

Abstract
Background
Adriamycin (ADR) is a drug used clinically for anticancer treatment; however, it causes adverse effects in the liver. The mechanism by which these adverse effects occur remains unclear, impeding efforts to enhance the therapeutic effects of ADR. Its hepatotoxicity might be related to increasing reactive oxygen species (ROS) and mitochondrial dysfunction. The interaction between ADR and the local renin-angiotensin system (RAS) in the liver is unclear. ADR might activate the RAS. Angiotensin-II (Ang-II) leads to ROS production and mitochondrial dysfunction. In the present study we investigated whether ADR’s hepatotoxicity interacts with local RAS in causing oxidative stress resulting from mitochondrial dysfunction in the rat liver.
Material/Methods
Rats were divided into 5 groups: control, ADR, co-treated ADR with captopril, co-treated ADR with Aliskiren, and co-treated ADR with both captopril and Aliskiren. Mitochondria and cytosol were separated from the liver, then biochemical measurements were made from them. Mitochondrial membrane potential (MMP) and ATP levels were evaluated.
Results
ADR remarkably decreased MMP and ATP in liver mitochondria (p<0.05). Co-administration with ADR and Aliskiren and captopril improved the dissipation of MMP (p<0.05). The decreased ATP level was restored by treatment with inhibitors of ACE and renin.
Conclusions
Angiotensin-II may contribute to hepatotoxicity of in the ADR via mitochondrial oxidative production, resulting in the attenuation of MMP and ATP production.
MeSH Keywords: Angiotensin II; Doxorubicin; Membrane Potential, Mitochondrial; Oxidative Stress
Background
During the last 50 years nearly 1000 drugs have been identified as possible causes of hepatic injury, termed drug-induced hepatotoxicity [1], especially adriamycin (ADR) [2]. Cancer is one of the most of common causes of death in the world [3], and the prevalence of ADR-induced hepatotoxicity, therefore, is increasing.
While ADR is the most effective drug in cancer tissue [3], it has, unfortunately, some adverse effects in noncancerous tissue, including liver, heart, kidney, brain, immune system, and testis. These adverse effects limit clinical use of the drug [3,4]. It is unclear why the liver is a target of ADR’s adverse effects. One reason may be that the liver is the primary involved in detoxification of exogenous and endogenous toxins, especially toxic chemicals, probably resulting in hepatotoxicity [5]. Hepatic damage is irreversible in a dose-dependent manner [4,6]. Another reason is that reactive oxygen species (ROS) are related toinvolved in adverse effects in normal liver tissue [5,6]. Multiple studies have reported that ADR’s toxic effects could relate to generating oxidative stress in the normal liver; therefore, some antioxidants (e.g., selenium, Vitamin E, and NAC) can decrease ADR-induced liver damage [3]. Also, it has been well documented that ADR drastically increases lipid oxidation and mitochondrial ROS content, and decreases liver antioxidant enzymes and mitochondrial function [7]. Other factors contributing to organ toxicity include ADR generation of the inflammatory cascade, and, eventually, programmed cellular death (apoptosis). To enhance the therapeutic effects and decrease the toxic adverse effects of ADR, it is essential to identify and characterize the mechanisms by which these effects occur in the liver.
ADR’s hepatotoxicity has been shown to be associated with oxidative stress linked to redox-cycling of the drug. The mechanism of generation of oxidative stress induced by ADR in liver tissue has not been completely defined. Two different mechanisms have been identified. One is that the ADR redox-cycling may be triggered by 1-electron reduction with the formation of ADR radical. The reaction is catalyzed by various enzymes (e.g., NADPH-cytochrome P450 reductases, NOS, NADPH oxidase, and catalase) [4,6]. Although reoxygenation is needed to convert it from its radical to its nonradical form, the superoxide radical is simultaneously manufactured. Subsequently, repeating this reaction many times could lead to oxidative stress. Moreover, those enzymes caused in ADR radical production have been shown to be highly concentrated in hepatocytes. The second mechanism is a reaction of ADR with iron. ADR radicals delocalized Fe2+ ADR from ferritin and generate H2O2, eventually causing oxidant injury in the liver [6]. It has been suggested that the liver is especially involved in ADR radical production [4].
The renin-angiotensin system is frequently activated in patients with chronic liver diseases, such as cirrhosis (which contributes to fibrosis progression), hepatocellular carcinoma [8–10], and liver inflammation [11]. Angiotensin II (Ang-II) is an important effector peptide of this system [10]. Ang-II has some crucial physiological functions, such as cell contraction, cell growth/hypertrophy, apoptosis, differentiation, and secretion of inflammatory cytokines [12,13]. Ang-II actions are mainly mediated by its 2 sub-receptors – the Ang-II type 1 receptor (AT-1) and the Ang-II type 2 receptor (AT-2). These 2 subunits mediate the effects of Ang-II on various organs, and AT-2 (mostly in fetal cells) is less common than AT-1 [10,12,14]. Increased systemic Ang-II was reported to augment and promote inflammation and oxidative stress [13]. In target cells, Ang-II induces free radical formation and oxidative stress, stimulating redox-sensitive intracellular pathways. In addition to systemic the RAS, virtually every organ system in the human body has been shown to possess a local Ang signaling system in peripheral tissues (e.g., liver, vasculature, kidneys, adrenal glands, heart, and immune cells [15]. The systemic RAS is activated in patients with cirrhosis and the local RAS is induced in fibrotic livers and activated hepatic stellate cells (HSCs) [13].
