

Abstract
Aim:
To investigate the mechanisms underlying the isomer-selective distribution of 3-n-butylphthalide (NBP) hydroxylated metabolites, 3-hydroxy-NBP (3-OH-NBP) and 10-hydroxy-NBP (10-OH-NBP), across the blood brain barrier (BBB).
Methods:
After oral administration of NBP (20 mg/kg) to rats, the pharmacokinetics of two major hydroxylated metabolites, 3-OH-NBP and 10-OH-NBP, in plasma and brains were investigated. Plasma and brain protein binding of 3-OH-NBP and 10-OH-NBP was also assessed. To evaluate the influences of major efflux transporters, rats were pretreated with the P-gp inhibitor tariquidar (10 mg/kg, iv) and BCRP inhibitor pantoprazole (40 mg/kg, iv), then received 3-OH-NBP (12 mg/kg, iv) or 10-OH-NBP (3 mg/kg, iv). The metabolic profile of NBP was investigated in rat brain homogenate.
Results:
After NBP administration, the plasma exposure of 3-OH-NBP was 4.64 times that of 10-OH-NBP, whereas the brain exposure of 3-OH-NBP was only 11.8% of 10-OH-NBP. In the rat plasma, 60%±5.2% of 10-OH-NBP was unbound to proteins versus only 22%±2.3% of 3-OH-NBP being unbound, whereas in the rat brain, free fractions of 3-OH-NBP and 10-OH-NBP were 100%±9.7% and 49.9%±14.1%, respectively. In the rats pretreated with tariquidar and pantoprazole, the unbound partition coefficient Kp,uu of 3-OH-NBP was significantly increased, while that of 10-OH-NBP showed a slight but not statistically significant increase. Incubation of rat brain homogenate with NBP yielded 3-OH-NBP but not 10-OH-NBP.
Conclusion:
The isomer-selective distribution of 10-OH-NBP and 3-OH-NBP across the BBB of rats is mainly attributed to the differences in plasma and brain protein binding and the efflux transport of 3-OH-NBP. The abundant 10-OH-NBP is not generated in rat brains.
Keywords: 3-n-butylphthalide (NBP), 3-OH-NBP, 10-OH-NBP, blood-brain barrier, isomer-selective, drug plasma protein binding, drug brain protein binding, P-gp/BCRP, cerebral ischemia
Introduction
3-n-Butylphthalide (NBP, (±)-3-butyl-1(3H)-isobenzofuranone) is an oral agent used for the treatment of cerebral ischemia. Racemic NBP was approved for marketing in China by the Chinese Food and Drug Administration (CFDA) in 20041,2. Pharmacological studies have revealed that NBP exerts neuroprotective effects by increasing blood flow in the cerebral ischemic area and inhibiting the release of 5-hydroxytryptamine, glutamate and cytochrome c through the c-Jun N-terminal kinase pathway3,4,5,6. Recent studies have also demonstrated that NBP attenuates oxidative stress and improves neutral morphology after chronic cerebral ischemia7.
We previously reported that NBP undergoes extensive metabolism in humans after a single dose of 200 mg NBP in soft capsules8. The major circulating metabolites found were 3-hydroxy-NBP (3-OH-NBP), 10-hydroxy-NBP (10-OH-NBP), 10-keto-NBP (10-CO-NBP), and NBP-11-oic acid8,9. The areas under the concentration-time curves (AUC) for 3-OH-NBP and 10-OH-NBP were 2.9 and 10.3 times greater, respectively, than that of NBP9. Because NBP is used to treat cerebral diseases, we also attempted to study the metabolite profile of NBP in rat brain. If the cerebral metabolite(s) possess greater or comparable pharmacological activities to NBP, the metabolite(s) could be developed as a new drug for treating cerebral ischemia. Indeed, some novel antiplatelet agents that are structurally based on NBP were reported to be superior to NBP and represent promising treatments for thrombosis-related ischemic stroke10.
Preliminary studies of NBP metabolites revealed that the exposure (AUC) of 3-OH-NBP was 4.64 times greater than that of 10-OH-NBP in rat plasma, whereas the exposure of 3-OH-NBP was only 11.8% that of 10-OH-NBP in rat brain. These findings indicate that 10-OH-NBP has a much greater ability to penetrate the blood–brain barrier (BBB) than 3-OH-NBP. This raises the question regarding what factors contribute to the unique distribution of 3-OH-NBP and 10-OH-NBP in rat plasma and brain.
Xenobiotic distribution to the brain is limited by the BBB, which acts as a self-defense mechanism that prevents drugs from entering11. The BBB expresses a number of effluxtransporters such as P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) and uptake transporters including OATP1A2 and 2B112,13. If active efflux and/or uptake transporters are involved in the distribution of 3-OH-NBP and 10-OH-NBP in the brain, these profiles might be isomer-selective. P-gp-mediated efflux transport across the BBB was reported to be responsible for enantiomer-selective brain penetration of cetirizine14,15.
