patient chart-based additional analysis revealed that 3 patients treated in the extension cohort continued their bortezomib+nelfinavir therapy post trial on an off label basis for a total of 7, 10 and 17 cycles. They experienced 10-, 12-, and 12-month intervals to next antimyeloma treatment, respectively.
Table 3.
Patients with bortezomib-refractory multiple myeloma treated in the extension cohort of the trial. Overview of the disease characteristics, response to prior therapies, number of treatment cycles and response achieved on an individual patient basis.
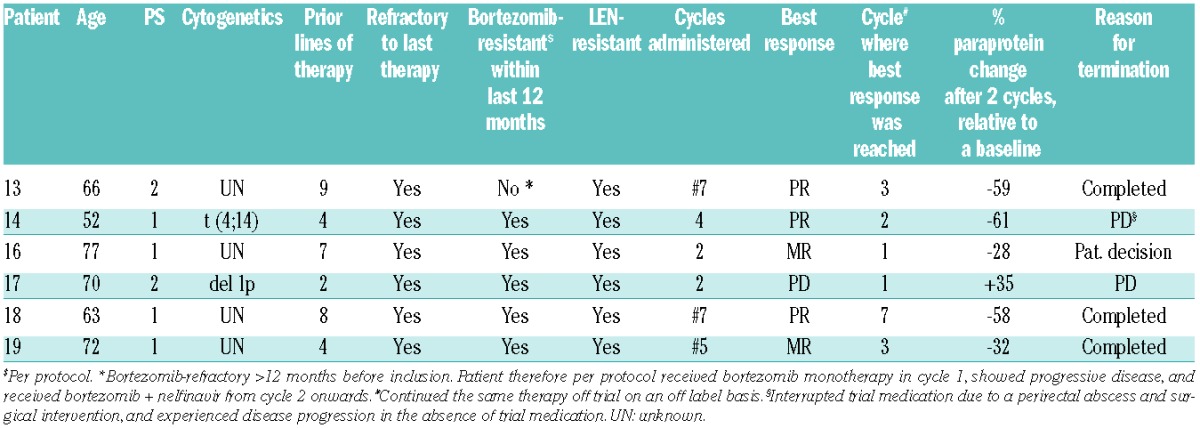
Table 4.
Details of last bortezomib-containing therapy [(regimen, duration, best treatment response, relapsed and bortezomib-refractory per IMWG criteria, primary bortezomib-refractory per IMWG criteria, time to next treatment (TNT), time between last and current bortezomib-containing regimen (months), treatment regimens and respective response between last and current bortezomib-containing regimen)], in conjunction with details of the current bortezomib+nelfinavir therapy [(best response, total number of bortezomib+nelfinavir cycles administered during trial and during subsequent off label therapy, time to next treatment (TNT)] for the 6 MM patients treated in the extension cohort of the trial.
Over all dose levels, of the 7 patients with bortezomib-refractory MM (according to IMWG criteria) treated in the trial, 4 achieved a PR, 2 an MR, and one PD. Of 11 patients with relapsed, bortezomib-resistant MM (according to trial criteria) treated, 4 achieved a PR, 2 an MR and 2 SD for 2 cycles or more with nelfinavir+bortezomib treatment (Figure 2), corresponding to a clinical benefit (PR+MR) rate of 55% in this small sample.
Figure 2.