It has been shown that Ang-II might modulate mitochondrial membrane potential and transcription of respiratory chain subunits, and may stimulate the production of ROS in mitochondria [16], thus leading to mitochondrial failure in cardiac, renal, and vascular smooth muscle cells [17,18]. Mitochondrial failure probably causes the massive production of ROS [11,18].
According to current understanding of ADR and RAS, ADR might crosstalk with the local RAS in liver tissue to amplify its adverse effects. The hypothesis in the present study, therefore, was that ADR cooperates with the local RAS in the liver tissue to generate free radical formation, resulting in oxidative stress and attenuating mitochondrial function. Hence, the aim of the study was to investigate the potential prophylactic effects of attenuated angiotensin-II production in the liver against mitochondrial dysfunction in rats with ADR-induced oxidative stress.
Material and Methods
Animals
All experimental procedures were carried out in strict accordance with the Animal Experimentation local Ethics Committee of the Erciyes University. Thirty-five male Sprague-Dawley rats were randomly divided into 5 treatment groups as shown Figure 1: Control (n=7), ADR (n=7), CAP (Captopril + ADR, n=7), AL (Aliskiren + ADR, n=7), and CAP + AL (Captopril + Aliskiren + ADR, n=7). ADR (Adriamycin HCl, Adriblastina vial 10 mg, Pharmacia) was administered in 4 equal injections (each intraperitoneal injection containing 4 mg/kg in saline, every other day, to reach a total dose of 16 mg/kg) during 7 days. On the same day the Control group received the same volume of physiological saline. Captopril was intragastrically administered (10 mg/kg, every day for 7 days). Aliskiren was intragastrically administered (50 mg/kg, every day for 7 days; Figure 1). After the last drug treatment, liver tissues were collected, and then kept at −80 oC until use; cytosol and mitochondria were separated by centrifugation for use in later biochemical assays.
Figure 1.
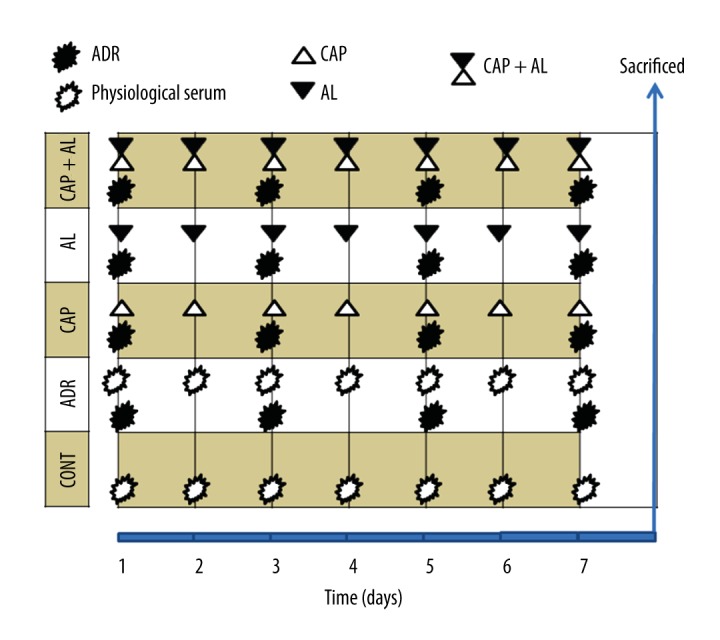
Experimental design of groups. CONT – Control group received only serum physiologic; ADR – Adriamycin group; CAP – Co-treatment ADR with ACE inhibition group by captopril; AL – Co-treatment ADR with Renin inhibition group by aliskiren; CAP + AL group – Co-treatment ADR with both ACE inhibition group by captopril and renin inhibition group by aliskiren; S – Physiological Serum. All animals were sacrificed on the 9th day (24 hours after the last dose).
Preparation of mitochondria and cytosol
Mitochondria from liver tissues were isolated as described before [19–21]. The mitochondria and cytosol were kept at −80°C until use.
Measurement of mitochondrial membrane potential in (MMP) mitochondria from liver
The fluorescent mitochondrial-specific cationic dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl benzimidazolylcarbocyanine iodide (JC-1) was used for assessment of MMP according to the manufacturer’s protocol.
Determination of ATP content in liver mitochondria
To measure ATP content and ADR-toxicity in the liver, a bioluminescent kit was used according to the manufacturer’s protocol. The kit is based upon the bioluminescence of ATP that is present in all metabolically active cells.
Biochemical Studies
Evaluation of total antioxidant status
Total antioxidant status (TAS) of the liver’s cytosol and mitochondria were measured according to manufacturer’s protocol based on colorimetric measurement (Rel Assay Kit, Turkey). The results are expressed in mmol Trolox equiv./L.
Evaluation of total oxidant status
Total oxidant status (TOS) of the liver’s cytosol and mitochondria were measured using a Rel Assay Kit (Rel Assay Kit, Turkey), based on colorimetric measurement. The results are expressed in μmol H2O2/L.
Calculation of oxidative stress index
The TOS-to-TAS ratio was used as the oxidative stress index (OSI). The OSI value was calculated as follows: OSI = (TOS)/(TAS) (μmol/L/ μmol Trolox equivalent/L).