In addition to active transport, protein binding in plasma and the brain also significantly affects the distribution of drugs to the brain11. It is believed that only small molecules with molecular weights of less than 400–600 Da may diffuse across the BBB, allowing them to act as effective neuro-pharmaceuticals16,17. Hence, only free unbound molecules can penetrate the BBB; 3-OH-NBP and 10-OH-NBP bound to plasma proteins would be blocked by the BBB. Plasma protein binding and brain protein binding have opposite effects on the penetration of molecules across the BBB. Greater plasma protein binding reduces the free fraction of molecules that can be exposed to the BBB, whereas greater brain protein binding facilitates equilibrium toward diffusing across the BBB. Other factors may also contribute to the distinct distribution of 3-OH-NBP and 10-OH-NBP, such as potential isomer-selective metabolism of NBP in rat brain.
In the present study, we aimed to elucidate the mechanism (s) underlying the unique distributions of 3-OH-NBP and 10-OH-NBP in rat plasma and brain, and provide a basis for developing novel anti-cerebral ischemic agents using the NBP structure as a potential foundation.
Materials and methods
Materials
NBP was kindly provided by the Shijiazhuang Pharmaceutical Company (Shijiazhuang, Hebei, China). 3-OH-NBP, d4-3-OH-NBP, 10-OH-NBP, and d4-10-OH-NBP were synthesized as previously described with some modifications18,19. Tariquidar was purchased from J&K Scientific (Beijing, China). Pantoprazole was kindly provided by Jiangsu Chia-Tai Tianqing Pharmaceutical Co, Ltd (Nanjing, Jiangsu, China).
Animals
Male Sprague-Dawley (SD) rats (180–220 g, 8–9 weeks old) were provided by the Animal Center of Shanghai Institute of Materia Medica, Chinese Academy of Sciences (Shanghai, China). Animals were kept in a specific pathogen-free room maintained at 25 °C with humidity of 50%±10% and a 12-h dark/light cycle. Rats had free access to water and food throughout the studies. All rats were kept for one week to acclimate to the environment and were fasted overnight the day before the experiments. All study protocols were approved by the Animal Ethics Committee of Shanghai Institute of Materia Medica, Chinese Academy of Sciences (Shanghai, China), and the welfare of the rats received the highest consideration during the study.
Identification of NBP metabolites in rat brain and plasma
SD rats (n=3) were orally administered 60 mg/kg NBP dissolved in soybean oil. The rats were euthanized 1 h after drug administration, and blood and brain tissue samples were collected. Plasma samples were obtained by centrifuging the blood at 3500×g for 5 min. Then, 100 μL of plasma was precipitated by adding 300 μL acetonitrile, vortexed for 1 min, and centrifuged at 11 000×g for 5 min. The supernatant was dried under nitrogen at 40 °C and reconstituted in 100 μL of methanol and 5 mmol/L ammonium acetate (10:90, v/v). Brain tissue samples were homogenized in 5 volumes of methanol (1 g of tissue in 5 mL of methanol). Then, 100 μL of the brain homogenate was pretreated as described for plasma. Next, 10 μL of the reconstituted brain or plasma samples was subjected to ultra-performance liquid chromatography-ultraviolet-quadrupole/time-of-flight mass spectrometry (UPLC-UV-Q/TOF MS; Waters, Milford, MA, USA) analysis, as described in our previous study8,20. The UV wavelength was set at 230 nm.
Pharmacokinetics of 3-OH-NBP and 10-OH-NBP in rat plasma and brain
To elucidate the disproportional distribution of 3-OH-NBP and 10-OH-NBP inside and outside the BBB, we quantitatively determined the pharmacokinetics of 3-OH-NBP and 10-OH-NBP in rat plasma and brain. SD rats (n=21, 180–220 g) were randomly divided into 7 groups with 3 rats in each group. Each rat was orally administered 20 mg/kg NBP dissolved in soybean oil. The rats were euthanized at 0.25, 0.5, 1, 2, 4, 8, or 24 h after drug administration, and blood and brain tissue were collected. Plasma samples were obtained by centrifuging the blood at 3500×g for 5 min, and brain tissue was homogenized in 5 volumes of methanol (1 g of tissue in 5 mL of methanol). Next, we added 100 μL of an internal standard (IS) solution (600 ng/mL d4-3-OH-NBP for 3-OH-NBP and 300 ng/mL d4-10-OH-NBP for 10-OH-NBP) to 100 μL of the plasma or brain homogenate. The samples were deproteinized with 200 μL acetonitrile, and the precipitate was removed by centrifugation at 11 000×g for 5 min. The supernatant was dried under nitrogen at 40 °C and reconstituted in 100 μL of methanol/acetonitrile/5 mmol/L ammonium acetate (1:2:9, v/v/v). Finally, 10 μL of the reconstituted solution was analyzed by LC-MS/MS. The concentrations of 3-OH-NBP and 10-OH-NBP were determined using a Shimadzu LC-20AD HPLC system (Kyoto, Japan) coupled to an API4000 triple quadrupole MS (Sciex, Ontario, Canada) that was equipped with a TurboIonSpray ion source. Chromatographic separation was achieved on a Zorbax XDB-C18 column (50×4.6 mm, id, 1.8 μm; Agilent, Santa Clara, CA, USA) under gradient elution as previously described9. The MS was operated in the negative-positive switch mode: 0–3.5 min, (−)ESI for 3-OH-NBP and (+)ESI for 10-OH-NBP from 3.5–8.0 min. In the (−)ESI mode, the MS parameters were as follows: curtain gas, 10 psi; nebulizer gas (GS1), 40 psi; turbo gas (GS2), 40 psi; ion spray voltage, −3500 V; declustering potential, −50 V; and source temperature, 400 °C. In the (+)ESI mode, the MS parameters were as follows: curtain gas, 10 psi; GS1, 40 psi; GS2, 40 psi; ion spray voltage, +4200 V; declustering potential, 33 V; and source temperature, 400 °C. The ion transitions for multiple reaction monitoring were m/z 205.1→m/z 161.1 for 3-OH-NBP, m/z 209.1→m/z 165.1 for d4-3-OH-NBP, m/z 207.1→m/z 128.1 for 10-OH-NBP, and m/z 211.1→m/z 132.1 for d4-10-OH-NBP. The respective collision energies for 3-OH-NBP, d4-3-OH-NBP, 10-OH-NBP, and d4-10-OH-NBP were set to −18, −18, 38, and 38 eV.
