

Abstract
Adiponectin is an adipocyte-specific factor, first described in 1995. Over the past two decades, numerous studies have elucidated the physiological functions of adiponectin in obesity, diabetes, inflammation, atherosclerosis, and cardiovascular disease. Adiponectin, elicited through cognate receptors, suppresses glucose production in the liver and enhances fatty acid oxidation in skeletal muscle, which together contribute to a beneficial metabolic action in whole body energy homeostasis. Beyond its role in metabolism, adiponectin also protects cells from apoptosis and reduces inflammation in various cell types via receptor-dependent mechanisms. Adiponectin, as a fat-derived hormone, therefore fulfills a critical role as an important messenger to communicate between adipose tissue and other organs. A better understanding of adiponectin actions, including the pros and cons, will advance our insights into basic mechanisms of metabolism and inflammation, and potentially pave the way toward novel means of pharmacological intervention to address pathophysiological changes associated with diabetes, atherosclerosis, and cardiometabolic disease.
Keywords: adiponectin, diabetes, obesity, metabolic syndrome
Introduction
Adiponectin, a fat-derived hormone, was discovered two decades ago. It attracted considerable attention due to its involvement as a critical messenger for the crosstalk between adipose tissue and other metabolic related organs. Over the past 20 years, the metabolism community has shown tremendous interest in adiponectin, and enormous efforts have been directed to dissect the molecular mechanisms of action (Figure 1). Adiponectin targets the liver, heart, pancreatic β cells, kidney, potentially muscle, and many other cell types in various tissues. Adiponectin strongly suppresses hepatic gluconeogenesis by inhibiting genes involved in glucose production. Through its local action in key metabolic tissues, adiponectin promotes insulin sensitization and therefore improves whole body energy homeostasis. Adiponectin exerts strong protection against a number of pathological events in various cells by suppressing cell death, inhibiting inflammation, and enhancing cell survival. The identification of adiponectin receptors has greatly facilitated our efforts to delineate adiponectin functions. Solving the crystal structures of both adiponectin and its receptors has provided critical insights toward a better understanding of the molecular mechanisms, which also enables the structure-guided design of adiponectin mimetics with potentially potent effects on diabetes, atherosclerosis, and cardiovascular disease. Moreover, the elucidation of the basic cellular mechanisms emerging from adiponectin research also helped establish the concept that adipose tissue serves as an important endocrine organ (Scherer, 2006; Deng and Scherer, 2010). Here, we provide a historic perspective on adiponectin research during the past two decades, discuss major findings along the way (Figure 2), and highlight existing and new questions about adiponectin and adiponectin-related therapeutic applications.
Figure 1.
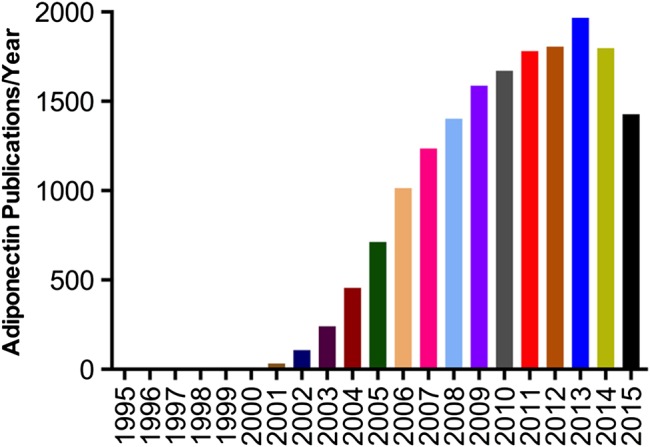
Publications on adiponectin year by year.
Figure 2.

Timeline of key findings in adiponectin research. Due to space limitation, only a partial list is shown.
The discovery
In 1995, we published the first report on adiponectin (at the time referred to as adipocyte complement-related protein of 30 kDa, Acrp30), identified from a subtractive cDNA library enriched in adipocyte-specific genes (Scherer et al., 1995). Mouse adiponectin encodes a 247-amino acid polypeptide with a N-terminal signal sequence, followed by a hyper-variable region, a collagenous domain of 22 G-X-Y repeats, and a globular domain (Figure 3A). At the protein level, the C-terminal globular domain of adiponectin exhibits striking similarities to the subunits of complement factor C1q, and therefore the original name Acrp30 was chosen.
Figure 3.

The biochemical properties of adiponectin. (A) The domain structure of mouse adiponectin. (B) Mouse adiponectin exists as a major 30 kDa band in reducing and denaturing SDS-PAGE. The minor upper band reflects a smaller pool with additional post-translational modifications of adiponectin. (C) Native adiponectin shows three distinct complex forms, high molecular weight (HMW, 12–18mer), middle molecular weight (MMW, hexamer), and low molecular weight (LMW, trimer).