Statistical data analysis
Statistical analyzes were conducted using Excel and SPSS version 21. All results are expressed as mean ±SEM. Comparison among different groups were made using multiple analyzes of variance (ANOVA), followed by a post hoc protected Tukey test p<0.05 was considered to be significant.
Result
Modulation of oxidative stress by inhibition of angiotensin-II production in liver tissue exposed to Adriamycin
ADR-induced toxicity is well known to be associated with increased oxidative stress, resulting from increased levels of reactive oxygen species and decreased levels of antioxidants. TAS and TOS are excellent indicators for determining OSI. They were measured as the type of mitochondria and cytosol from liver tissue in the present study. Mitochondrial and cytosolic TOS were significantly increased by ADR compared to CONT (p<0.05; Figure 2A, 2B). The inhibition of Ang-II in the liver was restored to mitochondrial at CAP and cytosolic TOS at CAP and CAP + AL, (p<0.05; Figure 2A, 2B). The elevation of TOS in mitochondria and cytosol could be related to Ang-II production in liver tissue.
Figure 2.

The effect of angiotensin-II on mitochondrial (A) and cytosolic (B) total oxidant status in rats with adriamycin-induced oxidative stress. TOS – Total oxidant status; CONT – Control group received only serum physiologic; ADR – Adriamycin group; CAP – Co-treatment ADR with ACE inhibition group by captopril; AL – Co-treatment ADR with renin inhibition group by aliskiren; CAP + AL group – Co-treatment ADR with both ACE inhibition group by captopril and Renin inhibition group by aliskiren. * p<0.05 vs. CONT; ** p<0.05 vs. ADR. All data are expressed as mean ±SEM.
ADR decreases TAS in mitochondria and cytosol, but not significantly compared to the Control group. The inhibition of Ang-II tended to increase of all groups, but not significantly (Figure 3A, 3B).
Figure 3.

The effect of angiotensin-II on mitochondrial (A) and cytosolic (B) total antioxidant status in rats with adriamycin-induced oxidative stress. TAS – Total antioxidant status; CONT – Control group received only serum physiologic; ADR – Adriamycin group; CAP – Co-treatment ADR with ACE inhibition group by captopril; AL – Co-treatment ADR with Renin inhibition group by aliskiren; CAP + AL group – Co-treatment ADR with both ACE inhibition group by captopril and renin inhibition group by aliskiren. All data are expressed as mean ±SEM.
The level of oxidative stress was calculated by using the ratio of TOS to TAS in mitochondria and cytosol. OSI in mitochondria and cytosol were significantly elevated in the ADR group (p<0.05 vs. CONT; Figure 4A, 4B). However, the inhibition of local Ang-II production by renin and/or ACE inhibitors led to a significant decrease in mitochondrial OSI at CAP and CAP + AL groups and cytosolic OSI at all groups, resulting from decreasing TOS (p<0.05 vs. ADR group, Figure 4A, 4B).
Figure 4.

Effect of angiotensin-II on mitochondrial (A) and cytosolic (B) oxidative stress index in rats with adriamycin-induced oxidative stress. OSI – Oxidative stress index; CONT – Control group received only serum physiologic; ADR – Adriamycin group, CAP – Co-treatment ADR with ACE inhibition group by captopril; AL – Co-treatment ADR with Renin inhibition group by aliskiren; CAP + AL group – Co-treatment ADR with both ACE inhibition group by captopril and renin inhibition group by aliskiren. * p<0.05 vs. CONT; ** p<0.05 vs. ADR. All data are expressed as mean ±SEM.
Restoration of mitochondrial dysfunction by inhibition of angiotensin-II production in liver tissue-exposed to Adriamycin
Mitochondria the main subcellular targets of ADR-toxicity (22). Attenuation of mitochondrial dysfunction might efficiently counteract ADR’s liver toxicity. Therefore, mitochondrial function was determined by measurement of MMP and ATP production. MMP was estimated via the ratio of red to green fluorescence by using a special mitochondrial cationic dye (JC-1) to demonstrate ADR’s toxicity in mitochondria. ADR caused sharp dissipation of MMP (p<0.05 vs. ADR group; Figure 5). In parallel, ADR’s effect on MMP significantly decreased ATP production (p<0.05 vs ADR group; Figure 6). However, attenuation of local Ang-II production by renin and/or ACE inhibitors significantly increased ATP production in liver mitochondria (p<0.05; Figure 6). These data demonstrate that angiotensin-II might be able to cooperate with ADR to derive oxidative, stress causing reduced mitochondrial membrane potential, resulting in minimizing ATP production in liver tissue.
Figure 5.
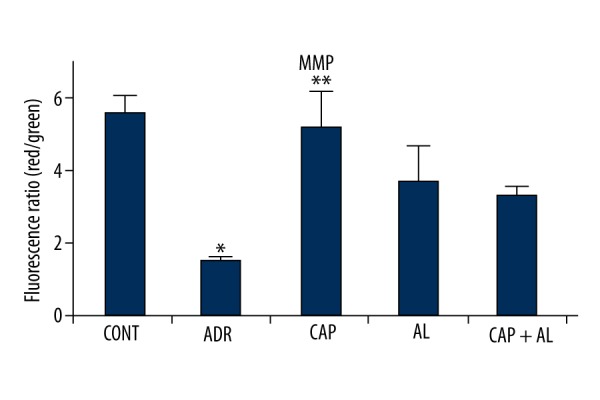
The effect of angiotensin-II on mitochondrial membrane potential in rats with oxidative stress induced by Adriamycin. MMP – Mitochondrial membrane potential; CONT – Control group received only serum physiologic; ADR – Adriamycin group; CAP – Co-treatment ADR with ACE inhibition group by captopril; AL – Co-treatment ADR with Renin inhibition group by aliskiren; CAP + AL group – Co-treatment ADR with both ACE inhibition group by captopril and renin inhibition group by aliskiren. * p<0.05 vs. CONT; ** p<0.05 vs. ADR. All data are expressed as mean ±SEM.