NBP metabolism in rat brain homogenate
An untreated SD rat was euthanized and blank brain homogenate was prepared by homogenizing brain tissue in 5 volumes of 100 mmol/L phosphate-buffered saline (PBS; 1 g of tissue in 5 mL of PBS). The metabolism of NBP was investigated by incubating the blank brain homogenate with NBP in the presence of the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH). The incubation mixture contained freshly prepared brain homogenate (50 μL), NADPH (2.0 mmol/L), and NBP (50 μmol/L) in a total volume of 200 μL. After 1 h of incubation at 37 °C, the mixture was quenched with an equal volume of ice-cold acetonitrile. Next, 100 μL of the sample was treated as described in “Identification of NBP metabolites in rat brain and plasma,” and UPLC-UV-Q/TOF MS was used to identify the metabolites of NBP.
Plasma and brain protein binding of 3-OH-NBP and 10-OH-NBP
Authentic plasma samples (2 samples at 0.5 h and 2 samples at 2 h) from “Pharmacokinetics of 3-OH-NBP and 10-OH-NBP in rat plasma and brain” were used to determine the plasma protein binding of 3-OH-NBP and 10-OH-NBP. The protein binding ratio was evaluated using the commonly used ultracentrifugation method21,22. All four samples were incubated in a water bath at 37 °C for 0.5 h and then centrifuged at 100 000×g for 16 h at 37 °C. The concentrations of 3-OH-NBP and 10-OH-NBP in the supernatant were determined by LC-MS/MS, and the unbound fraction (fu,pl) was calculated as Equation (1):
 |
where pl, conc, sup, and tot represent plasma, concentration, supernatant, and total, respectively.
To elucidate brain protein binding, fresh brain tissue was homogenized in 5 volumes of 100 mmol/L PBS (1 g of tissue in 5 mL of PBS) and was used as the matrix. A stock solution of 3-OH-NBP or 10-OH-NBP (1 mg/mL in methanol) was added to blank brain homogenate to final concentrations of 1000 and 200 ng/mL, respectively. The final concentration of methanol in the samples was <0.1% (v/v). The samples were incubated in a water bath at 37 °C for 0.5 h. Then, half of the sample was transferred to a new tube and frozen at −80 °C until analysis. The other half was centrifuged at 100 000×g for 16 h at 37 °C. The concentrations of 3-OH-NBP or 10-OH-NBP in the supernatants and frozen samples were determined by LC-MS/MS as described in “Pharmacokinetics of 3-OH-NBP and 10-OH-NBP in rat plasma and brain.” The unbound fraction in brain homogenate (fu,hom; hom, homogenate) was calculated as Concsup/Conctot, as described for fu,pl of plasma in Equation (1). Assuming there is a nonspecific binding relationship between the drug and tissue, the unbound fraction in non-diluted brain tissue (fu,br; br, brain) can be recalculated from the measured fu in brain homogenate (ie, fu,hom) by adjusting the dilution factor (Df) with Equation (2)23,24:
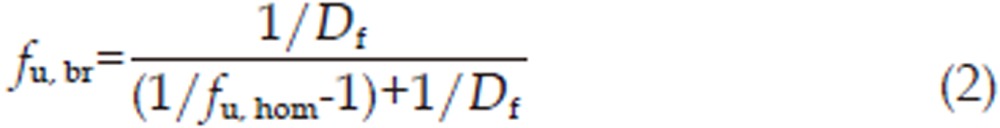 |
Intravenous administration of 3-OH-NBP and 10-OH-NBP
SD rats were randomly divided into 2 groups with 15 rats in each group. 3-OH-NBP and 10-OH-NBP were dissolved together in 5% DMSO and 95% of 40% β-cyclodextrin (40 g β-cyclodextrin in 100 mL of water). The control group (n=15) was intravenously administered 3-OH-NBP (12 mg/kg) and 10-OH-NBP (3 mg/kg). Rats were euthanized at 0.25, 0.5, 1, 3, and 6 h (3 rats per time point) after drug administration, and blood and brain tissue were collected. The experimental group (n=15) was intravenously administered the P-gp inhibitor tariquidar (10 mg/kg) and the BCRP inhibitor pantoprazole (40 mg/kg) 30 min before the intravenous administration of 3-OH-NBP (12 mg/kg) and 10-OH-NBP (3 mg/kg). Rats were treated and euthanized as described for the control group. The concentrations of 3-OH-NBP and 10-OH-NBP were determined using LC-MS/MS as described in “Pharmacokinetics of 3-OH-NBP and 10-OH-NBP in rat plasma and brain.” The Kp,uu value was calculated for each time point using Equation (3):
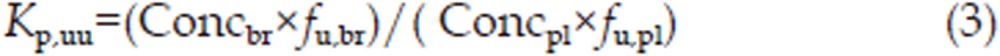 |
Basic relationships
The brain distribution of 3-OH-NBP and 10-OH-NBP was assessed using three different partition coefficients, Kp, Kp,u, and Kp,uu11. The partition coefficient Kp was calculated with Equation (4):
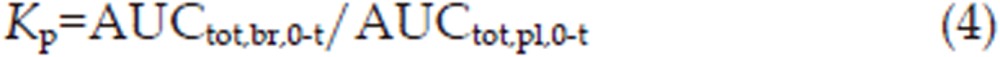 |
where AUCtot,br,0-t and AUCtot,pl,0-t are AUC for the total concentration in the brain and plasma, respectively.
