

Abstract
Resistance to HER2‐targeted therapies remains a major obstacle in the treatment of HER2‐overexpressing breast cancer. Understanding the molecular pathways that contribute to the development of drug resistance is needed to improve the clinical utility of novel agents, and to predict the success of targeted personalized therapy based on tumor‐specific mutations. Little is known about the clinical significance of HER family mutations in breast cancer. Because mutations within HER1/EGFR are predictive of response to tyrosine kinase inhibitors (TKI) in lung cancer, we investigated whether mutations in HER family kinase domains are predictive of response to targeted therapy in HER2‐overexpressing breast cancer. We sequenced the HER family kinase domains from 76 HER2‐overexpressing invasive carcinomas and identified 12 missense variants. Patients whose tumors carried any of these mutations did not respond to HER2 directed therapy in the metastatic setting. We developed mutant cell lines and used structural analyses to determine whether changes in protein conformation could explain the lack of response to therapy. We also functionally studied all HER2 mutants and showed that they conferred an aggressive phenotype and altered effects of the TKI lapatinib. Our data demonstrate that mutations in the finely tuned HER kinase domains play a critical function in breast cancer progression and may serve as prognostic and predictive markers.
Keywords: HER family mutation, Kinase domain, Lapatinib, Drug resistance, Biomarker
Highlights
Identification of new mutations in the kinase domain of EGFR, HER2, HER4.
Mutations can predict response to HER2‐targeted treatments.
Computational structure analysis predictions are validated experimentally.
HER2 mutations are associated with response to the tyrosine kinase inhibitor lapatinib.
HER2 mutants are more aggressive than HER2 wild type.
Abbreviations
- TKI
tyrosine kinase inhibitors
1. Introduction
Targeting HER2 has changed the natural history of HER2‐amplified breast cancer (Baselga et al., 2012; Geyer et al., 2006; Slamon et al., 2001). In the absence of HER2‐targeted therapy, patients whose tumors exhibit HER2 amplification have worse survival rates than patients with non‐amplified HER2 breast cancer (Piccart‐Gebhart et al., 2005; Romond et al., 2005; Slamon et al., 2011). Few mutations have been reported in HER2‐amplified and non‐amplified ovarian and breast cancers (Bose et al., 2013; Lee et al., 2006; Lin et al., 2011). However, little is known about the biological consequences, and prognostic and predictive roles of HER2 mutations in HER2‐amplified breast cancer.
The binding of specific growth factors to the extracellular regions of EGFR, HER3, and HER4 promotes an intracellular asymmetric dimerization of the kinase domains. In this dimer, the C‐terminal lobe of the ‘activator’ kinase binds to, and stabilizes, the active conformation of the N‐terminal lobe of the ‘receiver’ kinase (Monsey et al., 2010; Zhang et al., 2006). In abnormal conditions, receptor dimerization can also be induced by a high concentration of receptors or by kinase domain mutations, resulting in receptor activation by the transphosphorylation of tyrosine residues. Under HER2‐overexpressing conditions in particular, whereas the other family members usually dimerize upon ligand binding, HER2 exists mainly in its open conformation and can readily homo‐ or hetero‐dimerize in the absence of a ligand, making HER2 the preferred dimerization partner for all other family members (Brennan et al., 2000; Garrett et al., 2003; Ghosh et al., 2011; Graus‐Porta et al., 1997; Hynes and Stern, 1994; Junttila et al., 2009; Larson et al., 2010; Olayioye, 2001). HER2‐targeted therapies can block receptor dimerization and inhibit signal transduction pathways driving cancer cell growth and proliferation (Baselga, 2006; Hynes and Lane, 2005). Cancer cells adapt by activation of alternative survival pathways, resulting in the development of drug resistance (Esteva et al., 2010a; Huang et al., 2010; Liang et al., 2010; Morrow et al., 2011; Nagata et al., 2004; Nahta et al., 2005; Scaltriti et al., 2007; Zhang et al., 2011). Whether activating somatic mutations occur at the receptor level or at downstream molecules, no biomarker is currently available to identify those patients who are not likely to respond to HER2‐targeted therapies (Esteva et al., 2010b).
We hypothesized that the presence of a mutation in one or more HER family members can predict patient outcome and/or response to HER2‐targeted therapy. Because of their ability to phosphorylate their partners and because HER2 is the preferred partner, the EGFR and HER4 kinase domains were included in our sequencing study, as their kinase activity has the potential to influence the activity of HER2, which would reflect a differential response to HER2‐targeted therapy. For the same reason, although HER2‐HER3 is considered the most potent dimer of the family, HER3 lacks kinase activity and therefore was disregarded in our search for potentially functional mutations in the kinase domain. Here we describe the discovery of and clinical outcomes associated with 12 HER family kinase domain mutations. We also report the molecular characterization, biological activity, and mechanisms of response to HER2‐targeted therapy exhibited by the HER2 mutants.
2. Materials and methods
2.1. Materials
Reagents were obtained from the following sources: lapatinib from Selleck Chemicals; Dulbecco's modified Eagle's (DMEM)/F12, McCoy's 5A, RPMI, and fetal bovine serum (FBS) from Life Technologies; antibodies to phospho‐T308 and phospho‐Ser473 Akt, Akt, phospho‐Erk1/2, Erk1/2, and cleaved‐caspase 3 from Cell Signaling Technologies; laminin V from Sigma; HER2 from EMD Biosciences; c‐myc (9E11) from Santa Cruz Biotechnology; and Alexa‐conjugated antibodies from Invitrogen. The lentiviral expression plasmid pLVX puro and Lenti‐X Bicistronic Expression System were purchased from Clontech.
2.2. Patient selection
HER2‐positive primary breast cancers were obtained from 76 patients treated at The University of Texas MD Anderson Cancer Center for metastatic breast cancer between 1996 and 2006. All the patients had received trastuzumab‐based therapy for metastatic disease, and none of them had received trastuzumab in the adjuvant setting. HER2 positivity was defined as a score of 3+ using immunohistochemistry (IHC) and/or gene amplification detected by fluorescence in situ hybridization (FISH). The tissue collection protocol and access to clinical data were approved by the Institutional Review Board.
2.3. Cell lines and culture
SKBR3, MDA‐MB‐175, and MCF10A cells were obtained from the ATCC. BT474‐m1 cells were generously provided by Dr. Dihua Yu (MD Anderson Cancer Center). MDA‐MB‐175 cells were cultured in DMEM/F12 with 10% FBS. SKBR3 cells were cultured in McCoy's 5A medium with 10% FBS. BT474‐m1 cells were cultured in RPMI medium with 10% FBS. MCF10A cells were cultured in DMEM/F12 supplemented with 10 μg/ml insulin, 20 ng/ml epidermal growth factor, 0.5 μg/ml hydrocortisone, 100 ng/ml cholera toxin, 1 mM CaCl2, and 5% horse serum. All of the above cell lines were cultured at a density that allowed cell division throughout the course of the experiment. When indicated, cells were treated with lapatinib in growth medium for the indicated time.
2.4. Sequencing
FFPE tumor tissues from the HER2‐positive primary breast cancer patients treated at MD Anderson were obtained by macro‐dissection as directed by a pathologist. Tumor genomic DNA was isolated using the DNA: QIAamp DNA FFPE Tissue Kit protocol (Qiagen), as described by the manufacturer, and PCR reactions were used to amplify the DNA fragments of interest from genomic DNA using the high‐fidelity polymerase Platinum Taq DNA polymerase from Invitrogen. The proteinase K was incubated 72 h, with fresh enzyme added every 24 h. Isolated genomic DNA was diluted to 3 ng/μl in TE buffer (10 mM Tris–HCI, 0.1 mM EDTA, pH 8.0), and 6 ng of DNA was used to generate sequencing data on an ABI3730 DNA analyzer using dye primer sequencing chemistry. Each observed mutation was confirmed by two independent PCR amplification and sequencing reactions.
2.5. Structural analysis
EGFR kinase domain mutations were analyzed using active, ATP‐ and lapatinib‐bound crystal structures Protein Data Bank (PDB) accession numbers 1M14, 2GS6 and 1XKK, respectively. The lapatinib‐bound structure of HER4 (3BBT) was used for analyzing mutations of this kinase. Structural analysis of HER2 was based on 3PP0 (HER2 in an active‐like conformation bound to the inhibitor SYR127063). To obtain a structural model of HER2 in either an ATP‐bound active conformation or a lapatinib‐bound inactive conformation, we have additionally used ATP‐ and lapatinib‐bound structures of the ∼75% identical EGFR (2GS6 and 1XKK) as templates to build homology models of HER2 (wild‐type and mutants). Structural analysis of the wild‐type and L726F structures based on all these models gave very similar results. Homology models for mutants were built and loop regions missing in the experimental structures were completed using SWISS‐MODEL (Arnold et al., 2006). For computational analysis of the effects that neratinib has on Her2 mutants, the crystal structure of neratinib bound to the T790M/L858R EGFR (PDB 2JIV) was used as a template.
