

In chronic phase chronic myeloid leukemia (CP-CML), nilotinib (Tasigna®, Novartis, second-generation tyrosine kinase inhibitor, TKI2) as second-line therapy1 results in successful responses and tolerance in ~50% of patients in the long-term.2 As a consequence, some patients have been on nilotinib for approximately 9–10 years now. Recently, nilotinib, introduced as first-line treatment, induced superior cytogenetic and molecular responses and significantly decreased the risk of progression,3 and has been approved in this setting since 2010.
Despite an overall favorable safety profile, different studies4,5 suggest a specific association between nilotinib exposure and the increased risk of occlusive arterial disease (OAD), particularly in patients with pre-existing cardiovascular risk factors (CVRFs), but also in patients without risk factors. Cardiovascular events (CVEs) caused by a TKI are a rare but emerging phenomenon. The mechanism by which nilotinib may cause or aggravate pathological processes involved in OAD remains poorly understood.6,7 Retrospective and prospective studies have demonstrated that nilotinib can induce hyperglycemia and hypercholesterolemia,3,8,9 but this does not seem sufficient to totally explain the phenomenon of nilotinib related CVEs.
Homocysteine is a sulphur containing amino acid derived from methionine, independently associated with early arteriosclerosis, increased thromboembolic events and cardiovascular mortality.10 High blood pressure levels result in the proliferation of smooth muscle cells, endothelial cell dysfunction and increased collagen synthesis pro-inflammatory rearrangements, leading to an increased thromboembolic risk, with a linear relationship between homocysteine and the onset of CVEs.
We aimed, in a multicenter retrospective and prospective analysis, to determine whether hyperhomocysteinemia is associated with CVEs in CP-CML patients treated with nilotinib between September 2011 and February 2015, as compared with a group of imatinib-only treated patients. Standard clinical assessments, CVRF and the onset of any CVE were recorded. Patients were followed-up in 3 university centers, and all of them gave their consent. This study has been approved by the Comité Consultatif sur le Traitement de l’Information en matière de Recherche dans le domaine de la Santé (CCTIRS), and the Commission Nationale de L’informatique et des Libertés (CNIL), Paris, France.
In addition to routine physical exams, standard CML assessments,11 basic blood metabolic parameters, total plasma homocysteinemia (Bio-Rad kit on HPLC on Dionex Summit system, Thermo Fischer Scientific, France, (physiological levels = 5–15 μmol/l)) or with tandem mass spectrometry (MS/MS), and vitamin B9 and B12 levels were measured as they are involved in the homocysteine cycle. Patients were asked to come to the hospital fasting. Responses to TKI were analyzed according to classical definitions.11 We considered as a CVE any symptomatic arterial lesions (arterial stenosis, symptomatic thrombosis) detected clinically and proven by Doppler ultrasound and/or any means of arteriography, MRI or angioscanner.
We compared the cohort of nilotinib-treated patients with patients on imatinib first- or second-line, still on treatment, recruited randomly, because no clear association between imatinib and CVEs has been demonstrated to date.4 Comparisons were made with Pearson’s Chi-squared test for qualitative variables, Mann-Whitney or Student’s t-test for continuous variables, medians [min-max] were shown for variables with normal distribution, otherwise mean ±SD. The occurrence of CVEs was depicted with cumulative incidence curves, and the end of treatment as a competing risk, accompanied by the Gray test for univariate analysis. Multivariate analysis on CVEs from nilotinib initiation was performed according to the Fine and Gray regression model.