To compensate for differences in plasma protein binding, the partition coefficient Kp,u was used and was calculated with Equation (5):
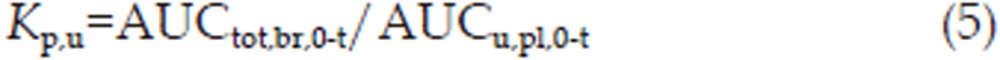 |
where AUCu,pl,0-t is the AUC of the unbound concentration in plasma and was determined as AUCtot,pl,0-t×fu,pl; fu,pl, which is the unbound fraction in plasma.
The ratio of the area under the unbound brain concentration-time profile (AUCu,br,0-t) and the area under the unbound plasma concentration-time profile (AUCu,pl,0-t) was also used to compensate for binding within the brain. Analogous to the partition coefficients described above, this ratio is named the unbound partition coefficient, Kp,uu, and directly describes equilibrium across the BBB25,26
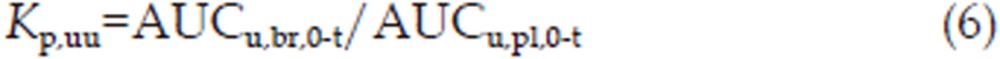 |
where AUCu,br,0-t=AUCtot,br,0-t×fu,br, and fu,br is the unbound fraction of the drug in brain tissue.
Statistical analysis
SPSS software (Statistical Package for the Social Sciences version 16.0 for Windows; SPSS Inc, Chicago, IL, USA) was used to determine significant differences between experimental groups using unpaired two-tailed Student's t-test. Differences of P<0.05 was considered statistically significant. The results are presented as the mean±SD.
Results
Identification of NBP metabolites in rat plasma and brain
UPLC-UV-Q/TOF MS analysis revealed 3 major metabolites in rat plasma at 1 h after oral administration of 60 mg/kg NBP. These metabolites were 10-CO-NBP, 3-OH-NBP, and 10-OH-NBP (Figure 1A). These three metabolites were also observed in rat brain in addition to the parent drug NBP (Figure 1B).
Figure 1.

Metabolic profiles of NBP in rat plasma (A) and brain homogenate (B) 1 h after oral administration of 60 mg/kg NBP as acquired by UPLC-UV.
It is interesting to note that the distribution of 3-OH-NBP and 10-OH-NBP in rat plasma and brain are truly unique. In rat plasma, the UPLC-UV peak area of 3-OH-NBP was approximately 3 times greater than that of 10-OH-NBP. However, in rat brain homogenate, the UPLC-UV peak area of 3-OH-NBP was small, accounting for just 11% that of 10-OH-NBP.
Pharmacokinetics of 3-OH-NBP and 10-OH-NBP in rat plasma and brain
3-OH-NBP and 10-OH-NBP concentrations in rat plasma and brain homogenate were determined after oral administration of 20 mg/kg NBP to rats. In rat plasma, the concentrations of 3-OH-NBP and 10-OH-NBP reached their peaks at 0.5–1.0 h after the dose (Figure 2A). The maximum concentration (Cmax) of 3-OH-NBP was 1472±358 ng/mL, and the Cmax of 10-OH-NBP was 242±163 ng/mL. The AUCtot,pl,0-24h of 3-OH-NBP was 5797±1010 ng·mL−1·h, which was 4.64 times greater than that of 10-OH-NBP (1250±188 ng·mL−1·h).
Figure 2.

Pharmacokinetics of 3-OH-NBP and 10-OH-NBP in rat plasma (A) and brain (B) after oral administration of 20 mg/kg NBP. Pl, plasma; br, brain.
The results were quite different in rat brain homogenate. As shown in Figure 2B, the Cmax of 3-OH-NBP was only 47.9±29.8 ng/g, whereas the Cmax of 10-OH-NBP was 213±160 ng/g. The AUCtot,br,0-24h of 3-OH-NBP (130±44 ng·g−1·h) was 11.8% that of 10-OH-NBP (1101±351 ng·g−1·h).