2.6. HER2 cloning and directed mutagenesis
Directed mutagenesis was performed using the Quick Change II XL site‐directed mutagenesis kit from Stratagene. The primers for mutagenesis were designed using the QuickChange Primer Design Program. The mutagenesis was performed on the pBabe plasmid encoding the myc‐tagged HER2 cDNA. HER2‐mutated fragments were introduced into the pLVX plasmid encoding myc‐tagged HER2‐wt. Every resulting product was validated by sequencing its whole length.
2.7. Retroviral vector, retroviral production, and infection
The Lenti‐X Bicistronic Expression System from Clontech was used for lentivirus production according to the manufacturer's instructions. Infection was performed in the presence of polybrene (8 mg/ml), and the cells were centrifuged at 1200 g for 60 min at 32 °C. Infected cells were selected with puromycin (2.5 mg/ml).
2.8. Cell survival and toxicity
Cells were plated in 96‐well plates and incubated with different concentrations of lapatinib (1000 cells/well for MDA‐MB‐175 and SBKR3; 2000 cells/well for BT474‐m1). After 3 days, a viability experiment was performed using the CellTiter‐Glo® Luminescent Cell Viability Assay (Promega), according to the manufacturer's instructions. The toxicity experiments were performed using the ATP‐based assay Toxilight (Roche) according to the manufacturer's instructions.
2.9. Soft‐agar assay
Single cells (5000 cells/well for all cell lines) were suspended in 2 ml of complete medium supplemented with 0.33% agar in six‐well plates. For the lapatinib response studies, cells were incubated with 0.1 μM lapatinib 1 h prior to plating. After 3–6 weeks, colonies were stained and counted using the GelCount platform from Oxford Optronix.
2.10. Acini formation assay
Acini formation assays were performed in two different ways using MCF10A cells. The morphology studies were performed following the protocol described by Debnath et al. (Debnath et al., 2003). Briefly, 20,000 single cells were seeded on a solidified layer of growth factor‐reduced Matrigel, overlayed by medium containing 2% Matrigel, and cultured for 10 days.
2.11. Cell invasion assay in Matrigel‐coated Boyden chambers
Twenty‐four‐well inserts (8‐μm pores) were coated with 30 μl of serum‐free medium containing 6% growth factor‐reduced Matrigel. Cells (1 × 105) in serum‐free medium were seeded in each insert. In the bottom well, complete growth medium was used as a chemoattractant. After 24 h of incubation, invading cells were fixed in ice‐cold methanol, stained, and counted.
2.12. Immunoprecipitation and kinase assay
Cells were lysed and sonicated using a lysis buffer containing 1% NP40. Immunoprecipitation was performed on 1 mg protein using 3 μg of c‐myc antibodies and 30 μl of G‐sepharose bead 50% slurry. The immunoprecipitated samples were then used to perform a kinase assay using the Tyrosine Kinase Assay kit from Sigma. Proteins were resolved by SDS‐PAGE and transferred to PVDF membranes. Immunoblotting was performed using the indicated antibodies, and protein bands were detected using the Odyssey Imaging System (Li‐Cor Biosciences) or electrochemoluminescence.
2.13. Immunofluorescence
MCF10A and MDA‐MB‐175 cells were plated in a four‐well Lab‐Tek Permanox plastic chamber slide system and cultured for 3 days. Cells were then fixed and submitted to the immunofluorescence protocol previously described (Boulbes et al., 2011) using anti‐c‐myc antibody. Slides were photographed using an Olympus DSU confocal microscope system coupled with an Orca II ER Camera. Images were analyzed by deconvolution using the software Fluoview 5.0 (http://www.olympusfluoview.com/java/colocalization/index.html).
2.14. Mass spectrometric analysis of HER2 binding partners
The immunoprecipitated proteins were boiled in Laemmli buffer, separated using 10% SDS‐PAGE and divided into ten fractions. After in‐gel trypsin digestion, the tryptic peptides of each fraction were injected into the linear ion trap‐Fourier transform ion cyclotron resonance mass spectrometer (LTQ‐FTICR MS). The full‐scan survey mass spectrometry experiment (m/z 320‐2000) was performed in LTQ‐FTICR MS with a mass resolution of 100,000 at m/z 400. The top ten most abundant multiply‐charged ions were sequentially isolated for tandem mass spectrometry by LTQ. The following Mascot search parameter settings were used: peptide tolerance, 5 ppm with 2+ and 3+ peptide charges, and tandem mass spectrometry tolerance, 0.5 Da. The significance threshold for the identification was set to p < 0.01.
2.15. Xenograft human tumor model in nude mice
Female, 5‐week‐old, nude mice received an estrogen pellet subcutaneously 4 days prior to cell injection. Eight million BT474‐m1 cells expressing either HER2‐wt or L726F in 50% Matrigel‐PBS were injected into the mammary fat pads. When the xenograft tumors reached an average size of 150 mm3, the mice were randomly divided into two groups of ten mice each: group 1 was treated with vehicle and group 2 was treated with lapatinib (150 mg/kg/day via oral gavage). The vehicle used was 0.5% (w/w) hydroxypropylmethylcellulose with 0.1% Tween 80 in water. The tumors were measured daily with calipers, and the tumor volumes were calculated as follows: volume = length × width2/2.
2.16. Statistical analyses
Cell survival, cell migration, colony formation, acini formation, and in vivo tumor formation by cells expressing pLVX or HER2 mutants were compared using a two‐tailed Student's t test. p < 0.05 was considered statistically significant. The following annotations were used: (*) p < 0.05; (**) p < 0.01; (***) p < 0.001.
3. Results
3.1. Novel HER family mutation discovered in patients correlates with outcome
Patient characteristics are summarized in Table 1. Using the Sanger method, we sequenced the kinase domains of EGFR, HER2, and HER4 in 76 invasive HER2‐positive primary breast cancers. Eleven tumors carried 12 mutations in the kinase domains of EGFR (6 mutations), HER2 (3 mutations), or HER4 (3 mutations) (Figure 1, Supplementary Figure S1). All of the tumor samples were macro‐dissected to enrich for tumor DNA before mutation screening. Two of the HER2 variants (see Supplementary Figure S9) appeared to be homozygous (i.e. ∼100% of the sequencing trace was mutant). These variants were clearly on the amplified HER2 allele, suggesting that they were somatic and/or the target of selection during tumor evolution. One tumor contained mutations in EGFR and HER4.
Table 1.
HER2‐positive tumors – patient characteristics.
| Characteristic | N (%) |
|---|---|
| Menopausal status | |
| Premenopausal | 34 (44.7) |
| Postmenopausal | 42 (55.3) |
| Race/Ethnicity | |
| White | 65 (85.5) |
| Asian | 1 (1.3) |
| Hispanic | 7 (9.2) |
| African American | 3 (4) |
| ER | |
| Negative | 39 (51.3) |
| Positive | 37 (48.7) |
| PR | |
| Negative | 40 (52.6) |
| Positive | 36 (47.4) |
| Response to trastuzumab‐based chemotherapy | |
| Stable disease | 29 (38.2) |
| Partial response | 21 (27.6) |
| Progressive disease | 26 (34.2) |
Figure 1.
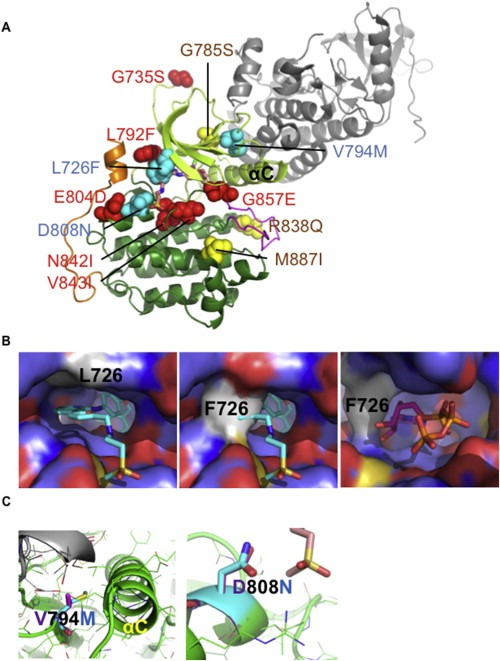
Structural analysis of the HER family mutants. A) Position of the 12 mutations on the kinase domain. All EGFR (red), HER2 (cyan), and HER4 (yellow) mutations were projected on the structure of the kinase domain (shown as the receiver kinase domain; the activator kinase domain is shown in gray). The kinase domain structure was taken from HER2 (PDB 3PP0) and missing residues were completed using homology modeling. The position of lapatinib (stick model with carbon atoms in beige) has been transferred from lapatinib‐bound EGFR (PDB 1XKK). The N‐terminal kinase lobe (light green), the C‐terminal lobe (dark green), the activation loop (magenta), and the C‐terminal extension (orange) are color coded. The αC helix is labeled. B) Structural models of HER2‐L726F. Zoom onto the lapatinib/ATP binding pocket. HER2 is shown as surface representation (color coded by atom type: C = light blue; N = dark blue; O = red; S = yellow). L726 or F726 is shown in light gray. Lapatinib (cyan carbons) and ATP (magenta carbons) are shown as stick models. In HER2‐wt, L726 forms part of the lapatinib‐binding pocket (left panel). F726 blocks binding of lapatinib to the closed kinase conformation (central panel). However, in the open, active kinase conformation, ATP can still bind (right panel). L726F may additionally stabilize the active state through interactions with a C‐terminal α helix (shown in gold in panel A). C) Structural models of HER2‐V794M and HER2‐D808M. Structural models for HER2‐wt (magenta) and mutants (cyan) are shown. V794M is predicted to influence kinase activity through altering asymmetric activation by the activator molecule (shown in gray) and the positioning of the αC helix (labeled). Left panel: Model based on activated EGFR kinase (2GS6). V794M may stabilize the αC helix in an active conformation and increase contacts between the activator molecule (gray) and the receiver molecule (green). Central panel: model built on HER2 in active‐like conformation (open conformation, but with the αC helix in an inactive conformation). Right panel: D808N is interacting with the methylsulfonylethylamino group of lapatinib (beige carbon atoms). For additional details, see Supplementary Material and Supplementary Figure S1.
