

Abstract
A series of 3-substituted 5-hydroxy-1,2,4-triazin-6(1H)-one derivatives were designed and synthesized as a new class of D-amino acid oxidase (DAAO) inhibitors. Some of the newly synthesized derivatives showed potent inhibitory activity against human DAAO with IC50 values in the nanomolar range. Among them, 6-hydroxy-3-phenethyl-1,2,4-triazin-5(2H)-one 6b and 3-((6-fluoronaphthalen-2-yl)methylthio)-6-hydroxy-1,2,4-triazin-5(2H)-one 6m were found to be metabolically stable in mouse liver microsomes. In addition, compound 6b was found to be orally available in mice and able to enhance plasma D-serine levels following its co-administration with D-serine compared to the oral administration of D-serine alone.
Keywords: d-Amino acid oxidase, Flavoenzyme, Pharmacokinetics, Schizophrenia, d-Serine
Graphical abstract

D-Amino acid oxidase (DAAO, EC 1.4.3.3) catalyzes the oxidative deamination of neutral D-amino acids including D-serine, a co-agonist at the glycine modulatory site of NMDA receptors. Because of the implication that hypofunction of NMDA receptors plays an important role in the pathophysiology of schizophrenia, there has been considerable efforts in developing DAAO inhibitors as a pharmacological approach for increasing D-serine and facilitating NMDA receptor-mediated neurotransmission. DAAO has been also implicated as a potential therapeutic target for the treatment of chronic pain since hydrogen peroxide, a reactive oxygen species co-generated by DAAO, is believed to contribute to pain hypersensitivity.1 In the past decade, a wide variety of DAAO inhibitors have been identified by several research groups.2, 3 As shown in Figure 1, newer scaffolds of DAAO inhibitors contain a branched chain, which occupies the secondary pocket adjacent to the active site of DAAO recognized by the crystal structure of human DAAO in complex with imino-DOPA.4 For example, our group exploited this secondary binding site using kojic acid derivatives represented by compound 1.5 Similarly, a group at Astellas reported potent DAAO inhibitors including 2a–b based on a 4-hydroxypyridazin-3(2H)-one scaffold with a phenethyl group extending to the secondary binding site.6 2-Substituted 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-diones such as 3 exhibited not only potent DAAO inhibitory activity but also improved metabolic stability compared to 1–2 in liver microsomes.7 This secondary binding site was also exploited by carboxylate-based DAAO inhibitors such as 48 and 5.9 In the search for a new class of DAAO inhibitors, we have examined a variety of scaffolds that may serve as a bioisostere for the carboxylic acid moiety interacting with the active site of DAAO.
Figure 1.

Representative known DAAO inhibitors 1–5 and 3-substituted 5-hydroxy-1,2,4-triazin-6(1H)-one derivatives 6.
We found that the 5-hydroxy-1,2,4-triazin-6(1H)-one moiety can serve as an effective carboxylate isostere and that its 3-position can be utilized to attach a branched chain that extends into the secondary binding site of DAAO. These derivatives of 6 were found to potently inhibit human DAAO with IC50 values in the nanomolar range. In this report, we describe the SAR for this new class of DAAO inhibitors as well as the pharmacological properties of selected compounds, including metabolic stability, oral pharmacokinetics, and effects on plasma levels of D-serine in mice following oral co-administration.