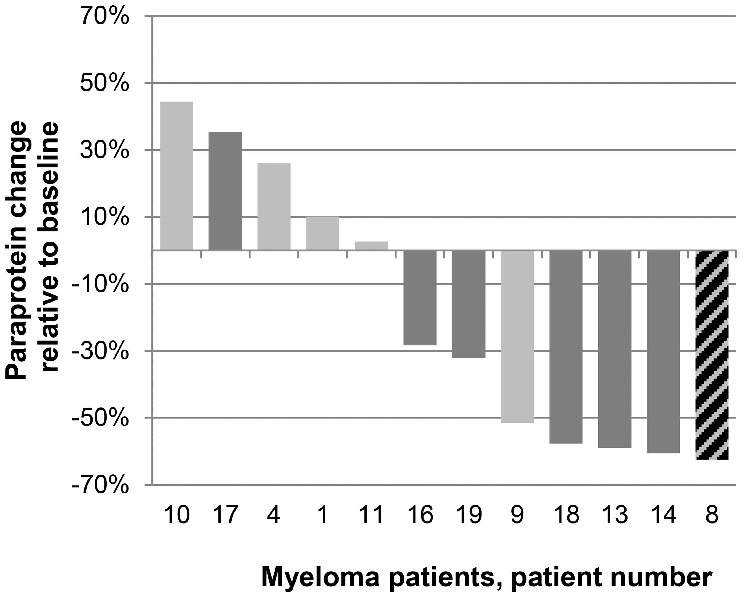
Overview of the best treatment response of all evaluable patients with multiple myeloma (MM) that have received trial treatment. The changes in paraprotein levels compared to baseline while on trial are displayed relative to baseline. Individual patients are represented by UPN numbers, patients of the phase I dose escalation cohort (UPN 1–12) in light gray, patients of the extension cohort (UPN 13–19) in dark gray. The streaked lines pattern denotes the only indivudual MM patient in the trial (UPN 08, dose escalation cohort) that was not bortezomib refractory by IMWG criteria before receiving bortezomib+nelfinavir therapy.
Pharmacodynamic analysis
Peripheral blood mononuclear cells (PBMC) (n=11, one technical failure) from the dose escalation phase of the trial were analyzed for protein expression indicative of UPR activation (PDI, BIP, CHOP, PARP), p-AKT, and proteasome activity (Figure 3). Comparison of protein expression between base-line (PK1) and peak nelfinavir plasma levels (PK2) revealed upregulation of CHOP and PARP (CHOP +56%, 95%CI: +17%–+95%, P=0.008; PARP + 57%, 95%CI: +2% – +112%, P=0.04; n=10), demonstrating induction of the UPR-mediated apoptotic machinery by nelfinavir monotherapy. PDI and BIP expression showed mean increases of 71% and 55% (P=0.19; P=0.17), also consistent with UPR activation by nelfinavir, while p-AKT decreased by 12% (P=0.37). Because activation of IRE1/XBP1 controls bortezomib-sensitivity of MM,4 we undertook a post hoc analysis of remaining PBMC lysate of one patient (UPN 04) for pIRE-1 protein by western blot, which suggestied pIRE1 induction by nelfinavir (Online Supplementary Figure S1).
Figure 3.
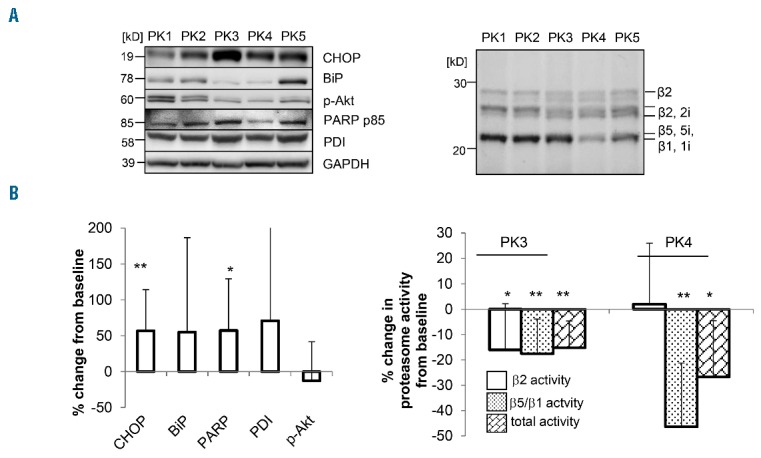
Expression of unfolded protein response (UPR)-related proteins and proteasome activity in peripheral blood mononuclear cells (PBMC) from treated patients PBMC were collected at the indicated time points PK1 (base-line predose), PK2 (nelfinavir 4 h postdose), PK3 (nelfinavir trough + bortezomib pre dose), PK4 (nelfinavir+bortezomib 4 h post dose), PK5 (nelfinavir trough, 24 h post bortezomib). Equal amounts of cellular protein from PBMC was resolved by 12.5% SDS PAGE. (A) exemplary changes in expression of UPR-related protein and proteasome activity in PBMC from one patient (UPN04). Upper left: protein representing the UPR and its related apoptotic machinery (BIP, CHOP, PDI, PARP), as well as pAKT, was assessed from PBMC collected at the time points PK1–PK5, and visualized by western blot, GAPDH served as a control. Upper right: Quantitative comparison of proteasome activity (seperately for the β2-type and β1/β5-type subunits) at the different time points PK1–PK5 was achieved with activity based proteasome probes in the same PBMC samples (see Methods). (B) Mean quantitative changes in expression of UPR-related proteins and proteasome activity in