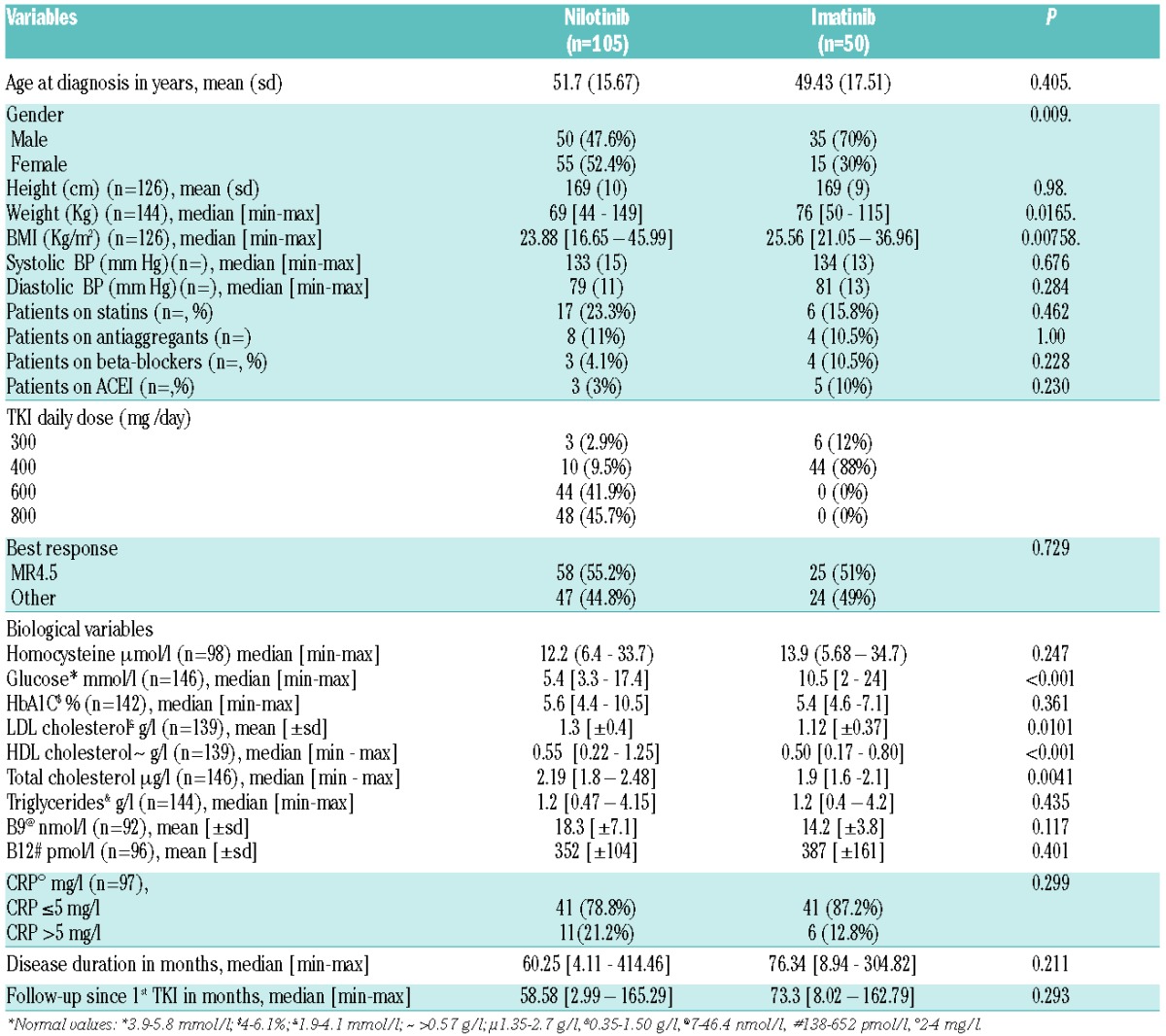
One hundred and five patients on nilotinib and 50 on imatinib were analyzed. There were no differences between the 2 groups except for gender, height and weight (Table 1). Total and LDL-cholesterol were higher in nilotinib patients, glycated hemoglobin levels were not different. Median follow-up and disease duration were not different. Importantly, co-medications with statins, antiaggregants, beta-blockers, ACEI, and blood pressure were not different between the 2 groups. Median homocysteine was 12.20 μmol/l in the nilotinib and 13.9 μmol/l in the Imatinib group (P=0.247). There was no relationship between homocysteinemia and the type of CVE (data not shown).
Table 1.
Clinical and biological parameters of nilotinib-treated CML patients and Imatinib-treated patients.
As normal levels of homocysteine were established in healthy subjects, we constructed a ROC curve in the nilotinib cohort to determine the homocysteine cut-off threshold for CVEs in such patients. The area under the ROC curve was 0.69 [0.55–0.85], and 13.95 μmol/l was the significant threshold, providing a sensitivity of 67% and a specificity of 71%.