We found that adiponectin is specifically expressed in adipose tissue and fully differentiated adipocytes in tissue culture (Scherer et al., 1995). Adiponectin can be readily detected in mouse serum using an antibody raised against the species-specific hyper-variable region. This antibody does not cross react to adiponectin from human, rabbit, or calf origin. Moreover, incubation of 3T3-L1 adipocytes with insulin leads to an enhanced release of adiponectin within 60 min, indicating that insulin may trigger adiponectin from regulated vesicles. Both size fractionation analysis and chemical cross-linking assays show that native adiponectin exists in multiple complexes, and our original studies raised the possibility that the biochemical properties of adiponectin may be differentially affected depending on how the subunits are complexed with each other.
Similarly, Spiegelman and colleagues identified mouse adiponectin from an mRNA differential display assay (and originally referred to it as AdipoQ) (Hu et al., 1996). They showed indeed that adiponectin transcription and translation are strongly induced by adipocyte differentiation. Further, mRNA levels for adiponectin are dramatically suppressed in obese mouse fat pads compared with controls. In contrast, the expression of aP2, another adipocyte-specific gene, is not affected under these conditions. Multiple lines of evidence in these early papers indicated that adiponectin may play a role in regulating whole body energy homeostasis.
In 1996, two Japanese groups reported the human homolog using different approaches. Maeda et al. (1996) constructed a cDNA library from human adipose tissue. They identified adiponectin as the most abundant transcript and named it adipose most abundant gene transcript 1 (apM1) (Maeda et al., 1996). Similar to the findings in rodents, northern blotting showed that adiponectin is highly enriched in human adipose tissue. In the other study, Nakano et al. (1996) employed a different method to identify adiponectin using gelatin-based affinity chromatography to isolate adiponectin from human plasma (originally referred to as gelatin-binding protein of 28 kDa, GBP28). After elution with high salt, they identified adiponectin by protein sequencing. Consistent with the findings in rodents by Scherer et al. (1995), Nakano et al. (1996) showed that adiponectin exists as multiple complexes with different molecular weights.
The biochemical properties
Mouse adiponectin contains 247 amino acids, which migrates at ∼30 kDa by gel electrophoresis (Scherer et al., 1995). Polyclonal antibodies against the hyper-variable region recognize a main band with a minor upper band, which is caused by a N-linked carbohydrate modification. Adiponectin is found in plasma at µg/ml levels, compared with ng/ml of conventional hormones of insulin and leptin (Pajvani et al., 2003). Adiponectin levels display a strong sexual dimorphism with females averaging twice the levels seen in males, imprinted early in the perinatal period through the action of testosterone (Combs et al., 2003). Testosterone may also have direct effects on the adult through the modulation of production, complex formation, and clearance of adiponectin (Xu et al., 2005).
The native status of adiponectin is in drastic contrast to the simple appearance under denaturing and reducing conditions seen in sodium dodecylsulphate–polyacrylamide gel electrophoresis (SDS-PAGE) (Figure 3B). By velocity sedimentation centrifugation, we have shown that adiponectin exists in two complexes as a hexamer and a high molecular weight form (Pajvani et al., 2003). Later on, with a more refined approach toward size fractionation, we could separate native adiponectin into three complexes; these observations were further elaborated on by us and others, such as Kadowaki and colleagues (Waki et al., 2003; Schraw et al., 2008). Although the nomenclature for the different complexes varies in different publications, the overall findings are entirely consistent. We refer to the different complexes as low molecular form (LMW, trimer), middle molecular form (MMW, hexamer), and high molecular form (HMW, 12–18mer) (Figure 3C). This is an important issue, since not only the total levels of adiponectin, but also the complex distribution may contribute to distinct downstream biological effects (Pajvani et al., 2004). For example, the difference of total levels of adiponectin in males vs. females may be simply due to the lower levels of HMW in males. Furthermore, we found that the ratio of HMW and total levels (SA) is a more reliable index for correlations with insulin sensitivity in both rodent and human studies. Indeed, HMW adiponectin elicits more potent glucose-lowering effects when injected to mice intraperitoneally (Pajvani et al., 2004). The anti-diabetic drug class belonging to the PPARγ agonists (the thiazolidinediones, TZDs) specifically increases HMW in adipocytes and in plasma.
In addition, Wang et al. (2002) identified four lysine sites for hydroxylation and following glycosylation (Lys68, 71, 80 and 104). They showed that the mutation of all four lysines to arginines completely abolishes the assembly and secretion of HMW, and hence reduces the biological function in vivo (Wang et al., 2006). Likewise, Waki et al. (2003) studied human mutations with an impact on HMW formation. The assembly of adiponectin hexamers from trimeric building blocks requires inter-trimeric disulfide bond formation at cysteine 39 (cysteine 36 in human adiponectin). We found that mutating this critical cysteine to an alanine abolishes hexamer formation and hence HMW formation. The Cys39Ala mutant adiponectin exists predominantly as trimers in both tissue culture studies and in a transgenic mouse model (Pajvani et al., 2003). These findings highlight the importance of adiponectin folding and assembly in regulation of plasma levels and complex distribution.