PBMC for all patients Botom left: effect of nelfinavir monotherapy on expression of UPR related protein in PBMC. The relative change in expression of the UPR-associated proteins was assessed for all patients by western blot as (as shown for UPN04 above), and quantified by flourescence scanning comparing the time points PK1 (baseline) versus PK2 (post-nelfinavir); n= 11 patients, one technical failure). The mean relative difference from a base-line signal and standard deviation of expression of the proteins indicated is displayed for PK1 versus PK2. Bottom right: changes in proteasome activity in PBMC for the entire dose escalation cohort are shown after nelfinavir monotherapy (PK1 vs. PK3) and after treatment with nelfinavir in combination with bortezomib (PK1 vs. PK4). The relative changes in proteasome activity in PBMC were quantified using affinity-based probes and flourescence scanning. Mean changes from 11 patients and standard deviation are presented.
During nelfinavir treatment, and prior to bortezomib injection (PK3), we observed a mean decrease in proteasome β2 and β1/β5 subunit activity of 16% and 17% (P=0.01 and P=0.002), respectively, compared to baseline. Four hours after bortezomib injection (PK4), mean reduction in β1/β5 proteasome activity was 46%, compared to baseline (P<0.0001), while β2 activity levels normalized (mean difference 2% from baseline). Nelfinavir treatment, therefore, has proteasome-inhibiting and UPR-inducing activity in vivo.
Pharmacokinetics
Nelfinavir plasma concentrations decreased during nelfinavir monotherapy for DL1 versus DL2 (PK2, mean plasma concentration DL1 13.3 μM vs. DL2 8.9 μM; P=0.08) (Figure 4). Peak plasma concentrations were higher during nelfinavir monotherapy compared to combination therapy with bortezomib (mean PK2 vs. PK4; 9.2 vs. 6.8 μM; P=0.002), suggesting induction of nelfinavir clearance either by autoinduction, concomitant bortezomib application, or both. A population PK model is consistent with autoinduction of the clearance of both nelfinavir (69% increase of nelfinavir clearence for time points >5 days after initiation of treatment), and the active metabolite M8 (120% increase of M8 clearence) (Online Supplementary Table S1). Covariate testing suggested no significant impact of patient age, gender, BSA, renal or liver function on the clearance of nelfinavir. Goodness-of-fit plots of model-predicted and observed nelfinavir and M8 concentration-time data support the adequacy of the model (Online Supplementary Figure S2).
Figure 4.
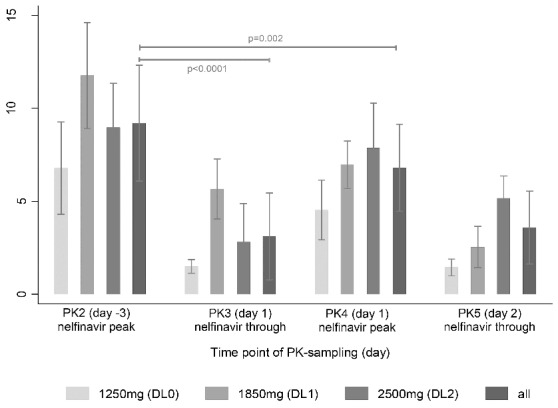
Mean nelfinavir plasma concentrations (μM) for the 4 pharmacokinetic sampling points and the 3 dosing cohorts (1250, 1850 and 2500 mg bid). Significance levels are given for the difference between mean NFV peak concentration (PK2) and either mean NFV through concnetration (PK3) or NFV peak concentration in combination with BTZ (PK4).
