

The development of Janus kinase (JAK) inhibitors has revolutionized the therapeutic landscape for the treatment of myeloproliferative neoplasms (MPN). Therefore, improving the efficacy and safety of JAK inhibitor therapy is a major prerequisite for reliable clinical use in the future. Ruxolitinib (RUX) is the first JAK inhibitor approved for the treatment of advanced stages of myelofibrosis (MF) and polycythemia vera (PV). RUX inhibits mutated JAK2-V617F kinase in hematopoietic cells as well as wild-type JAKs downstream of cytokine and growth factor receptors, especially in immune cells.1 Using this therapeutic strategy, inflammatory symptoms in MPN patients characterized by crippling splenomegaly, the release of inflammatory cytokines and the appearance of thrombosis, could be effectively reduced for the first time.2,3 Recently, we and others reported on RUX leading to impaired NK- and T-cell activation and function.4–6 JAK inhibitor treatment is also effective in reducing the severity of graft-versus-host disease in mice and patients post-allogeneic stem cell transplantation, confirming its effect on T-cell function in vivo.6,7 Along these lines, severe infectious complications such as Cryptococcus neoformans pneumonia, toxoplasmosis retinitis, hepatitis B virus reactivation, progressive multifocal leukoencephalopathy and disseminated tuberculosis have been reported in association with RUX treatment.8 A large international multicenter trial investigating RUX treatment in PV patients reported reactivation of Varicella zoster virus as being the most frequent infectious complication.2 Investigating the inhibitory effects of RUX on T-cell function, we found high heterogeneity among different donor samples. Moreover, intracellular availability has been described as a main determinant of kinase inhibitor function.9 ATP binding cassette (ABC) transporters are ubiquitously expressed molecules important for the translocation of different substrates across cell membranes. Many compounds used in clinical practice are substrates of ABC-transporters and their intracellular bioavailability can be severely influenced by differential expression of ABC-transporters or polymorphisms in ABC genes.10,11 In chronic myeloid leukemia (CML), ABCB1, ABCC1 and ABCG2 transporters have been described to substantially influence the development of tyrosine kinase inhibitor (TKI) resistance of CML cells, which has an impact on clinical responses.9,12–14 Moreover, momelotinib, another JAK1/2 inhibitor, has also been reported as a substrate of ABCB1 and ABCG2 transporters.15 Therefore, we hypothesized that the expression and function of ABC-transporter proteins may critically influence the inhibitory capacity of RUX on human T-cells.
To investigate the correlation of ABC-transporter expression with the modulation of RUX mediated T-cell function, we analyzed the expression of ABCB1, ABCC1 and ABCG2 as well as CD69, a global marker for T-cell activation, after stimulation with Phytohemagglutinin-L (PHA) on patient derived T-cells by flow cytometry. We isolated mononuclear cells (PBMCs) from the peripheral blood of 15 MPN patients who were being treated with RUX, and stimulated them with PHA 0,5% for 24h (Figure 1A). All blood samples were collected with informed consent according to the Declaration of Helsinki. We correlated the increase in CD69 expression in RUX-treated MPN patient samples following PHA-stimulation with the respective expression of ABCB1, ABCC1 and ABCG2 (Figure 1B–D). The extent of T-cell activation (CD69 expression) correlated closely with the expression of the respective drug transporters, indicating that reduced cellular elimination may correlate with increased intracellular efficacy of the drug. For ABCB1 the correlation was significant and for ABCC1 and ABCG2 there was a trend (Figure 1B–D).
Figure 1.