To more clearly see the differences between 3-OH-NBP and 10-OH-NBP, we reorganized the data in plasma and brain homogenate in terms of the target molecules. As shown in Figure 3A, the Kp (AUCtot,br,0-24h/AUCtot,pl,0-24h) value for 3-OH-NBP was 0.022, whereas the Kp value for 10-OH-NBP was 0.88 (Figure 3B), which indicates that the concentration of 10-OH-NBP was similar inside and outside the BBB, and the concentration of 3-OH-NBP was much lower inside the BBB than outside. The Kp value of 10-OH-NBP was 39.8 times greater than that of 3-OH-NBP (Figure 4), suggesting that 10-OH-NBP can easily cross the BBB into the brain compared to 3-OH-NBP.
Figure 3.

Re-organization of the pharmacokinetics of 3-OH-NBP (A) and 10-OH-NBP (B) in rat plasma and brain in terms of compounds. Mean±SD, n=3.
Figure 4.

The Kp, Kp,u, and Kp,uu values and ratios for 3-OH-NBP and 10-OH-NBP. Mean±SD, n=3.
Metabolism of NBP in the brain homogenate
After incubation of NBP in rat brain homogenate, 3-OH-NBP was detected as a major metabolite (Figure 5); on the contrary, 10-OH-NBP was not detected. This result excluded the possibility that abundant 10-OH-NBP in rat brain was generated by NBP metabolism in the brain.
Figure 5.

Metabolism of NBP in fresh rat brain homogenate in the presence of NADPH: (A) blank brain homogenate without NBP and (B) with NBP as substrate.
Plasma and brain protein binding of 3-OH-NBP and 10-OH-NBP
Ultracentrifugation was used to evaluate the plasma and brain protein binding ratios of 3-OH-NBP and 10-OH-NBP. The results indicated that most of the 3-OH-NBP was bound to rat plasma proteins, and only 22%±2.3% of the circulating concentration was unbound. In rat brain homogenate, all 3-OH-NBP (100%±9.7%) was unbound.
For 10-OH-NBP, the protein-binding properties differed from those of 3-OH-NBP. Overall, 60%±5.2% of 10-OH-NBP was unbound in rat plasma, whereas in rat brain homogenate, 82%±8.3% of 10-OH-NBP was unbound. The unbound fraction of non-diluted brain tissue recalculated from fu in brain homogenate was 49.9%±14.1% using Equation (2).
After accounting for the unbound fraction, the AUC ratio of unbound 3-OH-NBP to unbound 10-OH-NBP decreased from 4.64-fold to 1.70-fold in rat plasma and increased from 11.8% to 24% in rat brain.
The Kp, Kp,u, and Kp,uu values for 3-OH-NBP were 0.022±0.004, 0.101±0.017, and 0.101±0.017, respectively. The corresponding values for 10-OH-NBP were 0.881±0.123, 1.468±0.205, and 0.722±0.102. As shown in Figure 4, the Kp ratio of 10-OH-NBP to 3-OH-NBP was 39.8. After considering plasma protein binding, the Kp,u ratio of 10-OH-NBP to 3-OH-NBP reduced dramatically to 14.6. Furthermore, after considering brain protein binding, the Kp,uu ratio of 10-OH-NBP to 3-OH-NBP decreased to 7.2.
Intravenous administration of 3-OH-NBP and 10-OH-NBP
After intravenous administration of 3-OH-NBP and 10-OH-NBP, we calculated the Kp,uu at each time point. As shown in Figure 6A, the Kp,uu of 3-OH-NBP was between 0.08 and 0.15 in the control group. However, in the group pre-treated with P-gp and BCRP inhibitors, the Kp,uu of 3-OH-NBP significantly increased to 0.20, 0.41, and 0.40 at 0.25, 0.5, and 1.0 h, respectively (P<0.01). As shown in Figure 6B, the Kp,uu of 10-OH-NBP was between 0.44 and 1.11 in the control group. Pre-administration of P-gp and BCRP inhibitors increased the Kp,uu at each time point, however the values were not significantly different from those in the control group except at 0.5 h.
Figure 6.

Kp,uu values of 3-OH-NBP (A) and 10-OH-NBP (B) in rats treated with or without P-gp and BCRP inhibitors. Mean±SD, n=3. bP<0.05, cP<0.01 vs control.
Discussion
NBP is used for the treatment of cerebral ischemia. In our previous study investigating the metabolites of NBP in rat brain, we observed the following interesting phenomena: in rat plasma, exposure of 3-OH-NBP was 4.64 times greater than that of 10-OH-NBP, but exposure of 3-OH-NBP accounted for only 11.8% of 10-OH-NBP in rat brain. In the current study, we attempted to elucidate the mechanisms underlying this unique distribution of 3-OH-NBP and 10-OH-NBP across the BBB.
There are several hypotheses that may help to explain the marked differences in the distribution of 3-OH-NBP and 10-OH-NBP: (1) after NBP crosses the BBB, it is preferentially metabolized to 10-OH-NBP instead of 3-OH-NBP; (2) the plasma protein binding of 3-OH-NBP is much greater than that of 10-OH-NBP, lowering the unbound fraction of 3-OH-NBP and therefore reducing its exposure to the BBB; (3) the brain protein binding of 10-OH-NBP is greater than that of 3-OH-NBP, facilitating the equilibrium of unbound 10-OH-NBP towards penetrating the BBB; (4) efflux transporters prevent 3-OH-NBP from crossing the BBB.