Synthetic methods of 3-substituted 5-hydroxy-1,2,4-triazin-6(1H)-one derivatives 6a–o are illustrated in Scheme 1. 3-Arylalkyl derivatives 6a–c were prepared by first treating nitriles 7a–c with hydrogen chloride gas to form the corresponding acetimidate 8a–c.10 Subsequent reaction with methyl chlorooxoacetate in the presence of DIEA, followed by treatment with hydrazine afforded the desired products 6a–c. 3-Arylalkylthio derivatives 6d–f were prepared from thiosemicarbazide 9, which was condensed with glyoxylic acid and subsequently reacted with arylalkyl bromide to afford 3-arylalkylthio-1,2,4-triazin-5(4H)-ones 10d–f.11 Oxidation of the 6-carbon with bromine12 provided the final products 6d–f. The final oxidation step, however, produced sulfoxide derivatives as by-products that often made the purification of the desired products difficult. To this end, we developed an alternative route to 3-arylalkylthio derivatives, devoid of an oxidation step. Thiosemicarbazide 9 was condensed with diethyl oxalate to form 5-hydroxy-3-mercapto-1,2,4-triazin-6(1H)-one 11 in situ, which was reacted with alkyl bromide to obtain the final products 6g–o. Although the yields of the new synthetic procedure were poor across all the derivatives (8–21%), the one-pot preparative method was technically simpler and generated no inseparable by-products.
Scheme 1.

Synthesis of 3-substituted 5-hydroxy-1,2,4-triazin-6(1H)-one derivatives 6a–o. Reagents and conditions: (a) HCl (gas), MeOH/hexanes, 0 °C to rt, 86–97%; (b) (i) methyl chlorooxoacetate, DIEA, THF, 0 °C, (ii) NH2NH2 H2O, 0 °C, 13–96%; (c) (i) 80% aq. EtOH, 70 °C, (ii) glyoxylic acid, 80% aq. EtOH, 70 °C, (iii) R2Br, NaOH, 80 °C, 23–32%; (d) Br2, (3 drops), H2O, rt, 9–16%; (e) (i) NaOMe (25% soln in MeOH), diethyl oxalate, 65 °C, (ii) R2Br, rt, 50 °C, 8–21%.
The inhibitory potency of the synthesized compounds were determined using recombinant human DAAO as previously reported.13 In vitro DAAO inhibitory data are summarized in Table 1. 5-Hydroxy-1,2,4-triazin-6(1H)-one derivatives containing a variety of substituents at the 3-position displayed varying degrees of inhibitory potency against DAAO. All compounds but 6f were found to be potent DAAO inhibitors with IC50 values in the nanomolar range. In general, the SAR trends are similar to those of 4-hydroxypyridazin-3(2H)-one and 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione derivatives.6, 7 Two-unit chains, -CH2CH2- and -CH2S- appear to be very effective linkers connecting the aryl group and the 5-hydroxy-1,2,4-triazin-6(1H)-one moiety, providing some of the most potent DAAO inhibitors within the series.
Table 1.
Inhibition of Human DAAO by 3-substituted 5-hydroxy-1,2,4-triazin-6(1H)-one derivatives 6a–o.
| Cmpd | Structure | IC50 (µM)[a] |
|---|---|---|
| 6a | 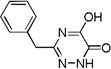 |
0.07 |
| 6b | 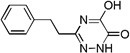 |
0.06 |
| 6c | 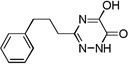 |
4.0 |
| 6d | 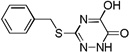 |
0.1 |
| 6e | 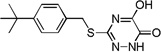 |
0.3 |
| 6f | 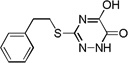 |
40.0 |
| 6g | 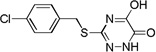 |
0.03 |
| 6h | 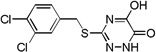 |
0.05 |
| 6i | 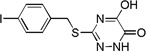 |
0.05 |
| 6j | 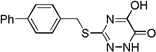 |
0.09 |
| 6k | 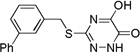 |
0.7 |
| 6l | 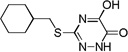 |
0.6 |
| 6m | 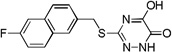 |
0.03 |
| 6n | 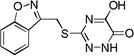 |
0.2 |
| 6o | 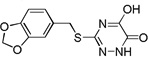 |
0.06 |
Assay methods are described in the Supporting Information.
