

Myeloproliferative neoplasms (MPN) are a phenotypically defined, heterogeneous group of blood cancers characterised by the proliferation of progenitor cells and the accumulation of mature myeloid cells, linked by a propensity to transform into myelofibrosis or acute myeloid leukaemia. The Jak2V617F mutation is commonly found in patients with MPN, specifically polycythaemia vera (>95%), essential thrombocythaemia and primary myelofibrosis (~50%).1–4 This mutation arises in the haematopoietic stem and progenitor cell (HSPC) compartment,5 and confers cytokine hypersensitivity and constitutive Jak2 tyrosine kinase signalling in the presence of an erythropoietin (EPO), thrombopoietin (TPO) or interleukin-3 (IL-3) receptor scaffold.2 Canonical Jak2 signalling relies on extracellular ligand binding to a membrane bound receptor to activate downstream pathways, in particular the phosphorylation of transcription factors Stat3 and Stat5, triggering nuclear translocation of these proteins and activation of multiple target genes. Stat5 loss prevents the development of Jak2V617F-induced MPN and is dispensable in normal haematopoiesis.6 Stat3 deletion in haematopoietic cells accelerates myeloproliferation.7 These findings highlight a critical role for Stat5 in MPN pathogenesis but a suppressive role for Stat3. Non-canonical nuclear roles for JAK2 have recently been shown,8 however Jak2V617F needs to bind to endogenous cytokine receptors for efficient signalling and MPN transformation.9 IL-3 signalling is dependent on downstream Jak2 activation, and IL-3R is expressed on HSPC populations, as is the receptor for TPO, MPL, identifying these cytokine signalling axes as potentially important in Jak2V617F MPN.
Recently, the first pharmacological inhibitor of Jak1/2 kinases, Ruxolitinib, has been approved for the treatment of patients with MPN. Ruxolitinib is effective at controlling the symptoms of MPN, but does not eliminate the MPN-initiating HSC population,10 which may favour resistant mutants. Conversely, treatments such as interferon-α may directly target HSPC in Jak2V617F-induced MPN,11 but remain poorly tolerated clinically. Other modalities to target Jak2V617F HSPC may include antibodies or small molecule inhibitors that selectively block cytokine signalling that are required for Jak2V617F pathogenesis.
As the JAK-STAT pathway and its upstream cytokine receptors have essential roles in MPN pathogenesis, dissecting the individual roles of these molecules is required to inform therapeutic strategies. In murine models, EpoR is not expressed on HSCs, but rather on lineage-committed erythroid precursors, and restricting Jak2V617F expression to EpoR expressing cells results in a markedly attenuated MPN phenotype,12 supporting the absence of a role for the EpoR in MPN initiation. Recently, TPO and its receptor MPL have demonstrated to be crucial for Jak2V617F-induced MPN development.13 Furthermore, IL-3R is not only expressed in HSPC populations, but is a critical regulator of white blood cell production,14 therefore it may be particularly relevant to the pathogenesis of polycythaemia vera (PV) which typically manifests with both elevated white blood cell counts and haematocrit. Interestingly, HSPC expressing high levels of IL-3Rα have been described in PV patients but not the progenitors of healthy individuals,5 providing further rationale to study IL-3 signalling in the pathogenesis of PV. In this work, we examine the requirement for IL-3 signalling in Jak2V617F MPN.
Using 6–8 week old conditional Jak2V617F (hereafter Jak2VF) knockin mice,10 we generated bone marrow chimeras by mixing 1×106 Jak2VF bone marrow (BM) cells expressing CD45.2 with 1×106 age-matched wild-type (WT) CD45.1 BM cells, and injected the cell mix into lethally irradiated C57BL/6 × Ptprca (CD45.1/CD45.2) mice (Figure 2G). We first sought to determine the cytokines responsible for driving JAK-STAT signal transduction in Jak2VF HSPC using phosphorylation-specific Stat5 antibody by flow cytometry after 10 minutes of stimulation with recombinant murine IL-3 (20ng/ml) or TPO (100ng/ml) (Figure 1A), as previously described.10 Either IL-3 or TPO activated Stat5 in Jak2VF myeloid-committed progenitors (LK; lineagelowc-Kit+Sca-1-), multipotent progenitors (LKS; lineagelowc-Kit+Sca-1+) and long-term haematopoietic stem cells (LT-HSC; LKS+CD150+CD48-). LK and LKS cells showed greater pStat5 transduction with IL-3 than with TPO, suggesting that these progenitor populations may be preferentially activated by IL-3. Conversely, Jak2VF LT-HSCs showed greater pStat5 stimulation with TPO. WT cells were similarly stimulated by IL-3 and TPO compared to Jak2VF cells (Figure 1B,D), consistent with the published data.10
Figure 2.