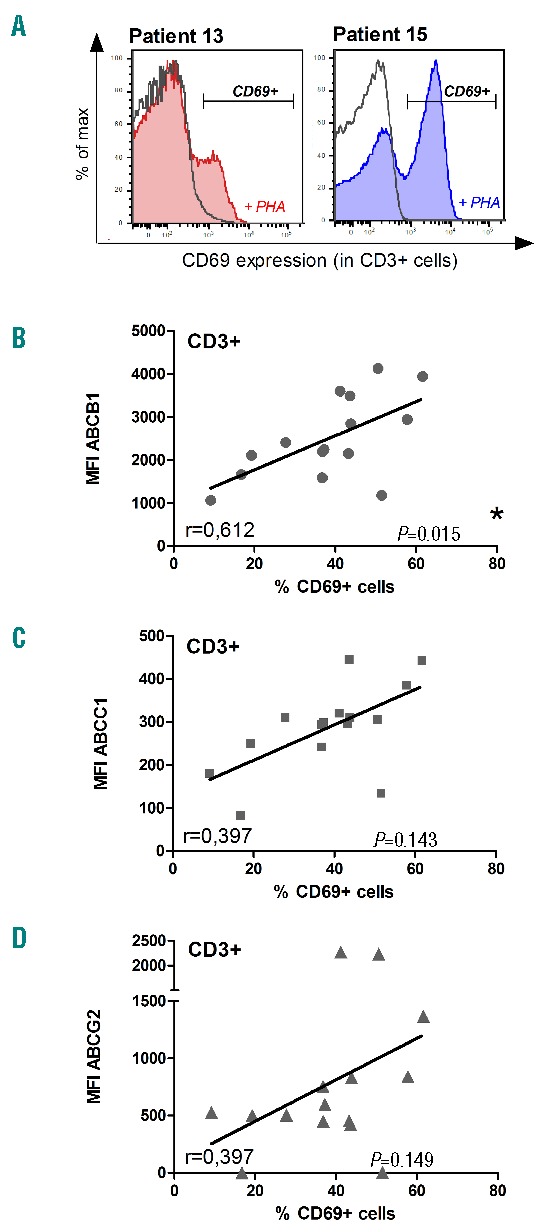
ABC-transporter expression is a marker for T-cell suppression in MPN patients under RUX treatment. (A) Representative examples of the gating strategy for the detection of CD69 positive cells in two patients from our cohort with a strong (left) or weak (right) T-cell inhibition while taking RUX medication. (B,C,D) The expression of ABC-transporters ABCB1 (B), ABCC1 (C) and ABCG2 (D) has been correlated to the expression of the activation marker CD69 on MPN patient T-cells (CD3+). PBMCs were isolated using Ficoll gradient from peripheral blood of MPN patients taking RUX medication. The cells were stimulated in vitro with PHA (0,5%) for 24h before staining (CD3, CD69, ABCB1, ABCC1, ABCG2, SYTOX®Blue Dead Cell Stain) and subsequent analysis by flow cytometry. Statistical analysis using data from 15 MPN patients receiving RUX treatment has been performed using Pearson correlation (Pearson correlation coefficient and P-value are indicated in the diagrams).
In order to corroborate these findings with higher numbers of donors, we decided to recapitulate our experiments using healthy donor (HD) T-cells. PBMCs from 50 HDs were pre-treated with RUX or DMSO (soluble control) for one hour followed by stimulation with PHA (0,5%). Flow cytometry analysis was performed 24h post-stimulation. Comparing RUX-treated cells to DMSO control we found a significant reduction of CD69 expression (Figure 2A) and proliferation as detected by CFSE staining (Figure 2B), confirming our previous findings in a large cohort of healthy donors.4 Among the ABC-transporters, ABCB1 appeared to be the predominantly expressed transporter protein on HD T-cells as analyzed by flow cytometry (Figure 2C). In order to correlate CD69 expression with the expression of the respective drug transporters, we used the relative values for CD69 expression (CD69 ratio: CD69RUX/CD69DMSO -Figure 2A, bottom) and proliferation (Figure 2B, bottom). We found significant correlation between T-cell activation (CD69 ratio) and expression of ABCB1, ABCC1 and ABCG2 in the subset of CD8 positive T-cells (Figure 2D–F). In contrast, no significant correlation could be observed for CD4 positive T-cells (data not shown). Of note, CD69 expression may not be suitable to assess for the complexity of T-cell activation in all subsets investigated.
Figure 2.