Four of the six EGFR mutations identified have already been described either in the literature or in Cosmic (Pallis et al., 2007; Shih et al., 2006; Tsao et al., 2005; Weber et al., 2005). The two other EGFR mutations (E804D and N842I), all three HER2 mutations, and all three HER4 mutations are novel mutations. In addition, the sequencing detected a thirteenth mutation in the HER2 transmembrane domain (I654V), which has previously been described both as a polymorphism and as an oncogenic variant (Breyer et al., 2009; Frank et al., 2005). None of the patients carrying a mutation in EGFR, HER2, and/or HER4 achieved a partial response to trastuzumab‐based chemotherapy in the metastatic setting. Twenty‐one (32%) of the 65 patients with no mutations in HER proteins achieved a partial response to treatment (two‐tailed Fisher's exact test, p = 0.029).
3.2. Computational analysis of HER family mutants
To obtain insights about the mechanisms of action of these 12 mutations, we used in silico structural analysis based on the published crystallographic structure of wild‐type EGFR, HER2, and HER4 kinase domains and computational homology models of the mutants. Based on the crystal structures, these mutations can be divided into three groups: those located in the ATP/tyrosine kinase inhibitor (TKI) binding site (EGFR‐L792F, N842I, V843I, and G857E; HER2‐L726F), those in the N‐terminal lobe (EGFR‐G735S; HER2‐V794M; HER4‐G785S), and those in the C‐terminal lobe (EGFR‐E804D; HER2‐D808N; HER4‐R838Q, M887I) (Figure 1, Supplementary Figure S1).
Since the mutations affect different kinase regions, it is unlikely that they all affect HER proteins by the same molecular mechanism. Computational structural analysis supports that these mutations act differently (our full computational structural analysis of all EGFR, HER2, and HER4 mutations can be found in the Supplementary Information and Supplementary Figure 1). For example EGFR‐N842I and EGFR‐G857E are both located in the ATP/TKI binding site. However, while EGFR‐N842I is predicted to affect binding of ATP/TKI and substrates, EGFR‐G857E is predicted to strongly promote the active (open) kinase conformation by sterically disfavoring the inactive conformation. Conversely, mutations in different positions may have similar effects. For example, EGFR‐G735S and HER2‐V794M are both likely to allosterically increase dimerization, and hence lateral activation of kinase activity. HER2‐V794M may also enhance kinase activity by stabilizing the active conformation of the αC helix (Figure 1C, left panel). Conversely, mutations such as HER2‐D808N may influence binding of ATP and/or substrates (Figure 1C, right panel), whereas HER4‐R838Q may function by altering interactions with regulatory partners and/or protein stability.
Although the tumors were not treated with the HER2‐targeted TKI lapatinib, our structural analysis predicted that some mutations affect the interaction with this TKI, albeit using different mechanisms. For example, HER2‐L726F is predicted to sterically clash with the binding of lapatinib (which binds HER2 in the inactive form) but not ATP (Figure 1C, central panel); HER2‐V794M may affect lapatinib binding by influencing the position of the αC helix, and HER2‐D808N by altering direct interactions between HER2 and the methylsulfonylethylamino group of lapatinib.
Our structural analysis highlights that the finely tuned structural framework of HER family kinases is vulnerable to deregulation in a variety of sites. Our analysis also suggests that some of the mutants may be predisposed to an altered interaction with TKIs.
3.3. HER2 mutations induce a more aggressive phenotype in MFC10A cells
After such strong correlation, and because HER2 is the family member most associated with human breast cancer, we decided to study further the HER2 mutations in vitro, as a proof of the concept presented in our hypothesis. We assessed the ability of the HER2 mutants to induce transformation. Mutant and wt HER2 were stably expressed in different cell lines: MCF10A, a non‐tumorigenic breast cell line, and the three HER2‐overexpressing cancer cell lines SKBR3, BT474‐m1, and MDA‐MB‐175 (Supplementary Figure S2). Our results showed that expression of HER2‐L726F, HER2‐V794M, and HER2‐D808N significantly increased invasion through Matrigel and growth factor‐independent and anchorage‐independent growth. Indeed, the number of invading MCF10A cells expressing any of the mutants was between 1.5‐ and 2‐fold higher than the number of invading MCF10A cells expressing HER2‐wt (Figure 2A). No significant difference in the proliferation rate was detected during the 24‐h duration of this experiment (Supplementary Figure S3A).
Figure 2.
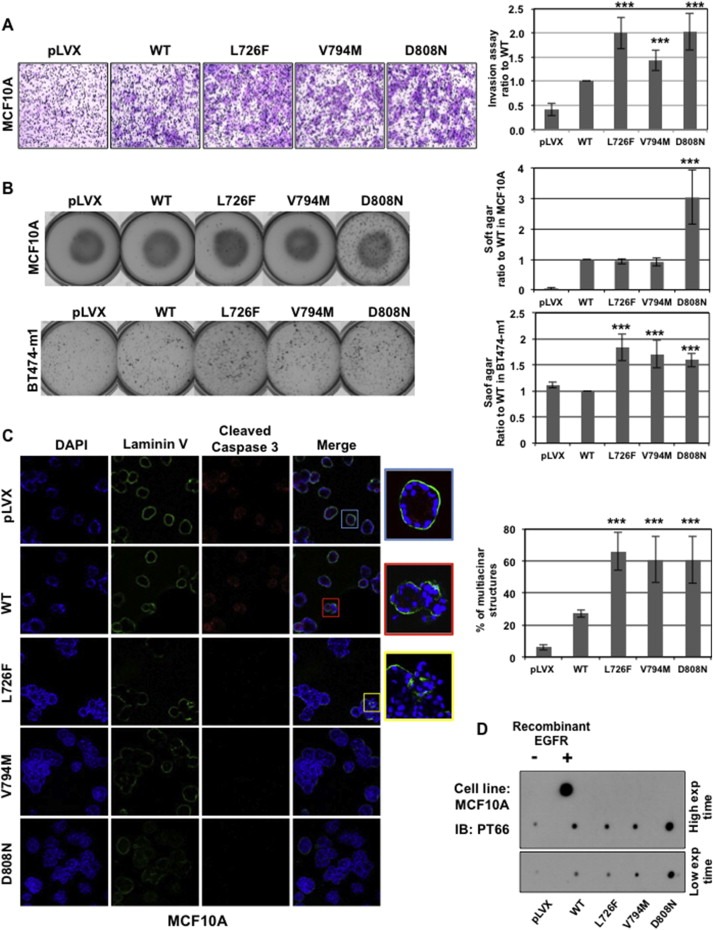
HER2 mutants induce a more aggressive phenotype. A) HER2 mutants increase MCF10A cell invasion. The graph (right panel) shows the proportion of HER2‐wt and L726F cells compared with control (mean value of 3 separate experiments) (***, p < 0.001). B) HER2 mutants increase anchorage independence. MCF10A (upper panel) or BT474‐m1 (lower panel) cells grown in soft agar were stained and counted (graphs) (***, p < 0.001). C) HER2 mutants impair formation of normal acini by MCF10A. The immunofluorescence experiment was performed on fixed acini using DAPI (blue), anti‐laminin V (green), and anti‐cleaved‐caspase 3 (red), and photos were taken using confocal microscopy (lower panel). Graph: Expression of the mutants correlates with an increase in the number of abnormal “grape‐like” multicentric acini formed by MCF10A cells. D) HER2 mutants have a similar or stronger kinase activity then HER2‐wt. Kinase assay was performed in vitro using c‐myc pulled‐down HER2‐wt and mutants (from MCF10A cells) on a non‐specific peptide. The resulting product was dot blotted onto a nitrocellulose membrane and labeled with PT66 phosphotyrosine targeting antibody. Gray levels were analyzed using NIH Image J software and normalized to 1 arbitrarily given the HER2‐wt.