IL-3 signalling does not affect bone marrow HSPCs of Jak2VF mice. (A) Example flow cytometry plots showing gating strategy of LK, LKS and LT-HSC populations in bone marrow (BM) cells. Enumerated frequencies of (B) LT-HSC, (C) LKS, (D) LK, (E) GMP, and (F) MEP cell populations, demonstrating overall unchanged proportions of these cells in WT C57BL/6, βc/βIL-3−/−, Jak2VF and Jak2VFβc/βIL-3−/− mice. (G) Schema of competitive transplant experiment. BM chimeras of Jak2VF or Jak2VFβc/βIL-3−/− (CD45.2) cells with WT (CD45.1) cells in equal numbers were generated in irradiated (IR) recipient mice (CD45.1/CD45.2). Mice were bled at four-week intervals to check peripheral blood chimerism. (H) Peripheral blood WBC and Hgb of Jak2VF or Jak2VFβc/βIL-3−/−BM chimeras at week 16 showed no difference in WBC levels or (I) spleen weight, but a slight increase was observed in Hgb in Jak2VFβc/βIL-3−/− mice compared to Jak2VF. (J) Peripheral blood chimerism between Jak2VF and Jak2VFβc/βIL-3−/− recipients over 16 weeks was unchanged in three replicate experiments. All statistical analyses performed with Mann-Whitney test (Graphpad Prism V6.0).
Figure 1.
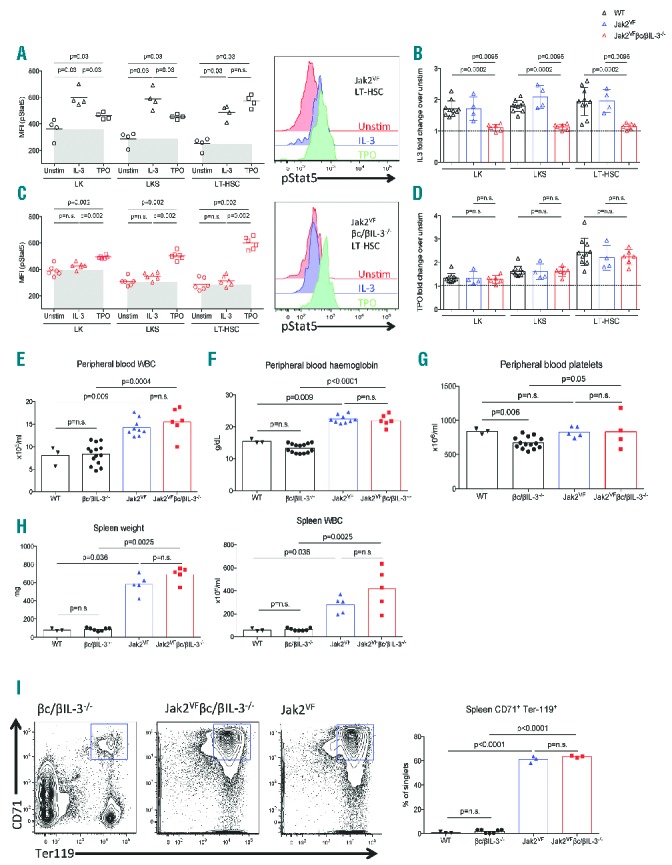
IL-3 signalling does not contribute to Jak2VF MPN pathogenesis. (A) Phospho-Stat5 (pStat5) levels in bone marrow (BM) LK, LKS and LT-HSC cells of Jak2VF mice after stimulation with IL-3 (20ng/ml), TPO (100ng/ml) or 1% BSA (Unstim), and representative flow cytometric analysis in Jak2VF BM LT-HSC. (B) Fold change difference of IL-3 stimulation of wild-type (WT), Jak2VF and Jak2VF βc/βIL-3−/− BM cells compared to unstimulated cells. (C) Phospho-Stat5 (pStat5) levels in bone marrow LK, LKS and LT-HSC cells of Jak2VF βc/βIL-3−/− mice after stimulation with IL-3, TPO or Unstim (as per panel A), showing IL-3 does not signal to Stat5 in these mice, and representative flow cytometric analysis in Jak2VFβc/βIL-3−/− BM LT-HSC. (D) Fold change difference of TPO stimulation of WT, Jak2VF and Jak2VFβc/βIL-3−/− BM cells compared to unstimulated cells. Peripheral blood counts of (E) white blood cells (WBC), (F) haemoglobin, and (G) platelets of WT C57BL/6, βc/βIL-3−/−, Jak2VF or Jak2VFβc/βIL-3−/− mice. (H) Spleen weight and WBC counts are elevated in Jak2VF and Jak2VFβc/βIL-3−/− mice, consistent with a PV phenotype. (I) Representative flow cytometry plots and quantitative data of spleen cells showing erythroid precursor expansion (CD71+Ter119+) in Jak2VF and Jak2VFβc/βIL-3−/− mice, but not in βc/βIL-3−/− mice. All statistical analyses performed with Mann-Whitney test (Graphpad Prism V6.0).