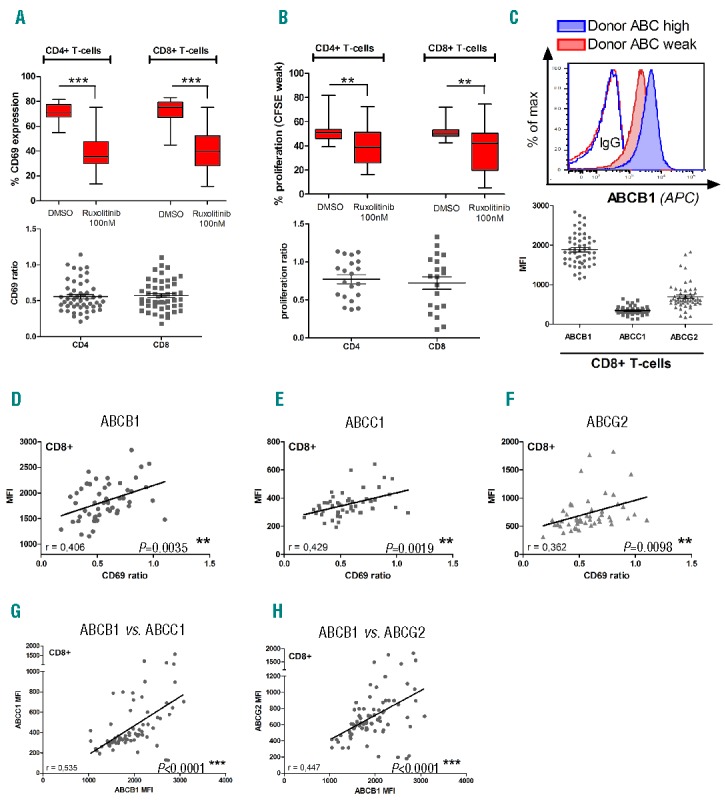
ABC-transporter expression predicts the degree of cytotoxic T-cell impairment after RUX treatment in healthy donor samples. (A) T-cell activation measured by CD69 expression upon RUX treatment in CD4+ and CD8+ T-cells. PBMCs were isolated from healthy donor peripheral blood by Ficoll, treated with RUX (100 nM) and subsequently stimulated with PHA (0,5%) or DMSO (soluble control). After 24h of incubation cells were analyzed by flow cytometry (CD3, CD4, CD8, CD69, ABCB1, ABCC1, ABCG2, SYTOX®Blue Dead Cell Stain). The CD69 ratio for each donor (bottom panel) was calculated from the absolute values for CD69 expression (top panel) after DMSO or RUX treatment. Statistical analysis was performed using Wilcoxon matched-pairs test (n=50; *P<0.05, **P<0.01, ***P<0.001). (B) T-cell proliferation was measured using CellTrace™ CFSE assay in CD4+ and CD8+ cells after RUX treatment (100 nM RUX, 0,5% PHA, 50 IU/ml IL2, 5 d of incubation, statistics: Wilcoxon matched-pairs test – n=20). (C) After staining T-cells with ABCB1 flow antibody a clear peak-shift between ABC “high” or “weak” expressing donors could be observed (top panel). (D,E,F) Correlation between ABC-transporter expression and CD69 ratio (experimental setting as described in A) in CD8+ T-cells. The mean fluorescence intensity (MFI) of ABCB1 (D), ABCC1 (E) and ABCG2 (F) has been correlated to the degree of T-cell inhibition (CD69 ratio) after in vitro RUX treatment. (G,H) Surface expression of ABCB1 and ABCC1 (G) as well as ABCB1 and ABCG2 (H) were correlated with each other. Statistical analysis has been performed using Pearson correlation (n=50; Pearson correlation coefficient and P-value indicated in the diagrams).
However, correlating the expression of ABCB1 with ABCC1 (Figure 2G) and ABCB1 with ABCG2 (Figure 2H) we could observe a significant correlation. In our cohort of healthy donors which we investigated, it seemed that individuals appear to either express high or low levels of the respective ABC-transporters on both CD4 and CD8 positive T-cells. Due to the current knowledge about ABC-transporter function one could assume synergy between these molecules concerning the transport of substrates.15 In order to functionally validate the influence of ABC-transporters on the efficacy of JAK inhibition, we sought to genetically inactivate ABCB1 using RNA interference (RNAi).