First, we investigated the metabolic profile of NBP with freshly prepared rat brain homogenate. Surprisingly, we found that NBP was preferentially metabolized to 3-OH-NBP; 10-OH-NBP was not detected in the homogenate incubation. This is in direct contrast to our hypothesis. The results exclude the likelihood that the abundant 10-OH-NBP detected in rat brain tissue was generated by the metabolism of NBP in the brain.
Then, we evaluated the plasma and brain tissue protein binding properties of 3-OH-NBP and 10-OH-NBP. The vast difference in the ability of these two molecules to cross the BBB is represented by the difference in the Kp values. The Kp ratio of 10-OH-NBP to 3-OH-NBP was 39.8, indicating that 10-OH-NBP possesses much higher permeability. After incorporating plasma protein binding into the Kp value, we obtained the Kp,u. The plasma protein binding of 3-OH-NBP and 10-OH-NBP behaved in an isomer-selective manner. The Kp,u ratio of 10-OH-NBP to 3-OH-NBP was 14.6. Furthermore, Kp,uu also accounts for binding to brain components. The Kp,uu ratio of 10-OH-NBP to 3-OH-NBP was 7.2. Therefore, it is clear that the dramatic decrease in values from Kp to Kp,u to Kp,uu was driven by differences in plasma and brain protein binding between 10-OH-NBP and 3-OH-NBP. The difference in plasma protein binding was also reported to be responsible for the stereoselective brain penetration of cetirizine11, disopyramide27, and propranolol28 enantiomers.
Kp,uu, calculated from the free concentrations of molecules inside and outside the BBB, represents equilibrium across the BBB11 and is independent of blood and brain protein binding29. The Kp,uu value is indicative of the involvement of molecular active transport across the BBB. A Kp,uu value >1.0 represents uptake across the BBB, whereas a Kp,uu value <1.0 represents efflux transport29. The Kp,uu values of 3-OH-NBP and 10-OH-NBP were 0.10 and 0.72, respectively, indicating that efflux transporters may be involved in the transport of these molecules. In our preliminary studies, we investigated whether 3-OH-NBP was a substrate of P-gp or BCRP in P-gp- and BCRP-overexpressing MDCK (Madin-Darby canine kidney) cell lines. The ER (efflux ratio) was approximately 1.4 and was not significantly different from the control group. We cannot yet rule out that 3-OH-NBP is not a P-gp or BCRP substrate because 3-OH-NBP has high membrane permeability (4.4–8.7×10−6 cm/s). It is usually difficult to confirm whether a highly permeable compound is a P-gp or BCRP substrate in vitro30, and thus, we directly investigated the effect of P-gp/BCRP inhibitors on the 3-OH-NBP distribution in rats that were intravenously administered 3-OH-NBP and 10-OH-NBP in the absence and presence of P-gp and BCRP inhibitors. Pre-administration of the P-gp inhibitor tariquidar and the BCRP inhibitor pantoprazole significantly increased the Kp,uu of 3-OH-NBP from 0.12 to 0.41 at 0.5 h and from 0.15 to 0.40 at 1 h (P<0.001). The Kp,uu values at 3 h and 6 h were very similar to those in the control group, most likely because the inhibitors were eliminated from circulation31,32. Unlike 3-OH-NBP, the Kp,uu value for 10-OH-NBP was only slightly increased as a result of prior administration of P-gp and BCRP inhibitors, which implies that weak efflux is involved in the transport of 10-OH-NBP; however, the Kp,uu values were not significantly different from those in the control group, except at 0.5 h.
These results suggest that 3-OH-NBP is a much stronger substrate than 10-OH-NBP for the P-gp and BCRP efflux transporters across the BBB. The efflux transporters of P-gp and BCRP have many overlapping substrates, such as imatinib33, sorafenib34, and lapatinib35. Within the BBB, P-gp and BCRP work together and prevent xenobiotics from entering the brain. This synergistic effect was also observed for the efflux of lapatinib35, imatinib36, vandetanib37, and regorafenib38. In addition to P-gp and BCRP, multidrug resistance protein 4/5 (MRP4/5), another major efflux transporter in the BBB endothelium, is drawing increasing attention in recent years39,40,41. Although administration of P-gp and BCRP inhibitors increased the Kp,uu value of 3-OH-NBP to approximately 0.4, it was still <1.0. These findings suggest that the efflux transporters were not completely inhibited. It is also possible that 3-OH-NBP is a substrate for other efflux transporters, such as MRP4 and/or MRP5, which needs further investigation.
In conclusion, our study demonstrated that the isomer-selective distribution of 3-OH-NBP and 10-OH-NBP across the BBB was mainly caused by plasma and brain protein binding differences and the efflux transport of 3-OH-NBP. We also excluded the possibility that high exposure of 10-OH-NBP was generated by the metabolism of NBP in the rat brain.
Author contribution
Xing-xing DIAO and Xiao-yan CHEN were responsible for the research design, data analysis, and writing of this paper; Xing-xing DIAO, Kan ZHONG, Xiu-li LI performed experiments; Da-fang ZHONG, a senior advisor, provided valuable advice for this study.