Surprisingly, HER2‐L726F and HER2‐V794M failed to increase the colony formation of MCF10A cells, in contrast to HER2‐D808N (Figure 2B, upper panel). However, the HER2‐L726F and HER2‐V794M mutants increased by 70%–80% the number of colonies formed when expressed in BT474‐m1 cells, suggesting that these two mutants might not be sufficient to induce anchorage‐independent growth in a non‐cancerous background but would be sufficient in a cancerous background (Figure 2B, lower panel). When cultured in a serum‐free medium, the HER2‐D808N mutant kept proliferating 5 times faster than cells expressing the wild type (Supplementary Figure S3B). No significant differences were found in the other mutants, further corroborating that the mechanism of action of HER2‐D808N is different from that of HER2‐L726F or HER2‐V794M.
We then monitored the formation of acini by MCF10A cells grown in three‐dimensional culture, as described in Debnath et al. (Debnath et al., 2003) (Figure 2C). As expected, the MCF10A cells formed normal acini exhibiting a well‐polarized outer layer of cells in contact with the formed basal membrane (laminin V in green) along with an inner subset of cells, centrally located, undergoing apoptosis (cleaved‐caspase 3 in red), which coincides with the formation of a hollow lumen (blue square). Conversely, the expression of the oncogene HER2‐wt in MFC10A cells was associated with the presence of multiacinar structures among the normal‐looking spheroids, a typical marker of tumorigenicity (Wang et al., 2006) exhibiting a luminal filling. Cleaved‐caspase 3 was expressed, but disruption of the basal membrane occurred in some places (red square). Cells expressing HER2 mutants exhibited multiacinar structures, almost complete luminal filling (absence of hollow lumen), and no detectable expression of cleaved‐caspase 3. This phenotype usually occurs when an increase in cell proliferation is combined with the inhibition of apoptosis. Moreover, some structures showed cells escaping through a breach in the basal membrane (yellow square). Finally, the expression of the HER2 mutants also increased the number of “grape‐like” multiacinar structures (Figure 2C, graph).
Finally, we performed an in vitro kinase assay on all the mutant and wt HER2 (Figure 2D). The results showed that the HER2‐L726F mutant exhibited slightly lower kinase activity, HER2‐V794M showed slightly higher activity, and HER2‐D808N showed dramatically stronger kinase activity than HER2‐wt. These results are consistent with the results presented in the soft‐agar assay.
Collectively, these data show that all HER2 mutants induced a more aggressive phenotype than HER2‐wt in non‐tumorigenic MCF10A cells. Thus, these data confirm our in silico prediction for HER2‐L726F and HER2‐V794M and demonstrate increased activity for HER2‐D808N, in agreement with a mechanistic model in which D808N increases the affinity of HER2 for ATP and/or substrates.
3.4. HER2 mutations alter response to lapatinib
We then studied the response of each mutant to two HER2‐targeted therapies used in the clinic: the TKI lapatinib and the antibody trastuzumab.
As predicted by our structural analysis, all mutants exhibited an altered response to lapatinib when grown in soft agar (Figure 3A). The results showed an increase in the response to lapatinib by the mutant HER2‐V794M, which may be explained by V794M promoting an αC helix conformation more suitable for lapatinib binding. In agreement with a predicted interaction between D808N and the methylsulfonylethylamino moiety of lapatinib, our experiment also showed slight resistance to lapatinib of the mutant HER2‐D808N in the cell line MDA‐MB‐175 that does not carry HER2 gene amplification. HER2‐D808N did not display lapatinib resistance in BT474 and SKBR3 cells, which suggested that the partial resistance exhibited by HER2‐D808N is most likely hidden by the high proportion of endogenous HER2‐wt.
Figure 3.
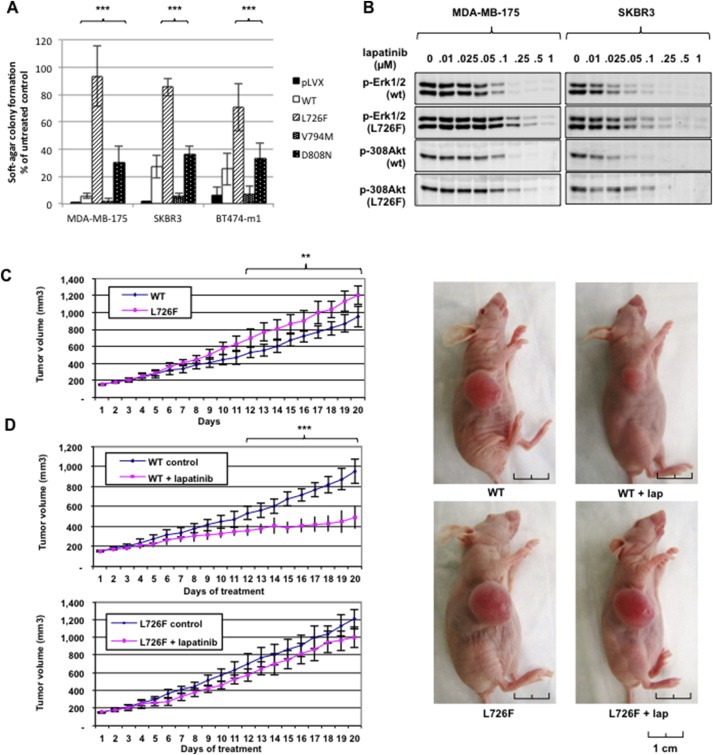
HER2 mutants alter response to lapatinib in vitro and in vivo. A) Response to lapatinib is altered in breast cancer cells expressing HER2 mutants. MDA‐MB‐175, SKBR3, and BT474‐m1 cells were incubated with 0.1 μM lapatinib, plated in 0.3% soft agar, and left growing for 2–4 weeks. Colonies were stained and counted using the GelCount platform (***, p < 0.001). B) ERK inhibition requires higher doses of lapatinib when cells express HER2 L726F. SKBR3 and MDA‐MB‐175 cells were incubated with lapatinib at the indicated concentrations, lysed, and the protein submitted for Western blot analysis. C) HER2 L726F forms larger tumors than HER2‐wt. BT474‐m1 cells expressing HER2‐wt or L726F were injected into nude mice, and the tumors formed were measured every day for 3 weeks. Each value represents the mean of tumors formed in at least 8 mice (**, p < 0.01). D) Tumors formed by cells expressing HER2 L726F are resistant to lapatinib treatment. Mice were treated with vehicle or vehicle plus lapatinib at 150 mg/kg/day via oral gavage for 3 weeks. Each value represents the mean of tumors formed in at least 8 mice (***, p < 0.001). Representative tumors were photographed after 3 weeks of lapatinib treatment.
More interestingly, HER2‐L726F exhibited a strong resistance to lapatinib confirmed by a viability experiment (Supplementary Figure S4A). Although the effect was less dramatic than for lapatinib, our experimental results and computational analysis confirmed a delayed response to neratinib, an irreversible TKI (Supplementary information and Supplementary Figure S4B and C), in particular in the cell line MDA‐MB‐175, suggesting that the mutation would impair the effect of any TKI. Also, the complete inhibition of the Akt and Erk pathways required a higher dose of lapatinib (∼1/2 log) when cells expressed the HER2‐L726F mutant rather than HER2‐wt (Figure 3B, levels of expression in Supplementary Figure S5).
Finally, we used BT474‐m1 cells expressing HER2‐L726F to perform xenograft experiments. The results showed that 1) the cells expressing the HER2‐L726F mutant formed significantly larger tumors over a period of 3 weeks than the cells expressing HER2‐wt (Figure 3C) and 2) the tumors formed by the cells expressing the mutant were more resistant to lapatinib treatment than were the tumors formed by the cells expressing HER2‐wt (Figure 3D).
Our study did not show any difference in response to trastuzumab (data not shown). Although this result might be explained by the fact that the mutations are not located in the extracellular domain targeted by the antibody, it is most likely explained by the high levels of endogenous HER2‐wt present in our cellular models.
Taken together, these results confirmed our structural predictions and showed that HER2 mutations located in the kinase domain have the potential to alter response to the HER2‐targeting TKI lapatinib.
3.5. ERK pathway is overactivated despite HER2‐L726F underphosphorylation
Because of the strong lapatinib resistance and aggressive phenotype of HER2‐L726F demonstrated in vivo, we decided to focus on the HER2‐L726F mutant and further investigate its downstream signaling and mechanism of action using MCF10A cells.
Surprisingly, although expression of HER2‐L726F was correlated with sustainable ERK1/2 activation in serum‐starved conditions (Figure 4A), HER2‐L726F also displayed underphosphorylation overall and in particular on the residue Y1248 (Figure 4B), which is commonly accepted to be a marker of HER2 kinase activity but has also been shown to be dispensable for ERK or Akt pathway activation (Deb et al., 2001; Hazan et al., 1990; Rexer et al., 2011). This underphosphorylation was also observed in HER2‐overexpressing cancer cell lines (Supplementary Figure S6) and could be explained by impaired kinase activity, homodimerization, or phosphatase activity.