Figure 6.
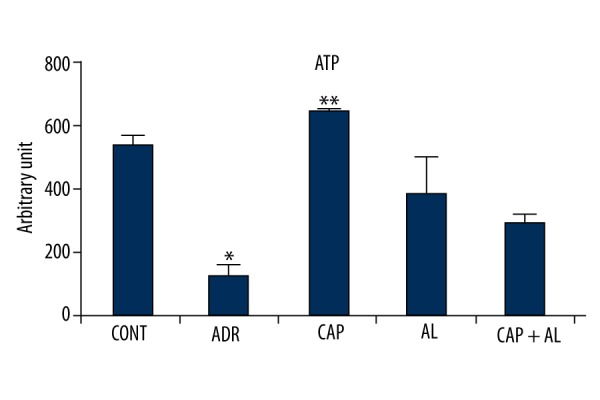
The effect of angiotensin-II on ATP level in rats with oxidative stress induced by Adriamycin. CONT – Control group received only serum physiologic; ADR – Adriamycin group; CAP – Co-treatment ADR with ACE inhibition group by captopril; AL – Co-treatment ADR with Renin inhibition group by aliskiren; CAP + AL group – Co-treatment ADR with both ACE inhibition group by captopril and renin inhibition group by aliskiren. * p<0.05 vs. CONT; ** p<0.05 vs. ADR. All data are expressed as mean ±SEM.
Discussion
Because both ADR and Ang-II play important roles in oxidative stress related to some liver pathologies [3], we hypothesized that ADR may cause liver damage via local Ang-II production. Several lines of evidence from our current study support this hypothesis: inhibition of Ang-II production attenuates several markers of ADR-induced liver injury and decreases TOS, resulting in decreased oxidative stress and increased MMP and ATP in rats exposed to ADR. These results suggest that Ang-II cooperates with ADR’s hepatotoxicity in rats, confirming the potential health benefit of inhibition of Ang-II in vivo against oxidative stress induced by ADR.
It is clear that ADR causes irreversible liver damage, triggering apoptotic processes in normal liver tissue. ADR-mediated hepatotoxicity has been shown to include focal damage in hepatocytes, vascular damage, and steatosis. Thus, its hepatotoxicity was ascribed to subcellular hepatic alterations, including polymorphic mitochondria and cytoplasmic vacuolization. Various compounds (most of them antioxidants, such as vitamin E, erdosteine, cystathionine, ator [5], and catechin in ADR-induced hepatotoxicity) have been reported to prevent liver injury induced by oxidative stress [6].
Recent studies have been focused on better understanding oxidative stress mediated by ADR’s toxicity and the outcome of adverse effects in heart, liver, and kidney [4]. During the ADR redox-cycling process, ADR gains an electron and is reduced into ADR radical-catalyzed by several endogenous enzymes, including P-450 [5]. So, it was not so surprisingly that the liver has more P-450 enzyme than other tissues due to its detoxification activity [5]. Then, ADR shifts to its original form by re-oxygenation, generating reactive oxygen radicals. Eventually, these radicals promote oxidative stress through altering the redox status of essential proteins [5]. In agreement with this, a previous study reported a significant increase in MDA levels, which is a good indicator of oxidative damage. The same study also showed significant attenuation of some antioxidant levels (e.g., SOD, GSH, and GSH-Px) in rat liver treated with ADR [23]. Another study reported that ADR increased ROS concentration and oxidative stress biomarkers, as well as decreasing antioxidant enzyme activities and mitochondrial function in the liver [7]. As shown Figures 2–4, all results are consistent with the current study’s results.
Redox cycling occurs in the cytoplasm, endoplasmic reticulum, and (especially) in mitochondria [4]. The compartmentalization of these kinds of subcellular oxidative stress is important for cell functions, especially because mitochondrial oxidative stress can destroy mitochondrial function [24]. We separately measured oxidative stress (OSI, TOS, and TAS) in cytosol and mitochondria compartments in the present study, because oxidant radicals may freely cross intracellular membranes; therefore, subcellular compartmentalization of antioxidants may significantly modulate the harmful activity of ROS in subcellular compartments. Therefore, the level of mitochondrial oxidative stress is probably more important than the oxidative stress in all other cell organelles [4]. According to this assumption, the mitochondrial redox status might affect cellular redox status [25]. Therefore, the deficiency of mitochondrial antioxidant defense might increase the release of oxidant from the mitochondrial matrix, which can consequently oxidize some cytoplasmic proteins and cell signaling, as well as membrane pumps proteins. Moreover, the disruption of the mitochondrial membrane potential also is important in regulating necrosis and apoptosis [4,26]. Our results show that the mitochondria are the dominant source of oxidative stress-induced by ADR in the liver. Mitochondrial dysfunction associated with ADR-related toxicities might cause the observed change in activities of typical markers of oxidative status in damaged liver tissue. The decrease of glutathione was reported after ADR administration, causing oxidative stress [6].