Next, to assess the contribution of the IL-3R in Jak2VF -mediated JAK-STAT signalling, Jak2VF mice were bred with mice lacking the common beta subunit of the IL-3 receptor that is responsible for signal transduction of IL-3, IL-5 and GM-CSF, as well as the murine-specific IL-3 receptor which signals mIL-3 (Jak2VF βc/βIL-3−/−). BM chimeras of Jak2VF βc/βIL-3−/− mice were generated as detailed above for Jak2VFchimeras (Figure 2G). pStat5 was unable to be stimulated by IL-3 in Jak2VFβc/βIL-3−/− cells, indicating total loss of functional IL-3R signalling, while TPO signalling to Stat5 remained intact (Figure 1C). Importantly, IL-3 signalling was preserved in WT cells taken from the same chimeras, demonstrating that the absence of IL-3 signalling of Stat5 is due to the cell intrinsic absence of βc/βIL-3−/− (Figure 1B,D).
Physiological expression of Jak2V617F in murine HSPC drives the expansion of committed myeloid progenitors that results in high haemoglobin (Hgb) levels, increased white blood cells (WBC) in the peripheral blood and spleen, and marked splenomegaly due to extramedullary haematopoiesis. We therefore analysed the phenotype of 6–10 week old Jak2VFβc/βIL-3−/− vs. Jak2VF, as well as age-matched control WT C57BL/6 and βc/βIL-3−/− mice. Both Jak2VFβc/βIL-3−/− and Jak2VF mice displayed increased WBC, Hgb and platelets compared to controls (Figure 1E–G) and, importantly, no differences were seen between Jak2VFβc/βIL-3−/− and Jak2VF, indicating that the peripheral blood phenotype of Jak2VF MPN is not dictated by IL-3 signalling. Similarly, Jak2VFβc/βIL-3−/− and Jak2VF mice demonstrated pronounced splenomegaly characterised by increased spleen weight and cellularity (Figure 1H). Flow cytometric analysis of splenocytes revealed an expansion of early erythroid progenitors (CD71+Ter119+) in both Jak2VFβc/βIL-3−/− and Jak2VF compared to non-Jak2VF controls (Fig 1I).
Next, we sought to determine whether IL-3R signalling affected the number or function of HSPC compartments in MPN mice. Using immunophenotypic markers to gate committed myeloid progenitors (LK), multipotent progenitors (LKS) and LT-HSC as previously described11 (Figure 2A), we observed similar LK and LT-HSC frequencies between WT, βc/βIL-3−/−, Jak2VF, and Jak2VFβc/βIL-3−/− cells (Figure 2B,D). There was a small but significant increase in LKS cells in Jak2VFβc/βIL-3−/− compared with βc/βIL-3−/− (Figure 2C), consistent with the observation that Jak2VF expands myeloid progenitor cells.12 Furthermore, immunophenotypically-defined granulocyte-macrophage progenitors (GMP; LK+FcG-R+CD34+, Figure 2A) remained unchanged in both cohorts of Jak2VF MPN mice (Figure 2E), while megakaryocyte-erythroid progenitors (MEP; LK+FcG-R-CD34-, Figure 2A) were significantly expanded in Jak2VF mice as previously reported10 (Figure 2F), although not significantly expanded in Jak2VFβc/βIL-3−/− mice. Altogether, these data reveal that the expansion of Jak2VF HSPC occurs independently of IL-3 signalling in Jak2VF-mediated MPN.