First, we aimed to investigate the effects of ABCB1 inactivation in a human cell line model. We used the human erythroleukemia cell line (HEL) harboring the JAK2-V617F mutation and performed genetic inactivation of ABCB1 by RNAi. In detail, cells were infected with lentiviral particles harboring either two different ABCB1 shRNAs or a non-targeting control (scrambled control; scr). Three days after infection, HEL cells were counted and plated on 96 well plates at a density of 20,000 cells per well and treated with RUX (12,5 nM – 1000 nM). Proliferation was analyzed using a MTS-assay 4 days after treatment. The efficacy of RUX treatment was increased after genetic inactivation of ABCB1 (Figure 3A). As HEL cells are strongly dependent on the mutated JAK2 kinase, they undergo apoptosis following RUX treatment. We found increased efficacy of RUX-induced apoptosis upon the genetic inactivation of ABCB1 (Figure 3C). Of note, the efficacy to sensitize cells to JAK inhibitor treatment appeared to be dependent on the extent of ABCB1 inactivation. The abrogation of ABCB1 expression (as achieved for shRNA1) resulted in the most pronounced effects (Figure 3B).
Figure 3.
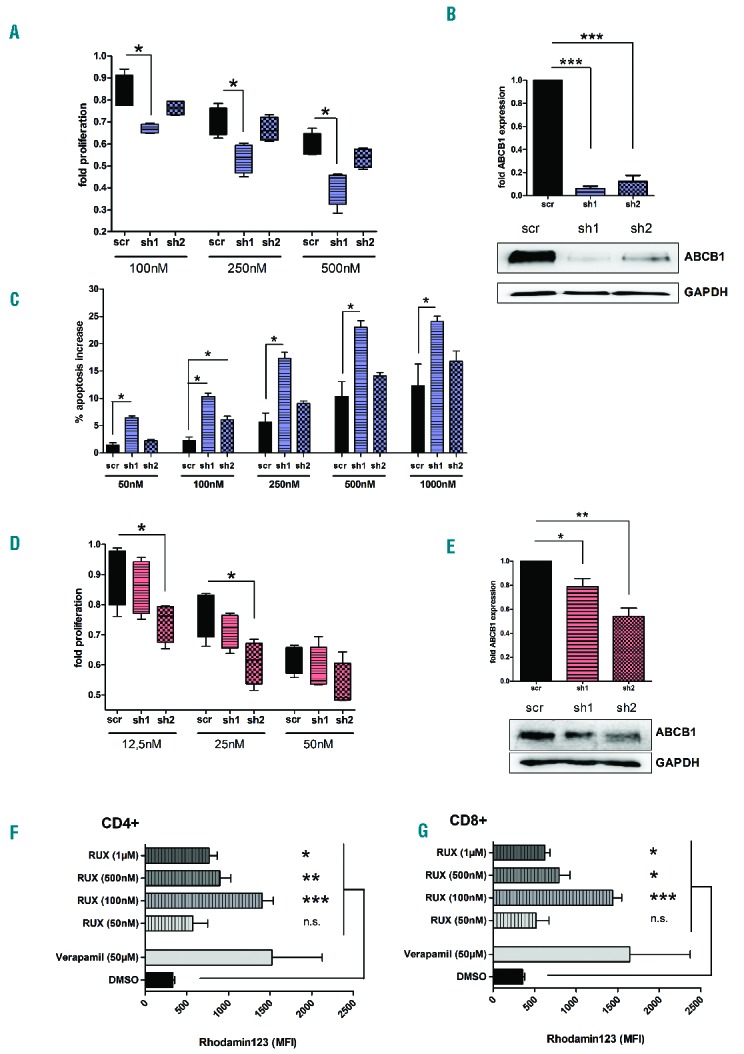
Inactivation of ABCB1 function leads to increased efficacy of RUX in JAK2 V617F-mutated cell lines and primary human T-cells (A) Proliferation of HEL cells treated with increasing concentrations of RUX measured by MTS assay after shRNA mediated knockdown of ABCB1. Statistical analysis was performed using unpaired t-test (n=4; *P<0.05, **P<0.01, ***P<0.001). (B) Efficacy of ABCB1 knockdown in HEL cells using western blot and densitometric analysis. (C) Measurement of apoptosis (Annexin V + SYTOX ®Blue Dead Cell Stain) in ABCB1-deficient HEL cells detected 48 h after RUX treatment (Wilcoxon-Mann-Whitney test, n=4). (D) Proliferation of human primary T-cells using a MTS assay after knockdown of ABCB1 and treatment with RUX. T-cells were isolated by magnetic linage depletion from HD-PBMCs and pre-stimulated with PHA (0,1%) for 15 h before infection (statistics: unpaired t-test, n=4). (E) Western blot analysis of ABCB1 knockdown efficacy in human T-cells. A representative western blot is shown (bottom panel); densitometric analysis is shown for four independent experiments (top panel). (F, G) Rhodamin 123 efflux assay performed on primary human T-cells. PBMCs were isolated using Ficoll gradient and stimulated with PHA (0,5%) for 24h. Cells were exposed to Rhodamin 