Hepatocytes are the likely targets of ROS attacks in the failing liver. Consequently, as an important source of ROS production, mitochondria could also be a primary target susceptible to ROS attacks. The defects in the mitochondrial structural design would lead to the adaptation of mitochondrial metabolism, resulting in decreased activity of mitochondrial enzymes in the liver exposed to ADR, hence becoming a principal contributor to intrinsic cell dysfunction. Mitochondrial degeneration and dysfunction have been reported in ADR administration. Increased levels of oxidative stress were observed in ADR-treated rats [6]. Free radicals also stimulate the pro-apoptotic protein (e.g., Bax), resulting in loss of hepatocyte [5]. The other reason hepatocytes are reduced by ADR is associated with redox alteration, causing irreversible liver toxicity and its changes are reported to be important in regulating hepatocyte division. The changing of redox potential may be common attributing for cells division the fact that ROS have been suggested to modulate the cell cycle in many cell lines [26]. Loss of hepatocytes probably leads to even worse liver toxicity of ADR, although it has been repored that some aspect of angiotensin signaling inhibition could be involved in alleviating adriamycin-induced cardiomyopathy [22] and renopathy [27].
Alterations in RAS have frequently been implicated in the pathophysiology of various diseases involving the heart, lung [28], kidney [27], and liver [10,29–31]. Ang-II, an essential member of the RAS, handles both physiologic [14] and pathophysiologic [13] effects of RAS. It is associated with liver fibrogenesis [31], cirrhosis, portal hypertension, and hepatic ischemia/reperfusion injury. Ang-II leads to apoptosis [32], tumor vessel growth, tumor invasion and metastasis, and immunosuppression, leading to the development of tumors [10]. In the other words, Ang-II also has undesirable effects, like ADR. Furthermore, recent studies have reported that AT-1 overexpression may be associated with hepatocellular, non-small cell lung, gastric, breast, ovarian, bladder, pancreatic, and prostate cancers [10,12]. Evidence shows that Ang-II plays a role in liver inflammation in humans and rodents (11). The importance of RAS has been shown in development of hepatic fibrosis, and the benefits of modulation of ACE gene expression have been reported [28]. Blockade of RAS can inhibit tumor growth. CAP, an angiotensin-converting enzyme blocker, was able to attenuate growth in a murine model of colorectal cancer liver metastases [33].
Besides the systemic RAS, many peripheral tissues can generate their own RAS components. This so-called local or tissue RAS has various roles, such as the promotion of inflammation and fibrosis. A recent study [11] reported that local RAS is present in the liver, heart, kidney, lacrimal gland, and lung [34]. Each kind of RAS expresses itself not only in the fibrotic liver [35], but also in liver parenchymal cells, including hepatocytes. Angiotensin-converting enzyme (ACE) is especially up-regulated. Activation of HSCs express the elements of RAS and thereby produces Ang-II [35]. Activation of the RAS is induced by a variety of local and systemic stimuli [36]. This local angiotensin is implicated in fibrotic pathogenesis in the liver [34,35]. Also, activated AT-1 expresses on the surface of HSCs and assembles into the hepatic fibrotic area [35]. Local or tissue RAS might be highly related to systemic RAS under pathophysiologic situations [11,13], or independent of the circulation system [37]. Therefore, in the current study we used 2 drugs – renin and ACE inhibitors – to prevent the local RAS, for evaluation of the relationship between ADR’s hepatotoxic actions and Ang-II. However, there are limited studies on neurohormonal activation of ADR on tissues. To the best of our knowledge, there is no published data on the association of ADR and Ang-II in liver mitochondrial dysfunction. Hence, it is not easy to explain the physiologic relevance or the underlying mechanism of the present observations; these deserve attention in future studies. However, previous research has suggested that increased intracellular Ang-II in diabetic rat heart was associated with enhanced cardiac myocyte apoptosis, fibrosis, and oxidative stress. The latter conditions were alleviated better by a renin inhibitor, which blocks the intracellular RAS, compared to Ang-II receptors and ACE inhibitors [37]. The renin inhibitor, aliskiren, was reported to enter the cell and inhibit the intracellular production of angiotensin II [38]. AL completely prevented hyperglycemia-induced Ang-II synthesis. The above findings suggest similar partitioning of aliskiren in the heart, which might have resulted in sufficiently high intracellular levels of aliskiren to inhibit rat renin in our studies. It is reported that a renin inhibitor prevents both intracellular and extracellular Ang-II synthesis [39]. Consistent with our previous study, we observed that aliskiren had some effective impacts on preventing oxidative stress in kidney-exposed to ADR’s toxicity [21]. An earlier study showed that hepatic fibrosis was significantly decreased in the AL group. The same study also reported attenuated progression of hepatic fibrosis by inhibition of activated hepatic stellate and Kupffer cells and by reducing oxidative stress in the AL group [40]. Additionally, Ang-II synthesis by cardiac fibroblasts, extracellular as well as intracellular, is catalyzed by ACE. Thus, ACE inhibitors can prevent Ang-II synthesis by cardiac fibroblasts, and Ang-II receptor inhibitors can block autocrine/paracrine effects of extracellular Ang-II [39]. Another study has been shown that inhibition of locally produced Ang-II could decrease inflammation and fibrosis in kidney, lung, and liver tissues. Because ACE and AT-1 plays a key role in liver fibrosis, RAS was reported to be highly expressed in activated human hepatic stellate cells [34]. Moreover, an AT-1 antagonist, losartan, also reduces the extent of liver damage [11].