Intracellular adiponectin folding and assembly involves a complex set of regulatory steps in the endoplasmic reticulum (ER) and Golgi (Xie et al., 2006; Wang and Scherer, 2008b). We found in a pulse-chase experiment that a significant amount of adiponectin is retained intracellularly. Administration of a reducing agent, such as dithiothreitol (DTT), acutely stimulates adiponectin release from 3T3-L1 adipocytes (Wang et al., 2007). We went further to show that an ER-resident chaperone ERp44 is responsible for the intracellular thiol-mediated retention of adiponectin for higher order complex formation. On the other hand, another ER chaperone, Ero1, can displace adiponectin from ERp44 and trigger disulfide bond formation to a hexamer followed by HMW assembly. Importantly, protein levels of both chaperones are subjected to pharmacological regulation with PPARγ agonists (TZDs), suggesting that TZDs may exert their impact by enhancing adiponectin levels in serum by increasing chaperone production and improving the folding environment in the ER (Wang et al., 2007). Likewise, Qiang et al. (2007) also reported that Ero1 is critical for adiponectin complex formation and secretion. Ero1 levels are increased over the course of adipocyte differentiation in vitro in parallel with the augmentation of adiponectin expression and secretion. Using a yeast-based two-hybrid screen, Liu and colleagues identified an additional adiponectin-interacting protein, DsbA-L, which harbors disulfide bond oxidoreductase activity (Liu et al., 2008). Both gain- and loss-of-function studies prove that DsbA-L is indispensable for HMW formation and secretion. The ER localization of DsbA-L is critical for adiponectin multimerization (Liu et al., 2015). More importantly, DsbA-L is reduced under obese conditions in both rodents and humans, consistent with the decreased levels of HMW adiponectin. Combined, both the total levels and the complex distribution of adiponectin are important indicators of obesity, diabetes, and the overall health of the adipocyte. Under some conditions, the ratio of HMW to total levels may even better reflect insulin sensitivity. The further characterization of the intracellular synthesis, folding, and assembly of adiponectin may still reveal novel findings and may represent a novel avenue for adiponectin targeting and diabetes intervention.
The structure
In 1998, Shapiro and Scherer solved the high resolution structure for adiponectin (Shapiro and Scherer, 1998). Due to the inevitable difficulties of crystalizing the hyper-variable region and the collagenous stalk, we chose the C-terminal globular domain for further characterization.
The tertiary structure of adiponectin reveals surprising similarities with the tumor necrosis factor (TNF) family (Shapiro and Scherer, 1998). The adiponectin structure shows an asymmetric trimer of three identical monomers. Many structural features can be superimposed on crystal structures for TNF family members, even though there is no homology at the primary amino acid level. The trimer of the adiponectin globular domain is stabilized by a central hydrophobic interface that excludes water molecules, located at the base of the structure. The collagenous stalk is predicted to protrude from the base of the trimer. On the other hand, the interface at the apex is largely hydrophilic, which may directly interact with receptor(s). The similarity between adiponectin/C1q family and the TNF family indicates that they may have arisen from a common primordial molecule during evolution.
Studies from multiple labs have shown that the trimeric adiponectin can lower plasma glucose by suppressing hepatic gluconeogenesis (Pajvani et al., 2003). Moreover, the half-life of trimeric adiponectin is much shorter than higher order of complexes. These findings raise an interesting possibility that a cell surface bound ‘reductase’ may be coupled with adiponectin receptor(s). The MMW or HMW adiponectin is firstly reduced to trimer, which then interacts with the receptor to exert downstream biological function. On the other hand, the bouquet shape of the HMW adiponectin complex may create additional novel interfaces for oligomerizing receptors. Further studies are required to identify the reductase or other surface interacting partners for adiponectin.
The physiological role
In 2001, two key papers demonstrated for the first time the physiological role of adiponectin and highlighted the adiponectin axis as a potential therapeutic area for diabetes treatment (Berg et al., 2001; Yamauchi et al., 2001). We purified native adiponectin from a mammalian expression system, aiming to avoid any complications from the lack of post-translational modifications seen in the bacterially expressed molecule (Berg et al., 2001). Mammalian-produced adiponectin retains all three complexes and behaves similarly as adiponectin from plasma. To evaluate the physiological role of adiponectin, we injected adiponectin into wild-type (WT) C57/BL6 mice and triggered a 4-fold increase in circulating adiponectin within 2 h. We observed a decrease in blood glucose levels at 4 h after administration, and this decrease is not associated with any changes of plasma insulin levels. The same injection of purified adiponectin can elicit glucose-lowering effects in both type II diabetic mice (such as the ob/ob mouse) and type I diabetic mice, such as the NOD mouse and streptozotocin-treated mouse models, in this case even with a prolonged effect (Berg et al., 2001). These findings suggest that adiponectin suppresses glucose production and/or stimulates glucose assimilation independent of insulin levels. Using an in vitro glucose production assay, adiponectin was shown to suppress gluconeogenesis, which was later confirmed by an in vivo clamp analysis (Combs et al., 2001). Adiponectin administration can achieve maximal inhibition of glucose production even in the presence of sub-physiological levels of insulin.