123 (200nM) for 1h. Subsequently the cells were washed of Rhodamin and incubated with RUX (50nM-1μM), Verapamil (50μM) as a positive control or DMSO as a negative control for 1h prior to surface staining (anti-CD3, anti-CD4, anti-CD8). RUX treated samples show a significant accumulation of Rhodamin 123 as compared to DMSO control in CD4 (F) and CD8 (G) positive T-cells (paired t-test; n=6).
Next, we aimed to recapitulate our findings in healthy donor T-cells. Purified HD T-cells were pre-stimulated with PHA (0,1%) and IL2 (10U/ml) and incubated for 15h at 37°C, followed by infection with lentiviral particles as described above. Pre-stimulation of the primary T-cells appeared to be necessary for effective transduction. T-cells were kept in the lentivirus-containing medium for 18h. As observed before, inactivation of ABCB1 increased the inhibitory capacity of RUX on T-cell proliferation at low RUX concentrations (Figure 3D). Similar to the results observed in HEL cells, the extent of ABCB1 inactivation correlated closely with the inhibitory capacity of RUX. About 50% reduction in ABCB1 expression appeared to be sufficient to sensitize primary human T-cells to JAK inhibitor treatment (Figure 3E). We observed the highest transduction efficiency in HEL cells (>90%) while 75–80% of primary T-cells were successfully transfected with no major differences between shRNA1 and2.
In order to provide evidence for the interaction between RUX and ABC-transporter function, we performed flow cytometric analysis (ABC efflux assay) using fluorescent dyes transported by ABCB1 (Rhodamin 123) and ABCG2 (Hoechst 33342). By applying increasing concentrations of RUX (50nM-1μM) we were able to show that the transport function of ABCB1 is significantly impaired by RUX treatment (>100nM) leading to the accumulation of Rhodamin 123 in CD4 (Figure 3F) and CD8 (Figure 3G) positive T-cells. Of note these concentrations can be reached as trough levels in patients with myeloproliferative neoplasia, as reported in early clinical trials.3 Verapamil, as a known substrate of ABCB1, served as a positive control in this experiment. In contrast, using Hoechst 33342 to detect changes is ABCG2 function we found no differences between RUX and DMSO treated T-cells (data not shown).
Taken together, we provide first evidence that expression of ABCB1, ABCC1 and ABCG2 on primary human T-cells correlates with the efficacy of JAK inhibitor treatment in regard to T-cell activation and proliferation.