Figure 4.
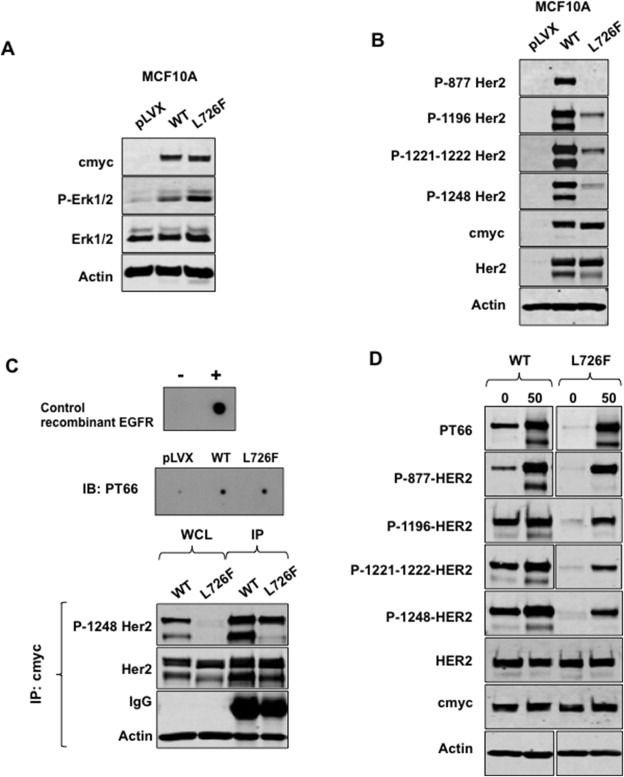
HER2 L726F conserves its kinase activity despite dramatic dephosphorylation. A) Cells expressing HER2 L726F sustain ERK activation after 24 h of starvation. Western blot analysis showed that expression of HER2 L726F was correlated with sustainable ERK1/2 activation in MCF10A cells. B) HER2 L726F mutant is dramatically underphosphorylated. Western blot analysis using antibodies targeting HER2 phosphorylated sites shows phosphorylation of HER2‐wt but not L726F in MCF10A cells. C) HER2 L726F displays kinase activity similar to that of HER2‐wt. Kinase assay was performed in vitro using c‐myc pulled‐down HER2‐wt and mutants (from MCF10A cells) on a non‐specific peptide. The product was dot blotted onto a nitrocellulose membrane and labeled with PT66 phosphotyrosine targeting antibody (upper panel). After the kinase assay, c‐myc pulled‐down HER2 was analyzed by Western blotting using a phospho‐Y1248 HER2‐specific antibody (lower panel). WCL: whole cell lysate. D) HER2 L726F dephosphorylation is due to increased phosphatase activity. MCF10A cells were incubated with 50 μM Na3Vo4 for 24 h, lysed, and analyzed by Western blotting.
To investigate the mechanism of HER2‐L726F underphosphorylation we performed a series of experiments using MCF10A cells. 1) In vitro kinase activity assay showed that HER2‐L726F kinase activity was similar to that of HER2‐wt (Figure 4C, upper panel). More importantly, Western blot analysis on the pulled‐down HER2‐L726F and HER2‐wt used in the kinase assay showed that the autophosphorylation site Y1248 was strongly phosphorylated in both HER2‐L726F and HER2‐wt (Figure 4C, bottom panel). 2) Crosslinking experiments showed that HER2‐L726F was able to dimerize (Supplementary Figure S7). It is likely that HER2‐L726F dimerizes by stabilizing an open (active‐like) conformation, in a way reminiscent of the one described for EGFR‐L858R (Shan et al., 2012). Such promotion of an open conformation is in accord with our structural analysis. 3) We incubated the MCF10A cells with the tyrosine phosphatase inhibitor sodium orthovanadate (Na3VO4; 50 μM) (Figure 4D). After 24 h, the phosphorylation levels of HER2‐L726F and HER2‐wt were comparable, showing that HER2‐L726F underphosphorylation was due to increased activity of tyrosine phosphatases by a mechanism that remains to be elucidated. The lower band observed in the HER2 blot corresponds to a 170 Kd HER2 degradation product previously described in SKBR3 cells by Lehrer et al., in 1995 (Lehrer et al., 1995). As seen on supplemental figures S2, S5 and S6, this band appears as well in parental HER2‐overexpressing cancer cells. This band is usually cropped in most publications, although it is shown in others (Jin et al., 2010; Mahlknecht et al., 2013; Rafidi et al., 2013; Scaltriti et al., 2011). We chose not to crop this band in our figures. Regarding the lack of the 170 band phosphorylation in L726F‐expressing cells after incubation with the phosphatase inhibitor, we believe the restoration was just incomplete, hence the lack of phosphorylation of individual sites. However, the site‐unspecific antibody PT66 (which targets several phosphorylated tyrosines on HER2) did show a similar level of phosphorylation on the mutant and on the WT, most likely because of the signal accumulation of each individual site.
Since an alteration in the phosphorylation status of HER2 may be associated with altered intracellular localization of HER2 (Ouyang et al., 1998), we evaluated the localization of HER2 using immunofluorescence (Supplementary Figure S8). The result showed that HER2‐L726F cells exhibited perinuclear cytoplasmic localization of HER2 in both cell lines and the primary tumor from the patient who was carrying the HER2‐L726F mutation. This observation confirms the atypical behavior of the underphosphorylated HER2‐L726F mutant.
Together our results describe a mechanism whereby HER2‐L726F, by allosterically promoting an open conformation, promotes lateral signal propagation through kinase–kinase interactions and thus amplification of downstream signaling, explaining its aggressive behavior.
3.6. HER2‐L726F is associated with various heat shock proteins (HSPs)
Finally, with the goal of finding new therapeutic options for patients carrying the HER2‐L726F mutation, and by extension other mutations driving resistance to lapatinib, we searched for molecular partners of this mutant. Mass spectrometry experiments showed that HER2‐L726F was associated with several chaperone and co‐chaperone molecules in significantly larger amounts than HER2‐wt (Table 2). In particular, HSP90A was shown to bind preferentially to the mutant, as confirmed by immunoprecipitation (Figure 5A). This partnership is interesting because 1) HSP90 is known to stabilize and potentiate HER2 activity (Solit et al., 2002), 2) the new HSP90 inhibitor 17‐demethoxygeldanamycin (17AAG) is undergoing clinical testing in combination with trastuzumab (Pacey et al., 2012), and 3) HSP90 has been shown to regulate the ERK pathway (Babchia et al., 2008). We also detected an association with macrophage migration inhibitory factor (MIF), which has recently been shown to be a novel HSP90 client and to correlate with clinical aggressiveness in HER2‐overexpressing breast cancer (Schulz et al., 2012).
Table 2.
Major interacting proteins with increased binding to the HER2 mutants compared to HER2‐wt.
| Protein identified | Name | Function |
|---|---|---|
| HSP90A | Heat shock 90 kDa protein alpha | Chaperone |
| HSP70 ½ | Heat shock 70 kDa protein 1/2 | Chaperone |
| HSP27 | Heat shock protein 27 | Chaperone |
| HSP40 | DnaJ (Hsp40) homolog, subfamily C, member10 | Co‐chaperone |
| CDC37 | HSP90‐cochaperone CDC37 | Co‐chaperone |
| BAG2 | BAG family molecular chaperone regulator 2 | Co‐chaperone |
| MIF | Macrophage migration inhibitory factor | Cytokine |
a Exogenous c‐myc‐tagged HER2 over‐expressed in MCF10A cells was pulled‐down with a c‐myc antibody and then analyzed using mass spectrometry.
Figure 5.
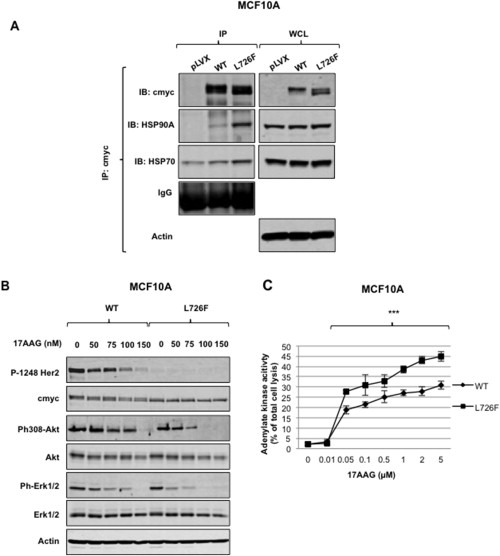
HER2 L726F associates with HSP90A. A) HER2 L726F associates with heat shock protein 90A and 70. MCF10A cells were lysed and proteins subjected to co‐immunoprecipitation using the indicated antibodies. Pulled‐down proteins were then analyzed by Western blotting. WCL: whole cell lysate. B) ERK activation in HER2 L726F is dependent on its association with HSP90. MCF10A cells were incubated with the indicated concentrations of the HSP90 inhibitor 17AAG for 24 h. Cells were lysed and proteins submitted for Western blot analysis. C) Cells expressing HER2 L726F are more sensitive to the cytotoxic effect of the HSP90 inhibitor 17AAG. MCF10A cells were incubated with the indicated concentrations of the HSP90 inhibitor 17AAG for 3 days and a cytotoxicity assay was performed. Values were reported to the maximum of cell toxicity in each well, which corresponds to total cell lysis (***, p < 0.001).