In a different study, Kadowaki and colleagues showed that adiponectin levels are decreased in either genetically obese ob/ob or dietary obese high-fat-diet (HFD) mouse models (Yamauchi et al., 2001). Administration of adiponectin leads to an improvement in insulin sensitivity in db/db, HFD, or KKAy animal models. Mechanistically, they found that adiponectin can increase β-oxidation in skeletal muscle and suppress lipid accumulation in the liver. In a follow-up study, Kadowaki and colleagues went further to show that both the globular and full-length adiponectin molecules can stimulate 5′AMP-activated protein kinase (AMPK) activity in skeletal muscle, while only the full-length adiponectin does so in the liver (Yamauchi et al., 2002). More importantly, AMPK is required for the physiological role of adiponectin in many, though not all tissues. Collectively, adiponectin can enhance whole body insulin sensitivity, highlighting adiponectin as an important messenger to communicate between adipose tissue and other metabolically relevant organs.
Besides its potent effects in the periphery, adiponectin also displays central effects. Administration of adiponectin by intravenous injection leads to movement of the protein across the blood-brain barrier and raises the adiponectin levels in cerebrospinal fluid (Qi et al., 2004). On the other hand, intra-cerebroventricular injection of adiponectin causes a decrease in body weight and an increase in energy expenditure, which may be mediated by upregulation of hypothalamic corticotrophin-releasing hormone (CRH). Moreover, adiponectin has been found in human cerebrospinal fluid with a predominant distribution of the trimeric form (Kusminski et al., 2007). Further studies are warranted to examine the levels, complex distribution, and physiological actions of adiponectin in the brain under clinically relevant conditions in human.
The receptors
Shortly after the discovery of adiponectin as a fat-derived factor, the search for its receptor(s) began. Kadowaki and colleagues accomplished this goal in 2003 (Yamauchi et al., 2003). They generated a cDNA library of human skeletal muscle mRNAs in Ba/F3 cells. With a series of fluorescent activated cell sorting, a cDNA clone was found conveying the ability to serve as a strong binding partner to globular adiponectin. This molecule, termed AdipoR1, shows ubiquitous expression, including in skeletal muscle and the liver. Another highly similar sequence was subsequently isolated and is referred to as AdipoR2, whose expression is more restricted to the liver. These two receptors display a surprisingly high resemblance to each other (identity of 66.7% at the protein level). Both R1 and R2 are integral membrane proteins, with seven transmembrane domains of their N-terminus inside the cell and the C-terminus facing outwards. This topology is opposite to that reported for all known G-protein coupled receptors. Scatchard plot analysis and ligand binding assays revealed that AdipoR1 can act as a high-affinity receptor for globular adiponectin, but it can also bind full-length adiponectin, and AdipoR2 shows intermediate affinity for both globular and full-length adiponectin molecules (Yamauchi et al., 2003).
In the genetically obese ob/ob mouse model, both AdipoR1 and AdipoR2 show reduced expression in the liver. Forced overexpression of either AdipoR1 or AdipoR2 by adenovirus leads to significant improvements in whole body metabolism, which is abolished in adiponectin-deficient animals (Yamauchi et al., 2007). Mechanistically, the Kadowaki group reported that AdipoR1 restoration in ob/ob animals activates AMPK and causes reduction of gluconeogenic genes in the liver and, as a consequence, glucose production is significantly suppressed. On the other hand, AdipoR2 overexpression leads to activation of AMPK to enhance glucose uptake and PPARα to increase fatty acid β-oxidation. Consistent with these studies, the systemic disruption of AdipoR1 leads to increased glucose production, while AdipoR2 knockout mice display insulin resistance. More importantly, the administration of purified adiponectin does not lead to glucose-lowering effects when injected into AdipoR1/R2 double knockout mice, suggesting these two receptors may mediate the bulk of the physiological actions of adiponectin.
Additionally, a muscle-specific knockout of AdipoR1 leads to reduction in AMPK activity and decreased involuntary exercise capacity (Iwabu et al., 2010). At a mechanistic level, adiponectin administration in C2C12 myotubes causes AMPK phosphorylation and PGC1α activation, and this process requires intact signaling of AdipoR1 and CaMKKβ. Adiponectin treatment in C2C12 myotubes can trigger an increase of intracellular calcium levels, which is critical for CaMKKβ activation and downstream activation of mitochondrial biogenesis and metabolic improvements. Crystallization of AdipoR1 and AdipoR2 was recently achieved, leading to the description of the structure at high resolution. These receptors represent a novel class of structure (Tanabe et al., 2015). Furthermore, these structures indeed suggest that the interaction between the ligand and receptors takes place between the globular domain of adiponectin and the extracellular surface of the receptors.