Interference of Ang-II in Adriamycin hepatotoxicity is associated with stimulating ROS production in hepatocytes, mainly via AT-1 and mitochondria-derived pathway because losartan can inhibit Ang-II-stimulated ROS generation [11]. These results are in line with our findings in the present study that attenuation of Ang-II production in the liver can modulate ROS production, arising from attenuation of mitochondrial dysfunction in liver exposed to ADR.
According to our current results, Ang-II cooperates with ADR to produce more oxidant, causing its hepatotoxicity (Figure 2) but not attenuation of antioxidants (Figure 3). This discrepancy may be caused by 3 things. The first is that crosstalk between ADR and Ang-II might be based more on oxidant production than changing of antioxidant. Most hepatic damage by Ang-II is known to be related to elevated oxidants [13]. The second cause depends on the first cause. Since the liver plays an important role in pro-oxidants, detoxification is known to have the highest content of antioxidants [14], a decline antioxidant levels in the liver tissue may need enormous oxidant production to significant alternation of oxidant. Thirdly, our study investigated acute hepatotoxicity induced by ADR and determined that time is an important factor in ADR’s cytotoxicity [41]. More time may be needed to change the highest content of antioxidants in liver tissue, consistant with our results that antioxidant level was not significantly decreased in any experimental groups. Moreover, the role of the RAS in this experimental model suggests that intrahepatic RAS is markedly upregulated, and RAS inhibition attenuates ADR’s hepatotoxicity. Our data are in agreement with other reports [42,43]. Moreover, it is possible to have an interaction between the systemic and intrahepatic RAS [13]. Therefore, the present study used ACE and renin inhibitors to block RAS, especially local ones.
Since insufficient work has been done on mitochondria to clarify this issue, the full of molecular mechanisms underlying the link between ADR and Ang-II are still unclear. However, AngII has been shown to alter MMP [18,44] and to stimulate ROS production [16]. This overproduction of mitochondrial ROS by Ang-II has been implicated in renal toxicity of ADR [21] and also neurodegenerative diseases, ischemia-reperfusion injury, atherosclerosis, and aging. Therefore, if Ang-II production could be inhibited, the mitochondrial dysfunction might be restored. The rising evidence of the beneficial effect of Ang-II inhibition suggests that a parallel intra-mitochondrial RAS might be capable of AngII synthesis to modulate ROS activity [44]. Because of more exposure to a high rate of oxidative damage, mitochondria must be destroyed and replaced [44]. A previous study suggested that mitochondria might have their functional RAS, leading to the intra-mitochondrial generation of An-II. It has been shown to alter mitochondrial number [44] or traffic to the surface of rodent mitochondria as well. An intracellular RAS system might have a role as a local amplifier of RAS signaling for attenuating a certain amount of systemic Ang-II [16]. The importance of mitochondrial RAS function has been not been defined yet [16]. Ang-II has been suggested to have some role in development of mitochondrial failure, resulting in overloading Ca2+, depolarization of MMP, and reducing ATP production [16,44] in liver [16] and kidney exposed to Adriamycin toxicity [21]. Also, ADR was found to accumulate in the mitochondria [19,20,27]. Eventually, intracellularly-produced or imported Ang-II might directly effect mitochondria.
Conclusions
The present study provides a novel mechanism of ADR hepatotoxicity in which ADR might crosstalk with local RAS, especially mitochondria, in the rat the liver. Local Ang-II could amplify ADR’s adverse effect on liver tissue. Therefore, our current research strongly suggests that protecting against ADR’s adverse effect on liver tissue can pharmacologically antagonize local angiotensin-II pathological effects, including oxidative stress, decreasing mitochondrial membrane potential and ATP production. Further studies are necessary to test the beneficial effect of Ang-II inhibition on ADR’s hepatotoxicity and the other cytotoxicity in animal and human studies.
Acknowledgements
The authors wish to thank Associate Professor Mukerrem Betul Yerer Aycan for helping technical assistants.
Footnotes
Source of support: This study was supported financially by the Research Foundation of Erciyes University
Disclosures
The authors have no conflicts of interest to disclose.