Interestingly, incubation of MCF10A cells with 17AAG showed that both Akt and ERK pathways lost their phosphorylation faster in the cells expressing HER2‐L726F (Figure 5B). These data also suggest that the high ERK activity observed in serum‐starved cells in Figure 4A may be due to the HSP90A potentiation of HER2‐L726F kinase activity. Moreover, a 48‐h incubation showed that the HER2‐L726F mutant is less stable than HER2‐wt and that HSP90A protects it from being degraded (Supplementary Figure S10). Finally, a toxicity assay performed after 3 days of incubation showed higher toxicity in HER2‐L726F‐expressing MCF10A cells (Figure 5C), suggesting that HSP90A appears important for stabilizing the survival‐promoting activity of HER‐L726F.
Taken together, these data confirmed the preferential partnership between HSP90A and HER2‐L726F on a functional level and suggested an alternative treatment for patients carrying a mutation that would be structurally predicted to confer resistance to lapatinib.
4. Discussion
Breast cancer is a heterogeneous disease. The most common genetic alterations in invasive breast carcinomas are HER2 gene amplification and mutations in p53, PIK3CA, and GATA3 (Koboldt et al., 2012; Santarpia et al., 2012). In this study, we identified 12 HER family kinase domain mutations in human primary invasive HER2‐overexpressing breast cancer. Although these mutations localize to different regions of the kinase domain and function through different mechanisms, all the mutations correlated with aggressiveness in vitro and lack of response to therapy in the metastatic setting.
Only a few mutations have been reported in HER2‐amplified and HER2 non‐amplified ovarian and breast cancers (Bose et al., 2013; Lee et al., 2006; Lin et al., 2011). We believe that this lack of data underlines the importance of our results and supports further the novelty of our discovery. Although we do not believe these are germline variants, matched patient blood was not available to definitively rule out this possibility. However, the evidence that these are somatic variants is as follows: 1) there is no record of any of these variants in publically available SNP databases, 2) among the mutations we identified, four of six EGFR mutations have been detected by others and are annotated as tumor‐specific variants in the COSMIC database (Pallis et al., 2007; Shih et al., 2006; Tsao et al., 2005; Weber et al., 2005), and 3) others have reported somatic variants in the kinase domains of EGFR, HER2, and HER4, so although many of the variants reported here are unique, the overall observation that we identified potential activating variants in this gene family is not novel (for review, see (Herter‐Sprie et al., 2013)). Moreover, these variants were clearly on the amplified HER2 allele suggesting that they are somatic and/or the target of selection during tumor evolution. In either case, the evidence supports the hypothesis that they are activating variants. Finally, our structure‐function analysis highlights the importance of a finely tuned and highly interconnected structural and dynamic framework of the kinase domain that allows each HER family member to operate normally and to bind to targeted drugs. However, this strong structure–function relationship makes these kinase domains particularly vulnerable to mutations of any kind. Our analysis therefore cautions that if mutations are detected in the kinase domains of HER family proteins, then an abnormal phenotype may result and standard HER2‐targeted therapy is likely to fail.
Because HER2 is the family member most associated with human malignancies, we decided to study further the HER2 mutations in vitro, as a proof of the concept presented in our hypothesis. We confirmed experimentally that each of the HER2 mutations caused an aggressive phenotype, albeit with distinct characteristics. We also observed that the HER2 mutations altered the interactions of the HER2 kinase domain with the TKI lapatinib, even though the tumors were never exposed to TKIs. Its pronounced lapatinib resistance makes HER2‐L726F a particularly adverse mutation for breast cancer treatment. Moreover, we observed a number of significant changes in interactions and signaling for HER2‐L726F, which together explain the aggressive phenotype of this mutant. HER2‐L726F also enhanced the kinase's interaction with the molecular chaperones HSP90, HSP70, and HSP27, which are increased in diverse human cancers and correlated with resistance to chemotherapy (Modi et al., 2007). HSP90 is indispensable for maintained activity and stabilization of the fragile structures of many proteins (Babchia et al., 2008; Tikhomirov and Carpenter, 2003). As HER2‐L726F is not close to the αC‐β4 loop that was reported to be important for HSP90 interaction (Citri et al., 2004), the increased HER2:HSP90 interaction may therefore result from allosteric conformational changes introduced by HER2‐L726F, or be triggered by a decrease in the stability of this mutant. Although the interaction with HSP90 and HER2 kinase dimerization were reported to be mutually exclusive (Citri et al., 2004), HSP90 may help stabilize a sufficient number of correctly folded HER2 molecules and thus enhance kinase signaling. Clinical trials are ongoing to determine the safety and efficacy of HSP90 inhibitors in HER2‐positive metastatic breast cancer (Modi et al., 2007), with the hope that these inhibitors could be used to replace lapatinib in patients whose tumors carry the HER2‐L726F mutation. Our results confirm that HER2, like EGFR, does not require tyrosine phosphorylation in its activation loop to be active (Gotoh et al., 1992) and that HER2 can induce ERK and Akt activation in the absence of phosphorylation of the C‐terminal tail (Deb et al., 2001; Rexer et al., 2011).
5. Conclusions
Our clinical analysis shows that mutations in HER kinase domains correlate with tumor aggressiveness and success of therapy in breast cancer. Taken together, our functional and structural analyses investigate underlying mechanisms and explain this correlation by showing that these kinase domains are finely tuned allosteric enzymes that can be deregulated in a large variety of mutations in almost any part of the structure. These detailed analyses will help improve our understanding of the function of similar mutations that are likely to be discovered in the future. As next‐generation sequencing studies uncover novel somatic mutations in breast cancer, understanding their functions and predictive role in breast cancer response to targeted therapies will be critical for successful implementation of personalized cancer treatment. Our study suggests that sequencing of the HER family kinase domains alone may provide a quick and affordable method for a rapid prognosis and selection of therapy for patients with HER2‐positive breast cancers. Also, it is important to note that, even if HER2 status differs between primary tumors and metastases, HER gene mutations will most likely still exist in the DNA of metastases and circulating tumor cells. Circulating tumor cells are considered to be the origin of metastasis and are of significant prognostic relevance in metastatic breast cancer. Therefore, this study could be applicable to the sequencing of HER family [“genes”?] in circulating tumor cells. Indeed, the detection of a mutation in those cells could provide a non‐invasive diagnostic tool to select patients who would need closer monitoring during their treatment.
Conflicts of interest
The authors disclose no potential conflicts of interest.
Grant support
This research was funded by a Postdoctoral Fellowship grant from the Susan G. Komen for the Cure and a grant from the Breast Cancer Research Foundation. The work was supported in part by the MD Anderson Cancer Center Support Grant (CA016672) through the Flow Cytometry and Cellular Imaging Facility, the DNA Analysis Facility, and the Research Animal Support Facility.
The pBabe vector encoding cterm myc‐tagged full‐length HER2 was kindly provided by Dr. Carlos L. Arteaga. BT474‐m1 cells were generously provided by Dr. Dihua Yu.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Acknowledgments
The authors thank Dawn Chalaire from the Department of Scientific Publications at MD Anderson Cancer Center for editorial assistance. Research by S.T.A was supported by the King Abdullah University of Science and Technology (KAUST).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.10.011.
Boulbes Delphine R., Arold Stefan T., Chauhan Gaurav B., Blachno Korina V., Deng Nanfu, Chang Wei-Chao, Jin Quanri, Huang Tzu-Hsuan, Hsu Jung-Mao, Brady Samuel W., Bartholomeusz Chandra, Ladbury John E., Stone Steve, Yu Dihua, Hung Mien-Chie, Esteva Francisco J., (2015), HER family kinase domain mutations promote tumor progression and can predict response to treatment in human breast cancer, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.10.011.