To elucidate the intracellular signaling of adiponectin and adiponectin receptors, Dong and colleagues conducted a yeast two-hybrid screening using the intracellular domain of AdipoR1 as the bait (Mao et al., 2006). They identified an adaptor protein, referred to as adaptor protein containing pleckstrin homology domain, phosphotyrosine binding domain and leucine zipper motif (APPL1). APPL1 interacts with both AdipoR1 and R2 and mediates the downstream effects in AMPK activation, glucose uptake, and lipid oxidation. The Dong group also demonstrated that APPL1 directly enhances LKB1 cytosolic translocation and consequently activates AMPK in muscle cells (Zhou et al., 2009). On the other hand, APPL1 forms a complex with IRS1/2 under basal conditions (Ryu et al., 2014). After stimulation by either insulin or adiponectin, the APPL1/IRS1/2 complex is recruited to insulin receptor and conveys insulin signaling. APPL1 expression is significantly reduced in the obese state, and the complete deficiency of APPL1 leads to strong systemic insulin resistance. These results suggest that APPL1 is a critical mediator of adiponectin action in whole body energy homeostasis.
Following the discovery of AdipoR1 and AdipoR2, Hug et al. (2004) identified T-cadherin as an additional receptor for adiponectin. Lodish and colleagues employed receptor cloning using a similar strategy as Kadowaki and colleagues. T-cadherin specifically binds to MMW and HMW forms of adiponectin. Interestingly, there is no detectable interaction between T-cadherin and either globular or bacterial-produced adiponectin, highlighting the importance of high order complexes and eukaryotic post-translational modifications of adiponectin for proper biological function.
T-cadherin shows abundant expression in endothelial cells and smooth muscle cells (Hug et al., 2004). T-cadherin also plays critical roles in adiponectin-mediated revascularization after chronic ischemia (Parker-Duffen et al., 2013). Moreover, Denzel et al. (2010) found that T-cadherin is expressed in cardiac myocytes and mediates the anti-hypertrophic role of adiponectin. Deficiency of T-cadherin also shows increased infarct size by ischemia/reperfusion and reduced induction of AMPK. However, T-cadherin is attached to the plasma membrane via a glycosyl phosphatidylinositol (GPI) anchor. The intracellular signaling relay for adiponectin may therefore require other unidentified co-receptors or AdipoR1/2.
The physiological role rediscovered: a unifying mechanism
Adiponectin exerts pleiotropic actions, i.e. it promotes insulin sensitivity, inhibits cell death, and suppresses inflammation. Although the receptors have been identified, much needs to be learned regarding downstream molecules and events. A unifying mechanism explaining the versatile roles of adiponectin under various conditions was lacking. Using multiple animal models, both gain- and loss-of-function, we found that an adiponectin receptor-associated ceramidase activity may be the underlying key mechanism explaining many of the adiponectin-related phenotypes (Holland et al., 2011).
Ceramides are members of a diverse class of lipids that are involved in insulin resistance, cell death, inflammation, and atherosclerosis (Chaurasia and Summers, 2015). Ceramidase is an enzyme converting harmful ceramides into a beneficial class of lipids, the sphinganines and sphingosines, including the sphingosine 1-phosphate (S1P) subclass. The ratio of ceramides to S1P is very important and has been implicated in multiple biological processes. We found that ceramide content in the liver is increased in either genetically obese ob/ob mice or HFD-induced obese animal models (Holland et al., 2011). Adiponectin administration leads to a potent decrease of hepatic ceramide levels and hence insulin sensitization. Moreover, in HFD-fed animals, genetically-induced increasing of adiponectin level is associated with improved glucose tolerance and decreased ceramide content. Importantly, these effects are independent of AMPK, as adiponectin injection elicits similar insulin sensitization and ceramide lowering in LKB1-deficient mice. This mechanism is well preserved in cardiomyocytes and pancreatic β cells in vivo and in vitro. At the molecular level, we found that adiponectin receptors convey ceramidase activity. Overexpression of the receptors by adenovirus leads to enhanced conversion of ceramide to S1P and consequently confers protection against insulin resistance. Indeed, ablation of both receptors in mouse embryonic fibroblasts diminishes adiponectin-induced ceramidase activity and again, these effects are AMPK independent. We proposed a universal model of action for adiponectin, which involves ceramidase activity as the central, receptor proximal component. More recently, we found that genetically enhancing acid ceramidase activity in either the liver or adipose tissue (through overexpression of acid ceramidase) is sufficient to trigger an improvement in insulin signaling and whole body energy homeostasis, highlighting again the importance of fine-tuning the regulation of ceramides for metabolic health (Xia et al., 2015).