Overall, 23 patients (22%) presented a new or worsened pre-existing CVE on nilotinib after a median of 51.5 months (7 carotid atheromatosis, 7 coronary events, 6 PAOD, 1 cerebrovascular event and 2 others). These CVEs occurred after a median of 47 (7.5–82.3) months with a regular increase over the years, 2 patients died in this group (1 stroke and 1 sudden death in a patient with a history of myocardial infarction). Only 4 (8%) patients presented a CVE in the imatinib group (3 coronary events and 1 other), with no deaths. The cumulative incidence of CVEs in the nilotinib group was 3.06% (2.72–3.4) at 12 months, 5.1% (4.66–5.54) at 24 months, 6.12% (5.64–6.6) at 36 months, 10.2% (9.6–10.81) at 60 months, compared to 0% at each of these time points in the imatinib group (P<0.001, Figure 1A). The cumulative incidence of CVEs was higher (P=0.005) in patients on nilotinib 800 mg (38%) versus nilotinib 600 mg daily or less (10%), with no plateau (Figure 1B). When we looked at the cumulative incidence of CVEs in the nilotinib group, independently of the dose of nilotinib, we demonstrated that the homocysteine threshold could efficiently discriminate high-risk incidence patients. Additionally, CVEs were more frequent on nilotinib according to homocysteinemia (P=0.0026), with a significant increase of CVEs when homocysteine was ≥13.95 μmol/l (P=0.005) (Figure 1C).
Figure 1.
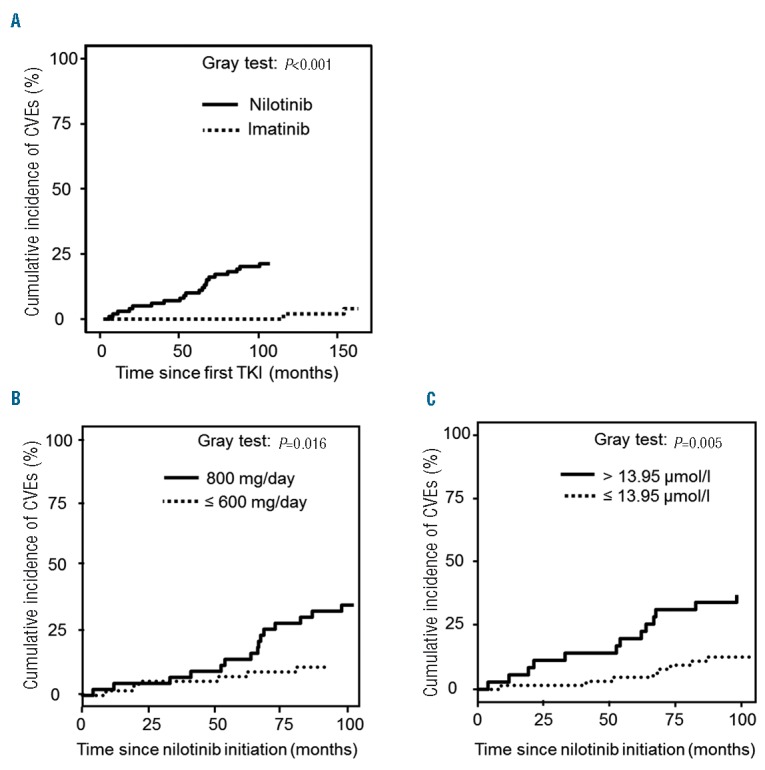
(A) Cumulative incidence of cardiovascular events (CVEs) in nilotinib and imatinib-treated patients. (B) Cumulative incidence of cardiovascular events (CVEs) according to the dose of nilotinib. C. Cumulative incidence of cardiovascular events (CVEs) according to homocysteine levels in the blood.