Many of the derivatives containing a benzylthio group at the 3-position were particularly potent DAAO inhibitors (IC50 < 100 nM) as represented by 6g–j, 6m, and 6o. Substitutions at the phenyl ring of these compounds are generally well tolerated, reflecting the adequate space available at the secondary binding site. There seems to be a preference for linear substituents at this binding site. For instance, para-phenyl derivative 6j was 8-fold more potent than the corresponding meta-phenyl derivative 6k. Further, incorporation of a fused ring such as naphthyl (6m) benzisoxazole (6n), or 1,3-benzodioxole (6o) also produced potent DAAO inhibitors, further underscoring the high steric tolerance at the secondary binding site.
The proposed binding mode of 6b (white) to the active site of DAAO is illustrated in Figure 2 using the crystal structure of DAAO in complex with 2a (cyan)6 as a template. A key hydrogen bond is observed between Tyr228 and Arg283 and the α-hydroxycarbonyl moiety of 6b. An additional interaction is observed with the nitrogen at the 1-position of the 5-hydroxy-1,2,4-triazin-6(1H)-one moiety and the carbonyl of Gly313. A similar interaction exists in the crystal structure of human DAAO with 2a. The high inhibitory potency retained by compounds 6j and 6m–o bearing a sterically hindered substituent at the 3-position can be explained by the spacious pocket created as a result of the movement of Tyr224 away from the active site seen in the crystal structure of DAAO in complex with 2a.6
Figure 2.

Proposed binding mode of 6b (white) to the active site of DAAO (PDB: 3W4K). Key residues and FAD are shown in green and yellow, respectively. Hydrogen-bonding interactions between 6b and the key residues are shown as grey dashed lines. Compound 2a (cyan) of 3W4K is also shown.
We have previously reported that some DAAO inhibitors possessing an α-hydroxycarbonyl moiety as a carboxylate bioisostere are prone to glucuronidation in liver microsomes in the presence of UDGPA.7, 14 For example, as shown in Table 2, compound 1 was extensively glucuronidated within 60 min. In contrast, compound 2b exhibited some degree of metabolic stability while compound 3 displayed complete resistance to glucuronidation. We tested the metabolic stability of two 5-hydroxy-1,2,4-triazin-6(1H)-one derivatives 6b and 6m in liver microsomes. As shown in Table 2, both DAAO inhibitors were found to be resistant to glucuronidation. This is in good agreement with the previous findings that that increased topological polar surface area (tPSA) of the carboxylate bioisostere moiety contributes to higher resistance to glucuronidation.7, 14 The susceptibility to glucuronidation, however, may also depend on other factors such as the extent of electron delocalization and keto-enol tautomerism. Both 6b and 6m were also stable in mouse liver microsomes in the presence of NADPH, a cofactor for CYP-dependent oxidation.
Table 2.
Metabolic Stability of DAAO Inhibitors in mouse Liver Microsomes
| Cmpd | tPSA (Å)a | % remaining after 60 min | |
|---|---|---|---|
| w/UDPGA | w/NADPH | ||
| 1 | 47 | 5 | NDb |
| 2b | 62 | 59 | 92 |
| 3 | 82 | >99 | >99 |
| 6b | 74 | 98 | 79 |
| 6m | 74 | 95 | 91 |
tPSA of the carboxylate bioisostere moiety.
Not determined.
The plasma levels of 6b over tine following oral administration (30 mg/kg) in mice are shown in Figure 3. Compound 6b was quickly absorbed and achieved its Cmax at 30 min after administration. The overall exposure calculated from the area under the curve (AUC0–6h) was 8.07 µg*h/mL. Binding of compound 6b to mouse plasma protein was found to be 74%. Considering the high inhibitory potency of 6b, sufficient unbound drug appears to be available for inhibition of peripheral DAAO at least for the first 3 hours.
Figure 3.

Plasma levels of 6b following oral administration of (30 mg/kg, po), in mice.