Additional target cells
Adiponectin, a fat-derived hormone, plays critical roles in metabolic regulation and maintenance for whole body energy homeostasis. The major target organs are the liver and skeletal muscle. Over the past two decades, numerous studies have shown that adiponectin exerts various effects on other organs under different contexts. Adiponectin protects kidney podocytes from cell death and is therefore involved in protecting kidney function under diabetic conditions (Rutkowski et al., 2013). Adiponectin can inhibit endothelial dysfunction and suppresses atherosclerosis (Wang and Scherer, 2008a). Moreover, adiponectin prevents macrophage death and thus reduces lesion formation in blood vessels. Adiponectin has also been found in the brain, where it regulates food intake and this central effect corroborates with peripheral actions to maintain energy homeostasis (Qi et al., 2004).
One of the most critical roles of adiponectin worthy a more extensive discussion is its role in the context of heart disease (Wang and Scherer, 2008a). Studies have shown that over two thirds of diabetic patients develop heart disease, ultimately also the primary cause of their death. It is therefore not surprising that adiponectin is involved in the regulation of cardiac function under diabetic conditions. Early studies by Walsh and colleagues found that adiponectin deficiency leads to exacerbated cardiac hypertrophy when subjected to pressure overloading (Shibata et al., 2004). At the molecular level, adiponectin stimulates AMPK and suppresses ERK activation in the heart. Overproduction of adiponectin by adenovirus leads to improvements of cardiac hypertrophy and cardiac function. Moreover, adiponectin administration in vitro directly reduces cardiac cell growth, highlighting the cell autonomous role of adiponectin in cardiomyocytes. The Walsh group went on to examine the role of adiponectin under cardiac ischemia/reperfusion (I/R) conditions, another common pathophysiological challenge for the heart (Shibata et al., 2005). I/R causes additional cardiac damage. The underlying mechanism is not entirely known. They showed that adiponectin knockout mice display enhanced infarction size compared with WT controls. Similarly, overexpression of adiponectin leads to significant cardioprotection against I/R injury. Adiponectin administration can stimulate AMPK and cyclooxygenase-2 activities, both of which antagonize apoptosis and TNFα production. These findings indicate that adiponectin may play important protective roles against heart disease.
However, a more nuanced assessment is needed here. A recent study indicates that adiponectin can directly stimulate MEF2 activation through p38 MAP Kinase signaling in cardiomyocytes (Dadson et al., 2015). Through this mechanism, adiponectin contributes to myocardial hypertrophic gene expression under pressure overloading. In the adiponectin transgenic mouse model, we found significant cardiac hypertrophy at older age (Holland et al., 2011). On the other hand, Ma and colleagues found that adiponectin protects the heart from I/R injury by inhibiting iNOS and suppression of ROS production (Tao et al., 2007), without the involvement of AMPK (Wang et al., 2009). They also showed that adiponectin inhibited TNFα-induced ICAM expression and resulting inflammation in endothelial cells (Wang et al., 2014). These actions require an increase of ceramidase activity, which can be recruited to the adipoRs via caveolin-1. Further studies are warranted to delineate the role of adiponectin in maintaining normal cardiometabolic health under challenging conditions.
Conclusions and perspectives
Over the past 20 years, adiponectin has gained considerable attention. Adiponectin plays versatile roles in many organs and it does so mainly via interaction with its two receptors. In the liver, adiponectin strongly suppresses hepatic gluconeogenesis, while enhancing nutrient utilization in skeletal muscle. The unique features of adiponectin make the pathway a plausible candidate to look for therapeutic strategies for diabetes. The progress made in the adiponectin field greatly helped us to better understand the role of adipose tissue. The many additional adipokines secreted from adipocytes carry metabolic clues from fat to other tissues to communicate the state of the adipocyte under various conditions.
Despite above, challenges remain and questions await answers. The unusually large size of the native adiponectin complex warrants further examination. It is entirely possible that the HMW form of adiponectin may carry a lipid-soluble factor. Adiponectin demonstrates strong insulin sensitization function, which may adversely lead to cardiac hypertrophy when constitutively over-supplied. The adiponectin message is already amongst the most abundant ones in adipocytes. Approaches to enhance the production may therefore need to focus on assembly and secretion. Adiponectin signaling involves not only the receptors, but also accessory molecules, which may all be targeted to enhance adiponectin sensitivity (Mao et al., 2006; Cheng et al., 2009). However, adiponectin resistance may also occur at different levels, which require further clarification. Elucidation of the role of adiponectin will lead us to identification of more specific and efficient means to target the metabolic disease.
Funding
This work was supported by a Scientist Development Grant (14SDG18440002) from the American Heart Association (Z.V.W.) and National Institutes of Health grants R01-DK55758, R01-DK099110, and P01-DK088761 (P.E.S.).