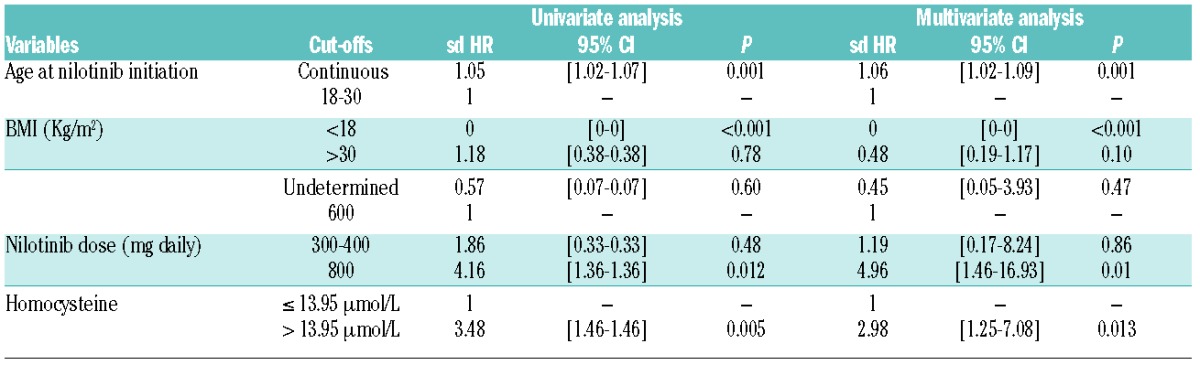
In order to further decipher the factors involved in CVEs we additionally compared the nilotinib subgroup of patients with CVEs (n=23) with the nilotinib subgroup of patients without CVEs (n=82) (Table 2). Interestingly, patients with CVEs were significantly older, with more males, in their majority on statins (as a consequence with total and LDL cholesterol at lower levels), on antiaggregants, on nilotinib 800 mg daily and with higher glycated hemoglobin levels and higher homocysteine levels (considered herein as a continuous variable, median 15.2 μmol/l versus 11.9, P=0.0128). Disease duration and median follow-up were longer in patients with CVEs on nilotinib (P=0.002 and P=0.001, respectively). By performing univariate analysis on the entire nilotinib population of patients, we identified factors which influence the onset of CVEs: Young age and low BMI (<18 Kg/m2) were associated with low rates of CVEs; whereas nilotinib 800 mg and homocysteine >13.95 μmol/l were linked to a high incidence of CVEs (P=0.012 and P=0.005 respectively, Table 3). Multivariate analysis adjusted on CVEs demonstrated that age at nilotinib initiation [P=0.001, HR=1.06 (95% CI: 1.02–1.09)], 800 mg daily dose of nilotinib [P=0.01, HR=4.96 (95% CI: 1.46–16.93)] and homocysteine levels >13.95 μmol/l [P=0.013, HR=2.98 (95% CI: 1.25–7.08)], were significant variables negatively influencing the onset of CVEs. Low BMI (<18) was correlated with a significantly lower incidence of CVEs (P<0.001, Table 3).
Table 2.
Clinical and biological parameters of nilotinib-treated CML patients with and without cardiovascular events.
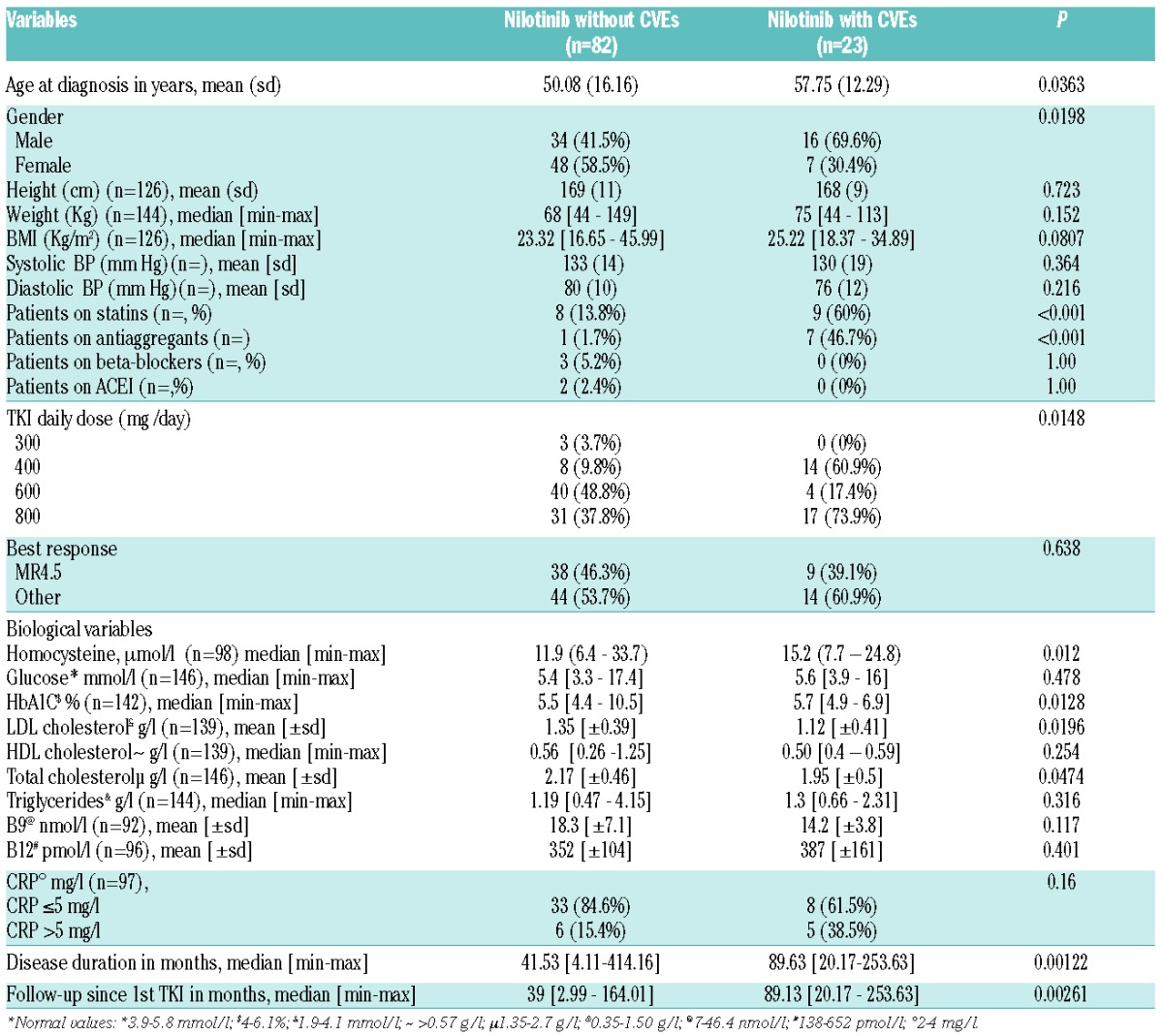
Table 3.