Discussion
This trial demonstrates that nelfinavir at a dose of 2500 mg b.i.d. can safely be added to the approved bortezomib therapy in patients with advanced hematologic cancers. Oral nelfinavir therapy induced activation of the UPR, including induction of pIRE1, consistent with pre-clinical data.22;24 The high fraction of bortezomib-refractory MM patients experiencing MM control after treatment with bortezomib+nelfinavir in our trial is consistent with the model that low activation levels of the UPR, and in particular of IRE1-XBP1, are a major determinator of proteasome inhibitor resistance in MM.4 We provide clinical proof of concept that the pharmacological induction of UPR activation can re-sensitize proteasome inhibitor refractory MM for proteasome inhibitor treatment.22 With response rates of bortezomib-refractory MM to next generation drugs (carfilzomib, pomalidomide) in the 20%–30% range,2,3 the signals of activity of bortezomib+nelfinavir observed in this patient group in our trial are encouraging: PR, including durable responses, was observed in 3 out of 5 patients with relapsed, bortezomib-refractory MM. Strikingly, all responding patients had experienced PD during bortezomib-containing combination therapy directly prior (< 60 days) to trial therapy, clearly indicating that the combination with nelfinavir is capable of overcoming bortezomib resistance in MM.
The low numbers of non-MM patients included in the trial limits an evaluation of potential activity in non-MM hematologic malignancies. One patient with relapsed, bortezomib-refractory mantle cell lymphoma with a clinically aggressive course achieved PR while another patient with relapsed ALL showed SD for 3 cycles. The addition of nelfinavir may, therfefore, also sensitize diseases other than MM for bortezomib treatment, consistent with preclinical data.
Trial design originated in 2010 allowed inclusion of MM patients who had relapsed within six months after bortezomib-containing therapy, while the IMWG criteria for bortezomib-refractory MM32 published in 2011 require disease progression within less than 60 days of such therapy. Although not required per protocol, all patients treated in the extension cohort of our trial fully met the current IMWG criteria for bortezomib-refractory MM, and in addition had lenalidomide-resistant MM. It is likely that a sizable fraction of the “bortezomib-resistant” patients from the dose escalation cohort of the study also matched the IMWG cirteria for bortezomib-refractory disease (i.e. had progressed <60 days after bortezomib-therapy, and not only after < 6 months); however, this was not captured in sufficient detail in the prospective data analysis plan designed before 2011. Panobinostat combined with bortezomib/dexamethasone had an overall response rate of 34% and a combined MR+PR rate of 52% in relapsed, bortezomib refractory MM.33 In our trial, 9 patients with relpased, bortezomib-resistant MM were treated with bortezomib+nelfinavir at the RP2D, rsulting in 4 PR (44%) and 6 patients with clinical benefit (MR+PR, 67%). There was no apparent additional hematologic toxicity from combining bortezomib with nelfinavir, in contrast to panobinostat, which may be important for treating a heaviliy pre-treated population of MM patients.
Dexamethasone is usually co-administered with bortezomib as anti-myeloma agent at doses between 40 and 160 mg weekly because of its synergistic activity. To apply the most stringent criteria for the identification of a clinical synergy between nelfinavir and bortezomib, our protocol did not allow dexamethasone co-administration in the dose escalation part of the trial, and also in the extension cohort, no dexamethasone co-administration was allowed until completion of cycle 3. After cycle 3 only, patients who did not achieve a MR were allowed to co-administer 8 mg dexamethasone with bortezomib, while higher doses were excluded due to potential interactions with nelfinavir through the Cyp3A4 system. All patients in the extension cohort, except UPN18, achieved their best response already during cycles 1–3, i.e. without the addition of dexamethasone, and UPN 13 achieved a PR with bortezomib-nelfinavir therapy after showing PD under bortezomib monotherapy.