References
- 1.Zimmerman HJ. Drug-induced liver disease. Clin Liver Dis. 2000;4(1):73–96. doi: 10.1016/s1089-3261(05)70097-0. [DOI] [PubMed] [Google Scholar]
- 2.Henninger C, Huelsenbeck J, Huelsenbeck S, et al. The lipid lowering drug lovastatin protects against doxorubicin-induced hepatotoxicity. Toxicol Appl Pharmacol. 2012;261(1):66–73. doi: 10.1016/j.taap.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 3.Wang B, Ma Y, Kong X, et al. NAD(+) administration decreases doxorubicin-induced liver damage of mice by enhancing antioxidation capacity and decreasing DNA damage. Chem Biol Interact. 2014;212:65–71. doi: 10.1016/j.cbi.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 4.Dudka J, Gieroba R, Korga A, et al. Different effects of resveratrol on dose-related Doxorubicin-induced heart and liver toxicity. Evid Based Complement Alternat Med. 2012;2012:606183. doi: 10.1155/2012/606183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Moselhy MA, El-Sheikh AA. Protective mechanisms of atorvastatin against doxorubicin-induced hepato-renal toxicity. Biomed Pharmacother. 2014;68(1):101–10. doi: 10.1016/j.biopha.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Injac R, Perse M, Obermajer N, et al. Potential hepatoprotective effects of fullerenol C60(OH)24 in doxorubicin-induced hepatotoxicity in rats with mammary carcinomas. Biomaterials. 2008;29(24–25):3451–60. doi: 10.1016/j.biomaterials.2008.04.048. [DOI] [PubMed] [Google Scholar]
- 7.Diamanti J, Mezzetti B, Giampieri F, et al. Doxorubicin-induced oxidative stress in rats is efficiently counteracted by dietary anthocyanin differently enriched strawberry (Fragaria x ananassa Duch.) J Agric Food Chem. 2014;62(18):3935–43. doi: 10.1021/jf405721d. [DOI] [PubMed] [Google Scholar]
- 8.Li YS, Ni SY, Meng Y, et al. Angiotensin II facilitates fibrogenic effect of TGF-beta1 through enhancing the down-regulation of BAMBI caused by LPS: a new pro-fibrotic mechanism of angiotensin II. PLoS One. 2013;8(10):e76289. doi: 10.1371/journal.pone.0076289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Granzow M, Schierwagen R, Klein S, et al. Angiotensin-II type 1 receptor-mediated Janus kinase 2 activation induces liver fibrosis. Hepatology. 2014;60(1):334–48. doi: 10.1002/hep.27117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oikawa H, Maesawa C, Tatemichi Y, et al. A disintegrin and metalloproteinase 17 (ADAM17) mediates epidermal growth factor receptor transactivation by angiotensin II on hepatic stellate cells. Life sciences. 2014;97(2):137–44. doi: 10.1016/j.lfs.2013.12.028. [DOI] [PubMed] [Google Scholar]
- 11.Zhao J, Liu J, Pang X, et al. Angiotensin II induces C-reactive protein expression via AT1-ROS-MAPK-NF-kappaB signal pathway in hepatocytes. Cell Physiol Biochem. 2013;32(3):569–80. doi: 10.1159/000354461. [DOI] [PubMed] [Google Scholar]
- 12.Duan YF, Li XD, Zhu F, Zhang F. Expression and clinical significance of angiotensin II type 1 receptor in human hepatocellular carcinoma. Exp Ther Med. 2014;7(2):323–28. doi: 10.3892/etm.2013.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bataller R, Gabele E, Parsons CJ, et al. Systemic infusion of angiotensin II exacerbates liver fibrosis in bile duct-ligated rats. Hepatology. 2005;41(5):1046–55. doi: 10.1002/hep.20665. [DOI] [PubMed] [Google Scholar]
- 14.Ali MA, Kazzam E, Amir N, et al. Effects of dehydration and blockade of angiotensin II AT1 receptor on stress hormones and anti-oxidants in the one-humped camel. BMC Vet Res. 2013;9:232. doi: 10.1186/1746-6148-9-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abadir PM, Foster DB, Crow M, et al. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci USA. 2011;108(36):14849–54. doi: 10.1073/pnas.1101507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Astin R, Bentham R, Djafarzadeh S, et al. No evidence for a local renin-angiotensin system in liver mitochondria. Sci Rep. 2013;3:2467. doi: 10.1038/srep02467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang GX, Lu XM, Kimura S, Nishiyama A. Role of mitochondria in angiotensin II-induced reactive oxygen species and mitogen-activated protein kinase activation. Cardiovasc Res. 2007;76(2):204–12. doi: 10.1016/j.cardiores.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 18.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102(4):488–96. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 19.Dursun N, Taskin E, Yerer Aycan MB, Sahin L. Selenium-mediated cardioprotection against adriamycin-induced mitochondrial damage. Drug Chem Toxicol. 2011;34(2):199–207. doi: 10.3109/01480545.2010.538693. [DOI] [PubMed] [Google Scholar]
- 20.Taskin E, Dursun N. The protection of selenium on adriamycin-induced mitochondrial damage in rat. Biol Trace Elem Res. 2012;147(1–3):165–71. doi: 10.1007/s12011-011-9273-9. [DOI] [PubMed] [Google Scholar]
- 21.Taskin E, Ozdogan K, Kunduz Kindap E, Dursun N. The restoration of kidney mitochondria function by inhibition of angiotensin-II production in rats with acute adriamycin-induced nephrotoxicity. Ren Fail. 2014;36(4):606–12. doi: 10.3109/0886022X.2014.882737. [DOI] [PubMed] [Google Scholar]