Acknowledgments
The authors thank Prof Hong-jian ZHANG (Soochow University, Suzhou, China) and Dr Zi-qiang (Zack) CHENG (AstraZeneca China, Shanghai, China) for helpful discussions. We also thank Ms Ye-dong WANG (Soochow University, Suzhou, China) for her help in conducting the in vitro experiments.
References
- 1NBP Pharmaceutical Co. L. Butylphthalide soft capsules package insert. http://www.nbp.com.cn/en/Produc_Show.asp?ProducID=197. 2009.
- 2Diao X, Ma Z, Lei P, Zhong D, Zhang Y, Chen X. Enantioselective determination of 3-n-butylphthalide (NBP) in human plasma by liquid chromatography on a teicoplanin-based chiral column coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2013; 939: 67–72. [DOI] [PubMed] [Google Scholar]
- 3Yan C, Feng Y, Zhang J. Effects of dl-3-n-butylphthalide on regional cerebral blood flow in right middle cerebral artery occlusion rats. Acta Pharmacol Sin 1998; 19: 117–20. [PubMed] [Google Scholar]
- 4Li J, Li Y, Ogle M, Zhou X, Song M, Yu SP, et al. DL-3-n-butylphthalide prevents neuronal cell death after focal cerebral ischemia in mice via the JNK pathway. Brain Res 2010; 1359: 216–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5Ji XC, Zhao WH, Cao DX, Shi QQ, Wang XL. Novel neuroprotectant chiral 3-n-butylphthalide inhibits tandem-pore-domain potassium channel TREK-1. Acta Pharmacol Sin 2011; 32: 182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6Chang Q, Wang XL. Effects of chiral 3-n-butylphthalide on apoptosis induced by transient focal cerebral ischemia in rats. Acta Pharmacol Sin 2003; 24: 796–804. [PubMed] [Google Scholar]
- 7Zhao W, Luo C, Wang J, Gong J, Li B, Gong Y, et al. 3-N-butylphthalide improves neuronal morphology after chronic cerebral ischemia. Neural Regen Res 2014; 9: 719–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8Diao X, Deng P, Xie C, Li X, Zhong D, Zhang Y, et al. Metabolism and pharmacokinetics of 3-n-butylphthalide (NBP) in humans: the role of cytochrome P450s and alcohol dehydrogenase in biotransformation. Drug Metab Dispos 2013; 41: 430–44. [DOI] [PubMed] [Google Scholar]
- 9Diao X, Ma Z, Wang H, Zhong D, Zhang Y, Jin J, et al. Simultaneous quantitation of 3-n-butylphthalide (NBP) and its four major metabolites in human plasma by LC-MS/MS using deuterated internal standards. J Pharm Biomed Anal 2013; 78–79C: 19–26. [DOI] [PubMed] [Google Scholar]
- 10Wang X, Wang L, Huang Z, Sheng X, Li T, Ji H, et al. Synthesis and biological evaluation of nitric oxide releasing derivatives of 6-amino-3-n-butylphthalide as potential antiplatelet agents. Bioorg Med Chem Lett 2013; 23: 1985–8. [DOI] [PubMed] [Google Scholar]
- 11Gupta A, Chatelain P, Massingham R, Jonsson EN, Hammarlund-Udenaes M. Brain distribution of cetirizine enantiomers: comparison of three different tissue-to-plasma partition coefficients: Kp, Kp,u, and Kp,uu. Drug Metab Dispos 2006; 34: 318–23. [DOI] [PubMed] [Google Scholar]
- 12Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov 2010; 9: 215–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13Goldwirt L, Beccaria K, Carpentier A, Farinotti R, Fernandez C. Irinotecan and temozolomide brain distribution: a focus on ABCB1. Cancer Chemother Pharmacol 2014; 74: 185–93. [DOI] [PubMed] [Google Scholar]
- 14Chen C, Hanson E, Watson JW, Lee JS. P-glycoprotein limits the brain penetration of nonsedating but not sedating H1-antagonists. Drug Metab Dispos 2003; 31: 312–8. [DOI] [PubMed] [Google Scholar]
- 15Polli JW, Baughman TM, Humphreys JE, Jordan KH, Mote AL, Salisbury JA, et al. P-glycoprotein influences the brain concentrations of cetirizine (Zyrtec), a second-generation non-sedating antihistamine. J Pharm Sci 2003; 92: 2082–9. [DOI] [PubMed] [Google Scholar]
- 16Pardridge WM. Transport of small molecules through the blood-brain-barrier - biology and methodology. Adv Drug Del Rev 1995; 15: 5–36. [PubMed] [Google Scholar]
- 17Khawli LA, Prabhu S. Drug delivery across the blood-brain barrier. Mol Pharm 2013; 10: 1471–2. [DOI] [PubMed] [Google Scholar]
- 18Yang H, Hu GY, Chen J, Wang Y, Wang ZH. Synthesis, resolution, and antiplatelet activity of 3-substituted 1(3H)-isobenzofuranone. Bioorg Med Chem Lett 2007; 17: 5210–3. [DOI] [PubMed] [Google Scholar]
- 19Brenstrum TJ, Brimble MA, Stevenson RJ. Directed ortho metalations of tertiary benzamides using lactones as electrophiles. Tetrahedron 1994; 50: 4897–904. [Google Scholar]
- 20Diao X, Pang X, Xie C, Guo Z, Zhong D, Chen X. Bioactivation of 3-n-butylphthalide via sulfation of its major metabolite 3-hydroxy-NBP: mediated mainly by sulfotransferase 1A1. Drug Metab Dispos 2014; 42: 774–81. [DOI] [PubMed] [Google Scholar]