References
- Arnold, K. , Bordoli, L. , Kopp, J. , Schwede, T. , 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 22, 195–201. [DOI] [PubMed] [Google Scholar]
- Babchia, N. , Calipel, A. , Mouriaux, F. , Faussat, A.M. , Mascarelli, F. , 2008. 17-AAG and 17-DMAG-induced inhibition of cell proliferation through B-Raf downregulation in WT B-Raf-expressing uveal melanoma cell lines. Invest. Ophthalmol. Vis. Sci. 49, 2348–2356. [DOI] [PubMed] [Google Scholar]
- Baselga, J. , 2006. Targeting tyrosine kinases in cancer: the second wave. Science. 312, 1175–1178. [DOI] [PubMed] [Google Scholar]
- Baselga, J. , Cortes, J. , Kim, S.B. , Im, S.A. , Hegg, R. , Im, Y.H. , Roman, L. , Pedrini, J.L. , Pienkowski, T. , Knott, A. , 2012. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N. Engl. J. Med. 366, 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose, R. , Kavuri, S.M. , Searleman, A.C. , Shen, W. , Shen, D. , Koboldt, D.C. , Monsey, J. , Goel, N. , Aronson, A.B. , Li, S. , 2013. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 1–15. online first [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulbes, D.R. , Shaiken, T. , Sarbassov dos, D. , 2011. Endoplasmic reticulum is a main localization site of mTORC2. Biochem. Biophys. Res. Commun. 413, 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan, P.J. , Kumagai, T. , Berezov, A. , Murali, R. , Greene, M.I. , 2000. HER2/neu: mechanisms of dimerization/oligomerization. Oncogene. 19, 6093–6101. [DOI] [PubMed] [Google Scholar]
- Breyer, J.P. , Sanders, M.E. , Airey, D.C. , Cai, Q. , Yaspan, B.L. , Schuyler, P.A. , Dai, Q. , Boulos, F. , Olivares, M.G. , Bradley, K.M. , 2009. Heritable variation of ERBB2 and breast cancer risk. Cancer Epidemiol. Biomarkers Prev. 18, 1252–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri, A. , Gan, J. , Mosesson, Y. , Vereb, G. , Szollosi, J. , Yarden, Y. , 2004. Hsp90 restrains ErbB-2/HER2 signalling by limiting heterodimer formation. EMBO Rep. 5, 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb, T.B. , Su, L. , Wong, L. , Bonvini, E. , Wells, A. , David, M. , Johnson, G.R. , 2001. Epidermal growth factor (EGF) receptor kinase-independent signaling by EGF. J. Biol. Chem. 276, 15554–15560. [DOI] [PubMed] [Google Scholar]
- Debnath, J. , Muthuswamy, S.K. , Brugge, J.S. , 2003. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 30, 256–268. [DOI] [PubMed] [Google Scholar]
- Esteva, F.J. , Guo, H. , Zhang, S. , Santa-Maria, C. , Stone, S. , Lanchbury, J.S. , Sahin, A.A. , Hortobagyi, G.N. , Yu, D. , 2010. PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am. J. Pathol. 177, 1647–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteva, F.J. , Yu, D. , Hung, M.C. , Hortobagyi, G.N. , 2010. Molecular predictors of response to trastuzumab and lapatinib in breast cancer. Nat. Rev. Clin. Oncol. 7, 98–107. [DOI] [PubMed] [Google Scholar]
- Frank, B. , Hemminki, K. , Wirtenberger, M. , Bermejo, J.L. , Bugert, P. , Klaes, R. , Schmutzler, R.K. , Wappenschmidt, B. , Bartram, C.R. , Burwinkel, B. , 2005. The rare ERBB2 variant Ile654Val is associated with an increased familial breast cancer risk. Carcinogenesis. 26, 643–647. [DOI] [PubMed] [Google Scholar]
- Garrett, T.P. , McKern, N.M. , Lou, M. , Elleman, T.C. , Adams, T.E. , Lovrecz, G.O. , Kofler, M. , Jorissen, R.N. , Nice, E.C. , Burgess, A.W. , Ward, C.W. , 2003. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell. 11, 495–505. [DOI] [PubMed] [Google Scholar]
- Geyer, C.E. , Forster, J. , Lindquist, D. , Chan, S. , Romieu, C.G. , Pienkowski, T. , Jagiello-Gruszfeld, A. , Crown, J. , Chan, A. , Kaufman, B. , 2006. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 355, 2733–2743. [DOI] [PubMed] [Google Scholar]
- Ghosh, R. , Narasanna, A. , Wang, S.E. , Liu, S. , Chakrabarty, A. , Balko, J.M. , Gonzalez-Angulo, A.M. , Mills, G.B. , Penuel, E. , Winslow, J. , 2011. Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers. Cancer Res. 71, 1871–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh, N. , Tojo, A. , Hino, M. , Yazaki, Y. , Shibuya, M. , 1992. A highly conserved tyrosine residue at codon 845 within the kinase domain is not required for the transforming activity of human epidermal growth factor receptor. Biochem. Biophys. Res. Commun. 186, 768–774. [DOI] [PubMed] [Google Scholar]
- Graus-Porta, D. , Beerli, R.R. , Daly, J.M. , Hynes, N.E. , 1997. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 16, 1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazan, R. , Margolis, B. , Dombalagian, M. , Ullrich, A. , Zilberstein, A. , Schlessinger, J. , 1990. Identification of autophosphorylation sites of HER2/neu. Cell Growth Differ. 1, 3–7. [PubMed] [Google Scholar]
- Herter-Sprie, G.S. , Greulich, H. , Wong, K.K. , 2013. Activating mutations in ERBB2 and their impact on diagnostics and treatment. Front. Oncol. 3, 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Gao, L. , Wang, S. , McManaman, J.L. , Thor, A.D. , Yang, X. , Esteva, F.J. , Liu, B. , 2010. Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-i receptor in breast cancer cells resistant to herceptin. Cancer Res. 70, 1204–1214. [DOI] [PubMed] [Google Scholar]
- Hynes, N.E. , Lane, H.A. , 2005. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer. 5, 341–354. [DOI] [PubMed] [Google Scholar]
- Hynes, N.E. , Stern, D.F. , 1994. The biology of erbb-2/neu/her-2 and its role in cancer. Biochim. Biophys. Acta. 1198, 165–184. [DOI] [PubMed] [Google Scholar]
- Jin, Q. , Yuan, L.X. , Boulbes, D. , Baek, J.M. , Wang, Y.N. , Gomez-Cabello, D. , Hawke, D.H. , Yeung, S.C. , Lee, M.H. , Hortobagyi, G.N. , 2010. Fatty acid synthase phosphorylation: a novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 12, R96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila, T.T. , Akita, R.W. , Parsons, K. , Fields, C. , Lewis Phillips, G.D. , Friedman, L.S. , Sampath, D. , Sliwkowski, M.X. , 2009. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 15, 429–440. [DOI] [PubMed] [Google Scholar]
- Koboldt, D.C. , Fulton, R.S. , McLellan, M.D. , Schmidt, H. , Kalicki-Veizer, J. , McMichael, J.F. , Fulton, L.L. , Dooling, D.J. , Ding, L. , Mardis, E.R. , 2012. Comprehensive molecular portraits of human breast tumours. Nature. 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson, J.S. , Goodman, L.J. , Tan, Y. , Defazio-Eli, L. , Paquet, A.C. , Cook, J.W. , Rivera, A. , Frankson, K. , Bose, J. , Chen, L. , 2010. Analytical validation of a highly quantitative, sensitive, accurate, and reproducible assay (HERmark) for the measurement of HER2 total protein and HER2 homodimers in FFPE breast cancer tumor specimens. Pathol. Res. Int. 2010, 814176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.W. , Soung, Y.H. , Seo, S.H. , Kim, S.Y. , Park, C.H. , Wang, Y.P. , Park, K. , Nam, S.W. , Park, W.S. , Kim, S.H. , 2006. Somatic mutations of ERBB2 kinase domain in gastric, colorectal, and breast carcinomas. Clin. Cancer Res. 12, 57–61. [DOI] [PubMed] [Google Scholar]
- Lehrer, S. , Lee, P. , Tartter, P. , Shank, B. , Brower, S.T. , 1995. Breast cancer and family history: a multivariate analysis of levels of tumor HER2 protein and family history of cancer in women who have breast cancer. Mt. Sinai J. Med. 62, 415–418. [PubMed] [Google Scholar]
- Liang, K. , Esteva, F.J. , Albarracin, C. , Stemke-Hale, K. , Lu, Y. , Bianchini, G. , Yang, C.Y. , Li, Y. , Li, X. , Chen, C.T. , 2010. Recombinant human erythropoietin antagonizes trastuzumab treatment of breast cancer cells via Jak2-mediated Src activation and PTEN inactivation. Cancer Cell. 18, 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, W.L. , Kuo, W.H. , Chen, F.L. , Lee, M.Y. , Ruan, A. , Tyan, Y.S. , Hsu, J.D. , Chiang, H. , Han, C.P. , 2011. Identification of the coexisting HER2 gene amplification and novel mutations in the HER2 protein-overexpressed mucinous epithelial ovarian cancer. Ann. Surg. Oncol. 18, 2388–2394. [DOI] [PubMed] [Google Scholar]