This correlation was detectable in primary MPN patient T-cells who received JAK inhibitor therapy. Moreover, these effects could be observed in HD-PBMC samples treated with RUX ex vivo. Genetic inactivation of ABCB1 confirmed the involvement of ABC-transporters in mediating RUX sensitivity in a cell line and primary human T-cells. Finally, we provide the first evidence that RUX inhibits ABCB1 function, suggesting that RUX is either a substrate or allosteric inhibitor of ABCB1. In contrast, no effect was detectable on ABCG2.
Therefore, it is tempting to speculate that antibody-based detection of ABCB1 surface expression or the measurement of Rhodamin 123 efflux from T-cells could serve as a flow cytometric tool to identify MPN patients at risk for immunosuppressive complications upon JAK inhibitor therapy. The feasibility and predictive value of these assays need to be examined in future clinical trials.
Footnotes
Funding: this work was supported by a grant from Novartis Inc. (HINC033) to DW and FHH and by the DFG-Collaborative Research Cluster (CRC854) to TF and FHH (Project A20). All human samples were collected and stored in the Hematology Tissue-Bank Magdeburg (HTM) supported by a grant from the Jose-Carreras Foundation (SP12/04) to FHH.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Ghoreschi K, Gadina M. Jakpot! New small molecules in autoimmune and inflammatory diseases. Exp Dermatol. 2014;23(1):7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perner F, Schnöder TM, Ranjan S, et al. Specificity of JAK-kinase inhibition determines impact on human and murine T-cell function. Leukemia. 2015. August 5 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 5.Parampalli Yajnanarayana S, Stubig T, Cornez I, et al. JAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasms. Br J Haematol. 2015;169(6):824–833. [DOI] [PubMed] [Google Scholar]
- 6.Schonberg K, Rudolph J, Vonnahme M, et al. JAK Inhibition Impairs NK Cell Function in Myeloproliferative Neoplasms. Cancer Res. 2015;75(11):2187–2199. [DOI] [PubMed] [Google Scholar]
- 7.Spoerl S, Mathew NR, Bscheider M, et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood. 2014;123(24):3832–3842. [DOI] [PubMed] [Google Scholar]
- 8.Heine A, Brossart P, Wolf D. Ruxolitinib is a potent immunosuppressive compound: is it time for anti-infective prophylaxis? Blood. 2013;122(23):3843–3844. [DOI] [PubMed] [Google Scholar]
- 9.Lipka DB, Wagner M, Dziadosz M, et al. Intracellular retention of ABL kinase inhibitors determines commitment to apoptosis in CML cells. PloS One. 2012;7(7):e40853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruhn O, Cascorbi I. Polymorphisms of the drug transporters ABCB1, ABCG2, ABCC2 and ABCC3 and their impact on drug bioavailability and clinical relevance. Expert Opin Drug Metab Toxicol. 2014;10(10):1337–1354. [DOI] [PubMed] [Google Scholar]
- 11.Choi YH, Yu A. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr Pharm Des. 2014;20(5):793–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eadie LN, Hughes TP, White DL. Interaction of the efflux transporters ABCB1 and ABCG2 with imatinib, nilotinib, and dasatinib. Clin Pharmacol. Ther. 2014;95(3):294–306. [DOI] [PubMed] [Google Scholar]
- 13.Gromicho M, Dinis J, Magalhaes M, et al. Development of imatinib and dasatinib resistance: dynamics of expression of drug transporters ABCB1, ABCC1, ABCG2, MVP, and SLC22A1. Leuk Lymphoma. 2011;52(10):1980–1990. [DOI] [PubMed] [Google Scholar]
- 14.Agrawal M, Hanfstein B, Erben P, et al. MDR1 expression predicts outcome of Ph+ chronic phase CML patients on second-line nilotinib therapy after imatinib failure. Leukemia. 2014;28(7):1478–1485. [DOI] [PubMed] [Google Scholar]
- 15.Durmus S, Xu N, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. P-glycoprotein (MDR1/ABCB1) and breast cancer resistance protein (BCRP/ABCG2) restrict brain accumulation of the JAK1/2 inhibitor, CYT387. Pharmacol Res. 2013;76:9–16. [DOI] [PubMed] [Google Scholar]