Univariate and multivariate analyses (sd HR stands for subdistribution hazard ratio, CI for confidence interval).
TKIs represent considerable progress in CML treatment and provide high rates of long-term responses, thus preventing progression in a majority of patients. The disease probably remains sustained over time by a discrete surviving fraction of BCR-ABL+ primitive cells,12 requiring the indefinite continuation of TKI therapy, maybe for the entire life span of patients. Severe, retarded, and life-threatening TKI-related side effects such as pleural and pericardial effusions, pulmonary hypertension for dasatinib, cardiovascular and mainly arterial problems for nilotinib and ponatinib, appeared with the long-term use of TKI in nonnegligible cure fractions of patients. As have others,2,4,5 we also herein demonstrate that the frequency of CVEs is higher in nilotinib-treated patients than in their imatinib counterparts. Interestingly, these CVEs are delayed (more than half occur ≥50 months of treatment), and worryingly, seem to increase extensively over time. Moreover, there is a relationship between CVEs and higher doses of nilotinib (where the frequency of CVEs is twice as much as that found in lower doses, Figure 1C). As a subsequent perspective, dose optimization of nilotinib, once minimal residual disease is obtained, should be considered to reduce these CVEs in the long-term. These data suggest that the pathophysiology of these events is specifically drug-induced, as the 2 groups of patients are somewhat similar. They are dose-dependent, accumulate with time and might involve total and LDL hypercholesterolemia [significantly higher in the nilotinib group (Table 1)], but that’s not all.