Given the reasonable plasma exposure by the oral route, we subsequently evaluated the effects of 6b on D-serine plasma pharmacokinetics following oral co-administration (both at 30 mg/kg, po). As shown in Figure 4, the plasma clearance of D-serine was nearly arrested for the first 3 hours. This is consistent with the plasma pharmacokinetics of 6b in mice. After 3 hours, the plasma D-serine levels started to decline with a clearance rate similar to that of D-serine alone treatment, presumably due to the insufficient free fraction of 6b available for peripheral DAAO inhibition. Overall, co-administration of D-serine with DAAO inhibitor 6b resulted in a 1.7 fold increase in the D-serine AUC0–6hr (74.2 µg*hr/ml) compared with D-serine treatment alone (43.3 µg*hr/ml). Given the previously reported cross-species variation in IC50 values between human and rat forms of DAAO,15 however, caution needs to be taken in establishing the PK/PD relationship in mice as we have not measured inhibitory potency of 6b in mouse DAAO.
Figure 4.

Effect of compound 6b (30 mg/kg, p.o.) on plasma pharmacokinetics of oral D-serine (30 mg/kg, p.o.) in mice.
The 3-substituted derivatives of 5-hydroxy-1,2,4-triazin-6(1H)-one represent a new class of DAAO inhibitors with potent inhibitory activity and substantial microsomal metabolic stability. Additionally, we have demonstrated the oral co-administration of a new DAAO inhibitor 6b with D-serine can improve plasma exposure to D-serine compared with D-serine administration alone. The degree of enhancement in plasma D-serine levels (1.7-fold) was, however, lower than that of compound 3 based on a 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione scaffold (2.2-fold) previously reported.7 This is in a good agreement with the slightly lower AUC value for 6b (8.07 µg*h/mL) following oral administration relative to that of 3 (12.9 µg*h/mL).7 Nevertheless, the enhancement of plasma D-serine levels achieved by 6b should be sufficient to produce pharmacological effects of D-serine in the CNS as a similar plasma D-serine exposure enhancing effect of another DAAO inhibitor, 6-chloro-1,2-benzisoxazol-3(2H)-one (CBIO),16 resulted in attenuation of dizocilpine-induced prepulse inhibition deficits in a mouse model schizophirenia.17
The SAR data summarized in Table 1 highlight considerable steric tolerance for substituents at the 3-position that exists in the secondary biding site of DAAO, which can be further explored for improved potency. Additionally, recent findings by Terry-Lorenzo et al. suggest that the active-site lid formed by amino acids 216–224 has some degree of flexibility and undergoes conformational rearrangements depending on the ligand structures.9 Of particular interest is the engagement of Tyr55 in ligand interaction facilitated by the movement of the active-site lid as seen in the co-crystal structure of compound 5 bound to DAAO (PDB: 4QFD).9 Effective exploitation of this dynamic structural feature may lead to further improvement in inhibitory potency within the new series of DAAO inhibitors based on the 5-hydroxy-1,2,4-triazin-6(1H)-one scaffold.
Supplementary Material
Acknowledgments
This work was in part supported by the National Institutes of Health (R01MH091387 to T.T.) and the Johns Hopkins Brain Science Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.#####.