- Mahlknecht, G. , Maron, R. , Mancini, M. , Schechter, B. , Sela, M. , Yarden, Y. , 2013. Aptamer to ErbB-2/HER2 enhances degradation of the target and inhibits tumorigenic growth. Proc. Natl. Acad. Sci. U S A. 110, 8170–8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi, S. , Stopeck, A.T. , Gordon, M.S. , Mendelson, D. , Solit, D.B. , Bagatell, R. , Ma, W. , Wheler, J. , Rosen, N. , Norton, L. , 2007. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J. Clin. Oncol. 25, 5410–5417. [DOI] [PubMed] [Google Scholar]
- Monsey, J. , Shen, W. , Schlesinger, P. , Bose, R. , 2010. Her4 and Her2/neu tyrosine kinase domains dimerize and activate in a reconstituted in vitro system. J. Biol. Chem. 285, 7035–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow, P.K. , Wulf, G.M. , Ensor, J. , Booser, D.J. , Moore, J.A. , Flores, P.R. , Xiong, Y. , Zhang, S. , Krop, I.E. , Winer, E.P. , 2011. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J. Clin. Oncol. 29, 3126–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata, Y. , Lan, K.H. , Zhou, X. , Tan, M. , Esteva, F.J. , Sahin, A.A. , Klos, K.S. , Li, P. , Monia, B.P. , Nguyen, N.T. , 2004. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 6, 117–127. [DOI] [PubMed] [Google Scholar]
- Nahta, R. , Yuan, L.X. , Zhang, B. , Kobayashi, R. , Esteva, F.J. , 2005. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 65, 11118–11128. [DOI] [PubMed] [Google Scholar]
- Olayioye, M.A. , 2001. Update on HER-2 as a target for cancer therapy: intracellular signaling pathways of ErbB2/HER-2 and family members. Breast Cancer Res. 3, 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang, X. , Gulliford, T. , Epstein, R.J. , 1998. The duration of phorbol-inducible ErbB2 tyrosine dephosphorylation parallels that of receptor endocytosis rather than threonine-686 phosphorylation: implications for the physiological role of protein kinase C in growth factor receptor signalling. Carcinogenesis. 19, 2013–2019. [DOI] [PubMed] [Google Scholar]
- Pacey, S. , Gore, M. , Chao, D. , Banerji, U. , Larkin, J. , Sarker, S. , Owen, K. , Asad, Y. , Raynaud, F. , Walton, M. , 2012. A phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Invest. New Drugs. 30, 341–349. [DOI] [PubMed] [Google Scholar]
- Pallis, A.G. , Voutsina, A. , Kalikaki, A. , Souglakos, J. , Briasoulis, E. , Murray, S. , Koutsopoulos, A. , Tripaki, M. , Stathopoulos, E. , Mavroudis, D. , Georgoulias, V. , 2007. ‘Classical’ but not ‘other’ mutations of EGFR kinase domain are associated with clinical outcome in gefitinib-treated patients with non-small cell lung cancer. Br. J. Cancer. 97, 1560–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccart-Gebhart, M.J. , Procter, M. , Leyland-Jones, B. , Goldhirsch, A. , Untch, M. , Smith, I. , Gianni, L. , Baselga, J. , Bell, R. , Jackisch, C. , 2005. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 353, 1659–1672. [DOI] [PubMed] [Google Scholar]
- Rafidi, H. , Mercado, F. , Astudillo, M. , Fry, W.H. , Saldana, M. , Carraway, K.L. , Sweeney, C. , 2013. Leucine-rich repeat and immunoglobulin domain-containing protein-1 (Lrig1) negative regulatory action toward ErbB receptor tyrosine kinases is opposed by leucine-rich repeat and immunoglobulin domain-containing protein 3 (Lrig3). J. Biol. Chem. 288, 21593–21605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexer, B.N. , Ham, A.J. , Rinehart, C. , Hill, S. , Granja-Ingram Nde, M. , Gonzalez-Angulo, A.M. , Mills, G.B. , Dave, B. , Chang, J.C. , Liebler, D.C. , Arteaga, C.L. , 2011. Phosphoproteomic mass spectrometry profiling links Src family kinases to escape from HER2 tyrosine kinase inhibition. Oncogene. 30, 4163–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romond, E.H. , Perez, E.A. , Bryant, J. , Suman, V.J. , Geyer, C.E. , Davidson, N.E. , Tan-Chiu, E. , Martino, S. , Paik, S. , Kaufman, P.A. , 2005. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 353, 1673–1684. [DOI] [PubMed] [Google Scholar]
- Santarpia, L. , Qi, Y. , Stemke-Hale, K. , Wang, B. , Young, E.J. , Booser, D.J. , Holmes, F.A. , O'Shaughnessy, J. , Hellerstedt, B. , Pippen, J. , 2012 Jul. Mutation profiling identifies numerous rare drug targets and distinct mutation patterns in different clinical subtypes of breast cancers. Breast Cancer Res. Treat. 134, (1) 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaltriti, M. , Rojo, F. , Ocana, A. , Anido, J. , Guzman, M. , Cortes, J. , Di Cosimo, S. , Matias-Guiu, X. , Cajal, S. , Arribas, J. , Baselga, J. , 2007. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl. Cancer Inst. 99, 628–638. [DOI] [PubMed] [Google Scholar]
- Scaltriti, M. , Serra, V. , Normant, E. , Guzman, M. , Rodriguez, O. , Lim, A.R. , Slocum, K.L. , West, K.A. , Rodriguez, V. , Prudkin, L. , 2011. Antitumor activity of the Hsp90 inhibitor IPI-504 in HER2-positive trastuzumab-resistant breast cancer. Mol. Cancer Ther. 10, 817–824. [DOI] [PubMed] [Google Scholar]
- Schulz, R. , Marchenko, N.D. , Holembowski, L. , Fingerle-Rowson, G. , Pesic, M. , Zender, L. , Dobbelstein, M. , Moll, U.M. , 2012. Inhibiting the HSP90 chaperone destabilizes macrophage migration inhibitory factor and thereby inhibits breast tumor progression. J. Exp. Med. 209, 275–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan, Y. , Eastwood, M.P. , Zhang, X. , Kim, E.T. , Arkhipov, A. , Dror, R.O. , Jumper, J. , Kuriyan, J. , Shaw, D.E. , 2012. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell. 149, 860–870. [DOI] [PubMed] [Google Scholar]
- Shih, J.Y. , Gow, C.H. , Yu, C.J. , Yang, C.H. , Chang, Y.L. , Tsai, M.F. , Hsu, Y.C. , Chen, K.Y. , Su, W.P. , Yang, P.C. , 2006. Epidermal growth factor receptor mutations in needle biopsy/aspiration samples predict response to gefitinib therapy and survival of patients with advanced non small cell lung cancer. Int. J. Cancer. 118, 963–969. [DOI] [PubMed] [Google Scholar]
- Slamon, D. , Eiermann, W. , Robert, N. , Pienkowski, T. , Martin, M. , Press, M. , Mackey, J. , Glaspy, J. , Chan, A. , Pawlicki, M. , 2011. Adjuvant trastuzumab in HER2-positive breast cancer. N. Engl. J. Med. 365, 1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon, D.J. , Leyland-Jones, B. , Shak, S. , Fuchs, H. , Paton, V. , Bajamonde, A. , Fleming, T. , Eiermann, W. , Wolter, J. , Pegram, M. , 2001. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that over expresses HER2. N. Engl. J. Med. 344, 783–792. [DOI] [PubMed] [Google Scholar]
- Solit, D.B. , Zheng, F.Z.F. , Drobnjak, M. , Munster, P.N. , Higgins, B. , Verbel, D. , Heller, G. , Tong, W. , Cordon-Cardo, C. , Agus, D.B. , 2002. 17-allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin. Cancer Res. 8, 986–993. [PubMed] [Google Scholar]
- Tikhomirov, O. , Carpenter, G. , 2003. Identification of ErbB-2 kinase domain motifs required for geldanamycin-induced degradation. Cancer Res. 63, 39–43. [PubMed] [Google Scholar]
- Tsao, M.S. , Sakurada, A. , Cutz, J.C. , Zhu, C.Q. , Kamel-Reid, S. , Squire, J. , Lorimer, I. , Zhang, T. , Liu, N. , Daneshmand, M. , 2005. Erlotinib in lung cancer – molecular and clinical predictors of outcome. N. Engl. J. Med. 353, 133–144. [DOI] [PubMed] [Google Scholar]
- Wang, S.E. , Narasanna, A. , Perez-Torres, M. , Xiang, B. , Wu, F.Y. , Yang, S. , Carpenter, G. , Gazdar, A.F. , Muthuswamy, S.K. , Arteaga, C.L. , 2006. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 10, 25–38. [DOI] [PubMed] [Google Scholar]
- Weber, F. , Fukino, K. , Sawada, T. , Williams, N. , Sweet, K. , Brena, R.M. , Plass, C. , Caldes, T. , Mutter, G.L. , Villalona-Calero, M.A. , Eng, C. , 2005. Variability in organ-specific EGFR mutational spectra in tumour epithelium and stroma may be the biological basis for differential responses to tyrosine kinase inhibitors. Br. J. Cancer. 92, 1922–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. , Huang, W.C. , Li, P. , Guo, H. , Poh, S.B. , Brady, S.W. , Xiong, Y. , Tseng, L.M. , Li, S.H. , Ding, Z. , 2011. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 17, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Gureasko, J. , Shen, K. , Cole, P.A. , Kuriyan, J. , 2006. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 125, 1137–1149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data