The pathophysiology of nilotinib-induced CVEs remains obscure and is probably related to off-target effects involving endothelial specific damage,6 but it does not seem that nilotinib creates a prothrombotic state as the majority of patients do not show any CVEs, none of them had DVT and plasmatic clotting factors are not modified (data not shown). Severe hyperhomocysteinemia (>150 μmo/L) is seen in rare inherited metabolic diseases13 due to cystathionine beta-synthase deficiency, characterized by generalized vascular and thromboembolic complications. Less severe hyperhomocysteinemias are described in acquired diseases (renal insufficiency, autoimmune disorders), in vitamin B6/B9/B12 deficiencies and secondary to treatments (folate and cobalamin antagonists, steroids, diuretics, fibrates, statins). Strange similarities between the CVEs in homocysteinuria and those seen in nilotinib-treated patients prompted us to analyze CML patients, with or without CVEs, for homocysteinemia. The incidence of CVEs in the nilotinib group was associated with higher levels of homocysteine (defined as ≥13.95 μmol/l) in univariate and multivariate analyses. The cumulative incidence of CVEs on nilotinib was linked to homocysteine levels in the blood. Patients with high homocysteine levels were 3 times more frequent in the nilotinib group. Homocysteine increases with age, however aging is observed in both groups and was not different in either. In the nilotinib group, only one patient was on a folate substitution. Whether higher levels of homocysteine in nilotinib-treated patients are a cause or a consequence of nilotinib remains to be determined. As the elevation of homocysteinemia in nilotinib-treated patients is modest, it seems unlikely that this amino acid is involved per se in the pathogenesis of CVEs in this population.
In conclusion, nilotinib represents an additional cardiovascular risk factor in TKI-treated CML patients. An extensive baseline capture of cardiovascular risk factors, biochemical factors including homocysteinemia and the monitoring of these parameters is recommended for the optimal management and success of nilotinib therapy in CML patients in the long-term.
Acknowledgments
The authors thank Mr Jérémy Ruby for his assistance in collecting the data and carrying out administrative tasks, Mrs Anne-Claire Barale our consultant nurse for harvesting and measuring basic clinical parameters, and laboratory technician staff for their kind help. We thank the association “Anim’ Monbernier”, Ruy Monceau, France, and its president Mr Armand Glasson for their support in this CML study. Additionally we thank Mrs. Barbara White-Meunier, PhD, for editorial assistance.
Footnotes
Funding: this study is academic and has not been funded by any pharmaceutical company.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354(24):2542–2551. [DOI] [PubMed] [Google Scholar]
- 2.Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia. 2013;27(1):107–112. [DOI] [PubMed] [Google Scholar]
- 3.Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362(24):2251–2259. [DOI] [PubMed] [Google Scholar]
- 4.Le Coutre P, Rea D, Abruzzese E, et al. Severe peripheral arterial disease during nilotinib therapy. J Natl Cancer Inst. 2011;103(17):1347–1348. [DOI] [PubMed] [Google Scholar]
- 5.Kim TD, Rea D, Schwarz M, et al. Peripheral artery occlusive disease in chronic phase chronic myeloid leukemia patients treated with nilotinib or imatinib. Leukemia. 2013;27(6):1316–1321. [DOI] [PubMed] [Google Scholar]
- 6.Hadzijusufovic E, Albrecht-Schgoer K, Huber K. Nilotinib exerts direct pro-atherogenic and anti-angiogenic effects on vascular endothelial cells: a potential explanation for drug-iduced vasculopathy in CML. Blood. 2013;122(12):Abstract 257. [Google Scholar]
- 7.Valent P, Hadzijusufovic E, Schernthaner GH, Wolf D, Rea D, le Coutre P. Vascular safety issues in CML patients treated with BCR/ABL1 kinase inhibitors. Blood. 2015;125(6):901–906. [DOI] [PubMed] [Google Scholar]
- 8.Racil Z, Razga F, Drapalova J, et al. Mechanism of impaired glucose metabolism during nilotinib therapy in patients with chronic myelogenous leukemia. Haematologica. 2013;98(10):e124–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rea D, Mirault T, Cluzeau T, et al. Early onset hypercholesterolemia induced by the 2nd-generation tyrosine kinase inhibitor nilotinib in patients with chronic phase-chronic myeloid leukemia. Haematologica. 2014;99(7):1197–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nygard O, Nordrehaug JE, Refsum H, Ueland PM, Farstad M, Vollset SE. Plasma homocysteine levels and mortality in patients with coronary artery disease. N Engl J Med. 1997;337(4):230–236. [DOI] [PubMed] [Google Scholar]
- 11.Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graham SM, Jorgensen HG, Allan E, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–325. [DOI] [PubMed] [Google Scholar]
- 13.Kraus JP, Oliveriusova J, Sokolova J, et al. The human cystathionine beta-synthase (CBS) gene: complete sequence, alternative splicing, and polymorphisms. Genomics. 1998;52(3):312–324. [DOI] [PubMed] [Google Scholar]