References and notes
- 1.Lu JM, Gong N, Wang YC, Wang YX. Br. J. Pharmacol. 2012;165:1941. doi: 10.1111/j.1476-5381.2011.01680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferraris DV, Tsukamoto T. Curr. Pharm. Des. 2011;17:103. doi: 10.2174/138161211795049633. [DOI] [PubMed] [Google Scholar]
- 3.Sacchi S, Rosini E, Pollegioni L, Molla G. Curr. Pharm. Des. 2013;19:2499. doi: 10.2174/1381612811319140002. [DOI] [PubMed] [Google Scholar]
- 4.Kawazoe T, Tsuge H, Imagawa T, Aki K, Kuramitsu S, Fukui K. Biochem. Biophys. Res. Commun. 2007;355:385. doi: 10.1016/j.bbrc.2007.01.181. [DOI] [PubMed] [Google Scholar]
- 5.Raje M, Hin N, Duvall B, Ferraris DV, Berry JF, Thomas AG, Alt J, Rojas C, Slusher BS, Tsukamoto T. Bioorg. Med. Chem. Lett. 2013;23:3910. doi: 10.1016/j.bmcl.2013.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hondo T, Warizaya M, Niimi T, Namatame I, Yamaguchi T, Nakanishi K, Hamajima T, Harada K, Sakashita H, Matsumoto Y, Orita M, Takeuchi M. J. Med. Chem. 2013;56:3582. doi: 10.1021/jm400095b. [DOI] [PubMed] [Google Scholar]
- 7.Hin N, Duvall B, Ferraris D, Alt J, Thomas AG, Rais R, Rojas C, Wu Y, Wozniak KM, Slusher BS, Tsukamoto T. J. Med. Chem. 2015;58:7258. doi: 10.1021/acs.jmedchem.5b00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hopkins SC, Heffernan ML, Saraswat LD, Bowen CA, Melnick L, Hardy LW, Orsini MA, Allen MS, Koch P, Spear KL, Foglesong RJ, Soukri M, Chytil M, Fang QK, Jones SW, Varney MA, Panatier A, Oliet SH, Pollegioni L, Piubelli L, Molla G, Nardini M, Large TH. J. Med. Chem. 2013;56:3710. doi: 10.1021/jm4002583. [DOI] [PubMed] [Google Scholar]
- 9.Terry-Lorenzo RT, Chun LE, Brown SP, Heffernan ML, Fang QK, Orsini MA, Pollegioni L, Hardy LW, Spear KL, Large TH. Bioscience reports. 2014;34:487. doi: 10.1042/BSR20140071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connolly DJ, Lacey PM, McCarthy M, Saunders CP, Carroll AM, Goddard R, Guiry PJ. J. Org. Chem. 2004;69:6572. doi: 10.1021/jo049195+. [DOI] [PubMed] [Google Scholar]
- 11.Mizutani M, Sanemitsu Y, Tamaru Y, Yoshida Z. J. Org. Chem. 1983;48:4585. [Google Scholar]
- 12.Brown D, Jones R. Aust. J. Chem. 1972;25:2711. [Google Scholar]
- 13.Ferraris D, Duvall B, Ko YS, Thomas AG, Rojas C, Majer P, Hashimoto K, Tsukamoto T. J. Med. Chem. 2008;51:3357. doi: 10.1021/jm800200u. [DOI] [PubMed] [Google Scholar]
- 14.Zimmermann SC, Rais R, Alt J, Burzynski C, Slusher BS, Tsukamoto T. ACS Med. Chem. Lett. 2014;5:1251. doi: 10.1021/ml500335z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duplantier AJ, Becker SL, Bohanon MJ, Borzilleri KA, Chrunyk BA, Downs JT, Hu LY, El-Kattan A, James LC, Liu S, Lu J, Maklad N, Mansour MN, Mente S, Piotrowski MA, Sakya SM, Sheehan S, Steyn SJ, Strick CA, Williams VA, Zhang L. J. Med. Chem. 2009;52:3576. doi: 10.1021/jm900128w. [DOI] [PubMed] [Google Scholar]
- 16.Rais R, Thomas AG, Wozniak K, Wu Y, Jaaro-Peled H, Sawa A, Strick CA, Engle SJ, Brandon NJ, Rojas C, Slusher BS, Tsukamoto T. Drug Metab. Dispos. 2012;40:2067. doi: 10.1124/dmd.112.046482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hashimoto K, Fujita Y, Horio M, Kunitachi S, Iyo M, Ferraris D, Tsukamoto T. Biol. Psychiatry. 2009;65:1103. doi: 10.1016/j.biopsych.2009.01.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.