Conflict of interest: none declared.
Acknowledgements
We thank members of the Wang lab and Scherer lab for valuable discussions.
References
- Berg A.H., Combs T.P., Du X. et al. (2001). The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 7, 947–953. [DOI] [PubMed] [Google Scholar]
- Chaurasia B., Summers S.A. (2015). Ceramides—lipotoxic inducers of metabolic disorders. Trends Endocrinol. Metab. 26, 538–550. [DOI] [PubMed] [Google Scholar]
- Cheng K.K., Iglesias M.A., Lam K.S. et al. (2009). APPL1 potentiates insulin-mediated inhibition of hepatic glucose production and alleviates diabetes via Akt activation in mice. Cell Metab. 9, 417–427. [DOI] [PubMed] [Google Scholar]
- Combs T.P., Berg A.H., Obici S. et al. (2001). Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J. Clin. Invest. 108, 1875–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs T.P., Berg A.H., Rajala M.W. et al. (2003). Sexual differentiation, pregnancy, calorie restriction, and aging affect the adipocyte-specific secretory protein adiponectin. Diabetes 52, 268–276. [DOI] [PubMed] [Google Scholar]
- Dadson K., Turdi S., Hashemi S. et al. (2015). Adiponectin is required for cardiac MEF2 activation during pressure overload induced hypertrophy. J. Mol. Cell. Cardiol. 86, 102–109. [DOI] [PubMed] [Google Scholar]
- Deng Y., Scherer P.E. (2010). Adipokines as novel biomarkers and regulators of the metabolic syndrome. Ann. NY Acad. Sci. 1212, E1–E19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzel M.S., Scimia M.C., Zumstein P.M. et al. (2010). T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J. Clin. Invest. 120, 4342–4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland W.L., Miller R.A., Wang Z.V. et al. (2011). Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 17, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E., Liang P., Spiegelman B.M. (1996). AdipoQ is a novel adipose-specific gene dysregulated in obesity. J. Biol. Chem. 271, 10697–10703. [DOI] [PubMed] [Google Scholar]
- Hug C., Wang J., Ahmad N.S. et al. (2004). T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc. Natl Acad. Sci. USA 101, 10308–10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwabu M., Yamauchi T., Okada-Iwabu M. et al. (2010). Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca2+ and AMPK/SIRT1. Nature 464, 1313–1319. [DOI] [PubMed] [Google Scholar]
- Kusminski C.M., McTernan P.G., Schraw T. et al. (2007). Adiponectin complexes in human cerebrospinal fluid: distinct complex distribution from serum. Diabetologia 50, 634–642. [DOI] [PubMed] [Google Scholar]
- Liu M., Zhou L., Xu A. et al. (2008). A disulfide-bond A oxidoreductase-like protein (DsbA-L) regulates adiponectin multimerization. Proc. Natl Acad. Sci. USA 105, 18302–18307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M., Chen H., Wei L. et al. (2015). Endoplasmic reticulum (ER) localization is critical for DsbA-L protein to suppress ER stress and adiponectin down-regulation in adipocytes. J. Biol. Chem. 290, 10143–10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K., Okubo K., Shimomura I. et al. (1996). cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (adipose most abundant gene transcript 1). Biochem. Biophys. Res. Commun. 221, 286–289. [DOI] [PubMed] [Google Scholar]
- Mao X., Kikani C.K., Riojas R.A. et al. (2006). APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat. Cell Biol. 8, 516–523. [DOI] [PubMed] [Google Scholar]
- Nakano Y., Tobe T., Choi-Miura N.H. et al. (1996). Isolation and characterization of GBP28, a novel gelatin-binding protein purified from human plasma. J. Biochem. 120, 803–812. [DOI] [PubMed] [Google Scholar]
- Pajvani U.B., Du X., Combs T.P. et al. (2003). Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications fpr metabolic regulation and bioactivity. J. Biol. Chem. 278, 9073–9085. [DOI] [PubMed] [Google Scholar]
- Pajvani U.B., Hawkins M., Combs T.P. et al. (2004). Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione-mediated improvement in insulin sensitivity. J. Biol. Chem. 279, 12152–12162. [DOI] [PubMed] [Google Scholar]
- Parker-Duffen J.L., Nakamura K., Silver M. et al. (2013). T-cadherin is essential for adiponectin-mediated revascularization. J. Biol. Chem. 288, 24886–24897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y., Takahashi N., Hileman S.M. et al. (2004). Adiponectin acts in the brain to decrease body weight. Nat. Med. 10, 524–529. [DOI] [PubMed] [Google Scholar]
- Qiang L., Wang H., Farmer S.R. (2007). Adiponectin secretion is regulated by SIRT1 and the endoplasmic reticulum oxidoreductase Ero1-Lα. Mol. Cell. Biol. 27, 4698–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski J.M., Wang Z.V., Park A.S. et al. (2013). Adiponectin promotes functional recovery after podocyte ablation. J. Am. Soc. Nephrol. 24, 268–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J., Galan A.K., Xin X. et al. (2014). APPL1 potentiates insulin sensitivity by facilitating the binding of IRS1/2 to the insulin receptor. Cell Rep. 7, 1227–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer P.E. (2006). Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes 55, 1537–1545. [DOI] [PubMed] [Google Scholar]