One DLT (reversible CTC grade 4 AST elevation) was observed during the trial. No clinically significant hepatic toxicity was observed in subsequent therapy cycles or in the MM patients in the extension cohort where dexamethasone was added in some. Mild diarrhea was the most frequent adverse event; this was managed by supportive measures, including the early use of symptomatic loperamide suggested in the protocol in case of diarrhea, and did not result in treatment discontinuation. The patient experiencing DLT was transferred to follow up according to protocol, but was treated with the trial regimen at DL2 on an off label basis because she had experienced a PR and lacked alternative options. She received a total of 8 therapy cycles without hepatic toxicity before undergoing allogeneic stem cell transplant.
We used PBMC as surrogate tissue for pharmacodynamic assessment because proteasome inhibition in PBMC largely parallels that in bone marrow.34 Nelfinavir moderately reduced β2 proteasome activity that is unaffected by bortezomib.22,35 Co-inhibition of β2 sensitizes myeloma cells for treatment with the β5-targeted drugs bortezomib and carfilzomib.36 Bortezomib-resistant myeloma cells upregulate β2 activity,37 presumably as an adaptive mechanism, and β2 proteasome activity increases during bortezomib treatment.35 The co-inhibition of β2 proteasome activity may, therefore, contribute to the combined activity of nelfinavir+bortezomib against bortezomib-resistant myeloma. However, similar to nelfinavir, saquinavir and lopinavir have shown UPR-inducing and bortezomib-sensitizing features in vitro in the absence of significant intrinsic proteasome inhibition.22 The proteasome inhibition caused by nelfinavir is, therefore, unlikely to be the only mechanism driving UPR induction, and other targets in the UPR regulatory machinery may be involved.17 Peak nelfinavir plasma concentrations were consistent with synergistic activity with proteasome inhibitors in vitro,22;24;29 and with autoinduction of clearance by high drug concentrations,30 suggesting that co-medication with bortezomib may decrease nelfinavir plasma levels. Other proteasome inhibitors, such as carfilzomib, may improve synergistic activity.22
This trial demonstrates feasibility and safety of combining full-dose proteasome inhibitor therapy with pharmacological activation of the UPR, using nelfinavir, an approved drug with a known safety profile. Furthermore, it provides proof of concept that nelfinavir can be used as an innovative, biology-driven approach to re-sensitize patients with proteasome inhibitor-refractory MM for proteasome inhibitor treatment.4 Future research should focus on the clinical activity of this combination in the bortezomib-refractory setting, but also assess whether bortezomib may be replaced by next generation drugs, e.g. carfilzomib, to avoid drug interactions and improve activity, and whether nelfinavir may likewise be used in combination with novel oral proteasome inhibitors to boost their low single agent activity.38 Alternative HIV protease inhibitors with similar UPR-inducing activity like lopinavir may provide more favorable pharmacology. Likewise, UPR activation with nelfinavir combined with lenalidomide or pomalidomide, which modulate the ubiquitination process upstream of the proteasome through interaction with cereblon, an ubiquitin ligase in the ubiquitin-proteasome system, may allow similar re-sensitizing mechanisms to be exploited. This ultimately suggests exploring the addition of HIV protease inhibitors to established combinations of proteasome inhibitors with immunomodulatory drugs, for example, in the carfilzomib/lenalidomide/dexamethasone regimen, one of the most powerful and tolerable regimens available to date for advanced multiple myeloma. The activity of bortezomib+nelfinavir in combination with standard dose dexamethasone is presently being studied in an SAKK phase II trial of the Swiss Group for Clinical Cancer Research (SAKK) in patients with bortezomib-refractory myeloma while nelfinavir in combination with lenalidomide in lenalidomide-refractory patients is being tested in an SAKK phase I/II trial.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/3/346
Funding
The trial could be conducted through financial funding and support by the Swiss Group for Clinical Cancer Research (SAKK), the Swiss State Secretariat for Education, Research and Innovation (SERI), the Swiss Foundation for Clinical Cancer Research (SSKK), Janssen Cilag AG, and a translational research grant (31003A_143924) to CD from the Swiss National Research Foundation (SNF).