Finally, to assess the functional consequences of IL-3 signalling on Jak2VF LT-HSC, we analysed Jak2VF and Jak2VFβc/βIL-3−/− bone marrow chimeras (Figure 2G). Chimeric mice phenocopied the MPN disease of primary mice, however there were no major differences seen between Jak2VF and Jak2VFβc/βIL-3−/− chimeras (Figure 2H,I). Importantly, the equivalent engraftment of donor cells was observed between both groups (Figure 2J), indicating that LT-HSC function is not affected by loss of IL-3R signalling.
Altogether, we have shown that Jak2VF-expressing mice that are deficient in IL-3R signalling do not show alterations in MPN phenotypes, while bone marrow HSPCs are also unaffected. Although we cannot completely rule out that IL-3 signals through an unusual heterodimeric receptor incorporating IL-3Rα, we did not see any pStat5 induction with recombinant mIL-3 and therefore these data strongly suggest that neither the IL-3, nor IL-5 nor GM-CSF signalling axes play a significant role in MPN pathogenesis. Furthermore, our observations that TPO, not IL-3, is the key cytokine providing stimulation of LT-HSCs supports the work of others identifying MPL as a key mediator of Jak2VF MPN.13 These findings also suggest that TPO signalling may be a more relevant target than IL-3 signalling, thus suggesting caution regarding the extension of AML clinical trials examining the antibody blockade of IL-3 signalling (ClinicalTrials.gov identifier: NCT02159495) to MPN.15 This work provides further clarity regarding the role of dysregulated cytokine signalling in Jak2VF MPN and HSPC biology.
Acknowledgments
We wish to thank Dr. Ann Mullally for helpful comments and review of the manuscript, as well as for providing the Jak2VF mice.
Footnotes
Funding: TV, RA and SWL have received support from the Leukaemia Foundation. HSR, AFL and SWL received support from the NHMRC (SWL: 1026594), Cancer Australia and Leukaemia Foundation, HSR is supported by the Peter Nelson Leukaemia Research Fund.
The online version of this letter has a SupplementaryAppendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. [DOI] [PubMed] [Google Scholar]
- 2.James C, Ugo V, Le Couedic J-P, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. [DOI] [PubMed] [Google Scholar]
- 3.Kralovics R, Passamonti F, Buser AS, et al. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N Engl J Med. 2005;352(17):1779–1790. [DOI] [PubMed] [Google Scholar]
- 4.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. [DOI] [PubMed] [Google Scholar]
- 5.Jamieson CHM, Gotlib J, Durocher JA, et al. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci U S A. 2006;103(16):6224–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan D, Hutchison RE, Mohi G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood. 2012;119(15):3539–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grisouard J, Shimizu T, Duek A, et al. Deletion of Stat3 in hematopoietic cells enhances thrombocytosis and shortens survival in a JAK2-V617F mouse model of MPN. Blood. 2015;125(13):2131–2140. [DOI] [PubMed] [Google Scholar]
- 8.Dawson MA, Bannister AJ, Gottgens B, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1[agr] from chromatin. Nature. 2009;461(7265):819–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu X, Huang LJ-S, Lodish HF. Dimerization by a Cytokine Receptor Is Necessary for Constitutive Activation of JAK2V617F. J Biol Chem. 2008;283(9):5258–5266. [DOI] [PubMed] [Google Scholar]
- 10.Mullally A, Lane SW, Ball B, et al. Physiological Jak2V617F Expression Causes a Lethal Myeloproliferative Neoplasm with Differential Effects on Hematopoietic Stem and Progenitor Cells. Cancer Cell. 2010;17(6):584–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mullally A, Bruedigam C, Poveromo L, et al. Depletion of Jak2V617F myeloproliferative neoplasm-propagating stem cells by interferon-Œ± in a murine model of polycythemia vera. Blood. 2013;121(18):3692–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mullally A, Poveromo L, Schneider RK, et al. Distinct roles for long-term hematopoietic stem cells and erythroid precursor cells in a murine model of Jak2V617F-mediated polycythemia vera. Blood. 2012;120(1):166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sangkhae V, Etheridge SL, Kaushansky K, Hitchcock IS. The thrombopoietin receptor, MPL, is critical for development of a JAK2V617F-induced myeloproliferative neoplasm. Blood. 2014;124(26):3956–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weber GF, Chousterman BG, He S, et al. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science. 2015;347(6227):1260–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Testa U, Pelosi E, Frankel A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark Res. 2014;2(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]