- 22.Octavia Y, Tocchetti CG, Gabrielson KL, et al. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52(6):1213–25. doi: 10.1016/j.yjmcc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Tulubas F, Gurel A, Oran M, et al. The protective effects of omega-3 fatty acids on doxorubicin-induced hepatotoxicity and nephrotoxicity in rats. Toxicol Ind Health. 2015;31(7):638–44. doi: 10.1177/0748233713483203. [DOI] [PubMed] [Google Scholar]
- 24.Dong W, Cheng S, Huang F, et al. Mitochondrial dysfunction in long-term neuronal cultures mimics changes with aging. Med Sci Monit. 2011;17(4):BR91–96. doi: 10.12659/MSM.881706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stefano GB, Snyder C, Kream RM. Mitochondria, chloroplasts in animal and plant cells: Significance of conformational matching. Med Sci Monit. 2015;21:2073–78. doi: 10.12659/MSM.894758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han D, Hanawa N, Saberi B, Kaplowitz N. Mechanisms of liver injury. III. Role of glutathione redox status in liver injury. Am J Physiol Gastrointest Liver Physiol. 2006;291(1):G1–7. doi: 10.1152/ajpgi.00001.2006. [DOI] [PubMed] [Google Scholar]
- 27.Taskin E, Ozdogan K, Kunduz Kindap E, Dursun N. The restoration of kidney mitochondria function by inhibition of angiotensin-II production in rats with acute adriamycin-induced nephrotoxicity. Ren Fail. 2014;36(4):606–12. doi: 10.3109/0886022X.2014.882737. [DOI] [PubMed] [Google Scholar]
- 28.Shahid SM, Fatima SN, Mahboob T. Angiotensin converting enzyme (ACE) gene expression in experimentally induced liver cirrhosis in rats. Pak J Pharm Sci. 2013;26(5):853–57. [PubMed] [Google Scholar]
- 29.He C, Miao X, Li J, Qi H. Angiotensin II induces endothelin-1 expression in human hepatic stellate cells. Dig Dis Sci. 2013;58(9):2542–49. doi: 10.1007/s10620-013-2685-y. [DOI] [PubMed] [Google Scholar]
- 30.Yoshiji H, Noguchi R, Ikenaka Y, et al. Renin-angiotensin system inhibitors as therapeutic alternatives in the treatment of chronic liver diseases. Curr Med Chem. 2007;14(26):2749–54. doi: 10.2174/092986707782360169. [DOI] [PubMed] [Google Scholar]
- 31.Yoshiji H, Kuriyama S, Yoshii J, et al. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology. 2001;34(4 Pt 1):745–50. doi: 10.1053/jhep.2001.28231. [DOI] [PubMed] [Google Scholar]
- 32.Lai HS, Lin WH, Lai SL, et al. Interleukin-6 mediates angiotensinogen gene expression during liver regeneration. PLoS One. 2013;8(7):e67868. doi: 10.1371/journal.pone.0067868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wen SW, Ager EI, Neo J, Christophi C. The renin angiotensin system regulates Kupffer cells in colorectal liver metastases. Cancer Biol Ther. 2013;14(8):720–27. doi: 10.4161/cbt.25092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yaguchi S, Ogawa Y, Shimmura S, et al. Angiotensin II type 1 receptor antagonist attenuates lacrimal gland, lung, and liver fibrosis in a murine model of chronic graft-versus-host disease. PLoS One. 2013;8(6):e64724. doi: 10.1371/journal.pone.0064724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim TK, Kleszczynski K, Janjetovic Z, et al. Metabolism of melatonin and biological activity of intermediates of melatoninergic pathway in human skin cells. FASEB J. 2013;27(7):2742–55. doi: 10.1096/fj.12-224691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee CJ, Subeq YM, Lee RP, et al. Calcitriol decreases TGF-beta1 and angiotensin II production and protects against chlorhexide digluconate-induced liver peritoneal fibrosis in rats. Cytokine. 2014;65(1):105–18. doi: 10.1016/j.cyto.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 37.Kumar R, Thomas CM, Yong QC, et al. The intracrine renin-angiotensin system. Clin Sci. 2012;123(5):273–84. doi: 10.1042/CS20120089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Re R. Intracellular renin-angiotensin system: the tip of the intracrine physiology iceberg. Am J Physiol Heart Circ Physiol. 2007;293(2):H905–6. doi: 10.1152/ajpheart.00552.2007. [DOI] [PubMed] [Google Scholar]
- 39.Singh VP, Le B, Khode R, et al. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes. 2008;57(12):3297–306. doi: 10.2337/db08-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kishina M, Koda M, Kato J, et al. Therapeutic effects of the direct renin inhibitor, aliskiren, on non-alcoholic steatohepatitis in fatty liver Shionogi ob/ob male mice. Hepatol Res. 2014;44(8):888–96. doi: 10.1111/hepr.12186. [DOI] [PubMed] [Google Scholar]
- 41.Aversano RC, Boor PJ. Histochemical alterations of acute and chronic doxorubicin cardiotoxicity. J Mol Cell Cardiol. 1983;15(8):543–53. doi: 10.1016/0022-2828(83)90330-9. [DOI] [PubMed] [Google Scholar]
- 42.Abd El-Aziz MA, Othman AI, Amer M, El-Missiry MA. Potential protective role of angiotensin-converting enzyme inhibitors captopril and enalapril against adriamycin-induced acute cardiac and hepatic toxicity in rats. J Appl Toxicol. 2001;21(6):469–73. doi: 10.1002/jat.782. [DOI] [PubMed] [Google Scholar]
- 43.Toblli JE, Ferder L, Stella I, et al. Enalapril prevents fatty liver in nephrotic rats. J Nephrol. 2002;15(4):358–67. [PubMed] [Google Scholar]
- 44.Re RN, Cook JL. The mitochondrial component of intracrine action. Am J Physiol Heart Circ Physiol. 2010;299(3):H577–83. doi: 10.1152/ajpheart.00421.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]