References
- 1.Moreau P, Richardson PG, Cavo M, et al. Proteasome inhibitors in multiple myeloma: 10 years later. Blood. 2012;120(5):947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel DS, Martin T, Wang M, et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood. 2012;120(14):2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.San MJ, Weisel K, Moreau P, et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14(11):1055–1066. [DOI] [PubMed] [Google Scholar]
- 4.Leung-Hagesteijn C, Erdmann N, Cheung G, et al. Xbp1s-negative tumor B cells and preplasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell. 2013;24(3):289–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neznanov N, Komarov AP, Neznanova L, Stanhope-Baker P, Gudkov AV. Proteotoxic stress targeted therapy (PSTT): induction of protein misfolding enhances the antitumor effect of the proteasome inhibitor bortezomib. Oncotarget. 2011;2(3):209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Obeng EA, Carlson LM, Gutman DM, et al. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011; 334(6059):1081–1086. [DOI] [PubMed] [Google Scholar]
- 8.Mayor T. Navigating the ERAD interaction network. Nat Cell Biol. 2011;14(1):46–47. [DOI] [PubMed] [Google Scholar]
- 9.Orlowski RZ. Why proteasome inhibitors cannot ERADicate multiple myeloma. Cancer Cell. 2013;24(3):275–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ling SC, Lau EK, Al-Shabeeb A, et al. Response of myeloma to the proteasome inhibitor bortezomib is correlated with the unfolded protein response regulator XBP-1. Haematologica. 2012;97(1):64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reimold AM, Iwakoshi NN, Manis J, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412(6844):300–307. [DOI] [PubMed] [Google Scholar]
- 12.Taubenheim N, Tarlinton DM, Crawford S, et al. High rate of antibody secretion is not integral to plasma cell differentiation as revealed by XBP-1 deficiency. J Immunol. 2012;189(7):3328–3338. [DOI] [PubMed] [Google Scholar]
- 13.Papandreou I, Denko NC, Olson M, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117(4):1311–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernstein WB, Dennis PA. Repositioning HIV protease inhibitors as cancer therapeutics. Curr Opin HIV AIDS. 2008;3(6):666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gills JJ, Lopiccolo J, Tsurutani J, et al. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. 2007;13(17):5183–5194. [DOI] [PubMed] [Google Scholar]
- 16.Chow WA, Jiang C, Guan M. Anti-HIV drugs for cancer therapeutics: back to the future¿ Lancet Oncol. 2009;10(1):61–71. [DOI] [PubMed] [Google Scholar]
- 17.Guan M, Fousek K, Jiang C, et al. Nelfinavir induces liposarcoma apoptosis through inhibition of regulated intramembrane proteolysis of SREBP-1 and ATF6. Clin Cancer Res. 2011;17(7):1796–1806. [DOI] [PubMed] [Google Scholar]
- 18.Gupta AK, Cerniglia GJ, Mick R, McKenna WG, Muschel RJ. HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Res. 2005;65(18):8256–8265. [DOI] [PubMed] [Google Scholar]
- 19.Plastaras JP, Vapiwala N, Ahmed MS, et al. Validation and toxicity of PI3K/Akt pathway inhibition by HIV protease inhibitors in humans. Cancer Biol Ther. 2008;7(5): 628–635. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Ikezoe T, Nishioka C, et al. NFV, an HIV-1 protease inhibitor, induces growth arrest, reduced Akt signalling, apoptosis and docetaxel sensitisation in NSCLC cell lines. Br. J Cancer. 2006;95(12):1653–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruning A, Vogel M, Mylonas I, Friese K, Burges A. Bortezomib targets the caspase-like proteasome activity in cervical cancer cells, triggering apoptosis that can be enhanced by nelfinavir. Curr Cancer Drug Targets. 2011;11(7):799–809. [DOI] [PubMed] [Google Scholar]