- Scherer P.E., Williams S., Fogliano M. et al. (1995). A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 270, 26746–26749. [DOI] [PubMed] [Google Scholar]
- Schraw T., Wang Z.V., Halberg N. et al. (2008). Plasma adiponectin complexes have distinct biochemical characteristics. Endocrinology 149, 2270–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro L., Scherer P.E. (1998). The crystal structure of a complement-1q family protein suggests an evolutionary link to tumor necrosis factor. Curr. Biol. 8, 335–338. [DOI] [PubMed] [Google Scholar]
- Shibata R., Ouchi N., Ito M. et al. (2004). Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat. Med. 10, 1384–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata R., Sato K., Pimentel D.R. et al. (2005). Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat. Med. 11, 1096–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe H., Fujii Y., Okada-Iwabu M. et al. (2015). Crystal structures of the human adiponectin receptors. Nature 520, 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao L., Gao E., Jiao X. et al. (2007). Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation 115, 1408–1416. [DOI] [PubMed] [Google Scholar]
- Waki H., Yamauchi T., Kamon J. et al. (2003). Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J. Biol. Chem. 278, 40352–40363. [DOI] [PubMed] [Google Scholar]
- Wang Z.V., Scherer P.E. (2008a). Adiponectin, cardiovascular function, and hypertension. Hypertension 51, 8–14. [DOI] [PubMed] [Google Scholar]
- Wang Z.V., Scherer P.E. (2008b). DsbA-L is a versatile player in adiponectin secretion. Proc. Natl Acad. Sci. USA 105, 18077–18078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Xu A., Knight C. et al. (2002). Hydroxylation and glycosylation of the four conserved lysine residues in the collagenous domain of adiponectin. Potential role in the modulation of its insulin-sensitizing activity. J. Biol. Chem. 277, 19521–19529. [DOI] [PubMed] [Google Scholar]
- Wang Y., Lam K.S., Chan L. et al. (2006). Post-translational modifications of the four conserved lysine residues within the collagenous domain of adiponectin are required for the formation of its high molecular weight oligomeric complex. J. Biol. Chem. 281, 16391–16400. [DOI] [PubMed] [Google Scholar]
- Wang Z.V., Schraw T.D., Kim J.Y. et al. (2007). Secretion of the adipocyte-specific secretory protein adiponectin critically depends on thiol-mediated protein retention. Mol. Cell. Biol. 27, 3716–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Gao E., Tao L. et al. (2009). AMP-activated protein kinase deficiency enhances myocardial ischemia/reperfusion injury but has minimal effect on the antioxidant/antinitrative protection of adiponectin. Circulation 119, 835–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Wang X., Lau W.B. et al. (2014). Adiponectin inhibits tumor necrosis factor-α-induced vascular inflammatory response via caveolin-mediated ceramidase recruitment and activation. Circ. Res. 114, 792–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J.Y., Holland W.L., Kusminski C.M. et al. (2015). Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis. Cell Metab. 22, 266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L., Boyle D., Sanford D. et al. (2006). Intracellular trafficking and secretion of adiponectin is dependent on GGA-coated vesicles. J. Biol. Chem. 281, 7253–7259. [DOI] [PubMed] [Google Scholar]
- Xu A., Chan K.W., Hoo R.L. et al. (2005). Testosterone selectively reduces the high molecular weight form of adiponectin by inhibiting its secretion from adipocytes. J. Biol. Chem. 280, 18073–18080. [DOI] [PubMed] [Google Scholar]
- Yamauchi T., Kamon J., Waki H. et al. (2001). The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 7, 941–946. [DOI] [PubMed] [Google Scholar]
- Yamauchi T., Kamon J., Minokoshi Y. et al. (2002). Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 8, 1288–1295. [DOI] [PubMed] [Google Scholar]
- Yamauchi T., Kamon J., Ito Y. et al. (2003). Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423, 762–769. [DOI] [PubMed] [Google Scholar]
- Yamauchi T., Nio Y., Maki T. et al. (2007). Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 13, 332–339. [DOI] [PubMed] [Google Scholar]
- Zhou L., Deepa S.S., Etzler J.C. et al. (2009). Adiponectin activates AMP-activated protein kinase in muscle cells via APPL1/LKB1-dependent and phospholipase C/Ca2+/Ca2+/calmodulin-dependent protein kinase kinase-dependent pathways. J. Biol. Chem. 284, 22426–22435. [DOI] [PMC free article] [PubMed] [Google Scholar]