- 22.Kraus M, Bader J, Overkleeft H, Driessen C. Nelfinavir augments proteasome inhibition by bortezomib in myeloma cells and overcomes bortezomib and carfilzomib resistance. Blood Cancer J. 2013;3e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta AK, Li B, Cerniglia GJ, et al. The HIV protease inhibitor nelfinavir downregulates Akt phosphorylation by inhibiting proteasomal activity and inducing the unfolded protein response. Neoplasia. 2007;9(4):271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bono C, Karlin L, Harel S, et al. The HIV-1 protease inhibitor nelfinavir impairs proteasome activity and inhibits the multiple myeloma cells proliferation in vitro and in vivo. Haematologica. 2012;97(7):1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruning A, Rahmeh M, Gingelmaier A, Friese K. The mitochondria-independent cytotoxic effect of nelfinavir on leukemia cells can be enhanced by sorafenib-mediated mcl-1 downregulation and mitochondrial membrane destabilization. Mol Cancer. 2010;9:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikezoe T, Saito T, Bandobashi K, et al. HIV-1 protease inhibitor induces growth arrest and apoptosis of human multiple myeloma cells via inactivation of signal transducer and activator of transcription 3 and extracellular signal-regulated kinase 1/2. Mol Cancer Ther. 2004;3(4):473–479. [PubMed] [Google Scholar]
- 27.Shim JS, Rao R, Beebe K, et al. Selective inhibition of HER2-positive breast cancer cells by the HIV protease inhibitor nelfinavir. J Natl Cancer Inst. 2012; 104(20):1576–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Y, Ikezoe T, Takeuchi T, et al. HIV-1 protease inhibitor induces growth arrest and apoptosis of human prostate cancer LNCaP cells in vitro and in vivo in conjunction with blockade of androgen receptor STAT3 and AKT signaling. Cancer Sci. 2005;96(7):425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraus M, Muller-Ide H, Ruckrich T, et al. Ritonavir, nelfinavir, saquinavir and lopinavir induce proteotoxic stress in acute myeloid leukemia cells and sensitize them for proteasome inhibitor treatment at low micromolar drug concentrations. Leuk Res. 2014;38(3):383–392. [DOI] [PubMed] [Google Scholar]
- 30.Pan J, Mott M, Xi B, et al. Phase I study of nelfinavir in liposarcoma. Cancer Chemother Pharmacol. 2012;70(6):791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brunner TB, Geiger M, Grabenbauer GG, et al. Phase I trial of the human immunodeficiency virus protease inhibitor nelfinavir and chemoradiation for locally advanced pancreatic cancer. J Clin Oncol. 2008;26(16):2699–2706. [DOI] [PubMed] [Google Scholar]
- 32.Rajkumar SV, Harousseau JL, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood. 2011;117(18): 4691–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Durie BG, Harousseau JL, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006; 20(9):1467–1473. [DOI] [PubMed] [Google Scholar]
- 34.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579–586. [DOI] [PubMed] [Google Scholar]
- 35.Blade J, Samson D, Reece D, et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. Br J Haematol. 1998;102(5): 1115–1123. [DOI] [PubMed] [Google Scholar]
- 36.Li N, Kuo CL, Paniagua G, et al. Relative quantification of proteasome activity by activity-based protein profiling and LC-MS/MS. Nat Protoc. 2013;8(6):1155–1168. [DOI] [PubMed] [Google Scholar]
- 37.Demo SD, Kirk CJ, Aujay MA, et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007;67(13):6383–6391. [DOI] [PubMed] [Google Scholar]
- 38.Richardson PG, Schlossman RL, Alsina M, et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood. 2013;122(14):2331–2337. [DOI] [PubMed] [Google Scholar]
- 39.Berkers CR, Verdoes M, Lichtman E, et al. Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nat Methods. 2005;2(5):357–362. [DOI] [PubMed] [Google Scholar]
- 40.Mirabella AC, Pletnev AA, Downey SL, et al. Specific cell-permeable inhibitor of proteasome trypsin-like sites selectively sensitizes myeloma cells to bortezomib and carfilzomib. Chem Biol. 2011;18(5):608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruckrich T, Kraus M, Gogel J, et al. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia. 2009;23(6):1098–1105. [DOI] [PubMed] [Google Scholar]
- 42.Richardson PG, Baz R, Wang M, et al. Phase 1 study of twice-weekly ixazomib, an oral proteasome inhibitor, in relapsed/refractory multiple myeloma patients. Blood. 2014; 124(7):1038–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]