- 21Srikanth CH, Chaira T, Sampathi S, Sreekumar VB, Bambal RB. Correlation of in vitro and in vivo plasma protein binding using ultracentrifugation and UPLC-tandem mass spectrometry. Analyst 2013; 138: 6106–16. [DOI] [PubMed] [Google Scholar]
- 22Zhang F, Xue J, Shao J, Jia L. Compilation of 222 drugs' plasma protein binding data and guidance for study designs. Drug Discov Today 2012; 17: 475–85. [DOI] [PubMed] [Google Scholar]
- 23Wan H, Rehngren M, Giordanetto F, Bergstrom F, Tunek A. High-throughput screening of drug-brain tissue binding and in silico prediction for assessment of central nervous system drug delivery. J Med Chem 2007; 50: 4606–15. [DOI] [PubMed] [Google Scholar]
- 24Kalvass JC, Maurer TS. Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos 2002; 23: 327–38. [DOI] [PubMed] [Google Scholar]
- 25Liu X, Chen C. Strategies to optimize brain penetration in drug discovery. Curr Opin Drug Discov Devel 2005; 8: 505–12. [PubMed] [Google Scholar]
- 26Kalvass JC, Maurer TS, Pollack GM. Use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs: comparison of unbound concentration ratios to in vivo p-glycoprotein efflux ratios. Drug Metab Dispos 2007; 35: 660–6. [DOI] [PubMed] [Google Scholar]
- 27Hanada K, Akimoto S, Mitsui K, Mihara K, Ogata H. Enantioselective tissue distribution of the basic drugs disopyramide, flecainide and verapamil in rats: role of plasma protein and tissue phosphatidylserine binding. Pharm Res 1998; 15: 1250–6. [DOI] [PubMed] [Google Scholar]
- 28Takahashi H, Ogata H, Kanno S, Takeuchi H. Plasma protein binding of propranolol enantiomers as a major determinant of their stereoselective tissue distribution in rats. J Pharmacol Exp Ther 1990; 252: 272–8. [PubMed] [Google Scholar]
- 29Hammarlund-Udenaes M, Paalzow LK, de Lange EC. Drug equilibration across the blood-brain barrier--pharmacokinetic considerations based on the microdialysis method. Pharm Res 1997; 14: 128–34. [DOI] [PubMed] [Google Scholar]
- 30Shugarts S, Benet LZ. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm Res 2009; 26: 2039–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31Xie Z, Zhang Y, Xu H, Zhong D. Pharmacokinetic differences between pantoprazole enantiomers in rats. Pharm Res 2005; 22: 1678–84. [DOI] [PubMed] [Google Scholar]
- 32Bauer M, Zeitlinger M, Karch R, Matzneller P, Stanek J, Jager W, et al. Pgp-mediated interaction between (R)-11C]verapamil and tariquidar at the human blood-brain barrier: a comparison with rat data. Clin Pharmacol Ther 2012; 91: 227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33Graber-Maier A, Gutmann H, Drewe J. A new intestinal cell culture model to discriminate the relative contribution of P-gp and BCRP on transport of substrates such as imatinib. Mol Pharm 2010; 7: 1618–28. [DOI] [PubMed] [Google Scholar]
- 34Swift B, Nebot N, Lee JK, Han T, Proctor WR, Thakker DR, et al. Sorafenib hepatobiliary disposition: mechanisms of hepatic uptake and disposition of generated metabolites. Drug Metab Dispos 2013; 41: 1179–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, et al. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino }methyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab Dispos 2009; 37: 439–42. [DOI] [PubMed] [Google Scholar]
- 36Bihorel S, Camenisch G, Lemaire M, Scherrmann JM. Modulation of the brain distribution of imatinib and its metabolites in mice by valspodar, zosuquidar and elacridar. Pharm Res 2007; 24: 1720–8. [DOI] [PubMed] [Google Scholar]
- 37Minocha M, Khurana V, Qin B, Pal D, Mitra AK. Co-administration strategy to enhance brain accumulation of vandetanib by modulating P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp1/Abcg2) mediated efflux with m-TOR inhibitors. Int J Pharm 2012; 434: 306–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38Durmus S, Sparidans RW, van Esch A, Wagenaar E, Beijnen JH, Schinkel AH. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) restrict oral availability and brain accumulation of the PARP inhibitor rucaparib (AG-014699). Pharm Res 2015; 32: 37–46. [DOI] [PubMed] [Google Scholar]
- 39Hillgren KM, Keppler D, Zur AA, Giacomini KM, Stieger B, Cass CE, et al. Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther 2013; 94: 52–63. [DOI] [PubMed] [Google Scholar]
- 40Russel FG, Koenderink JB, Masereeuw R. Multidrug resistance protein 4 (MRP4/ABCC4): a versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol Sci 2008; 29: 200–7. [DOI] [PubMed] [Google Scholar]
- 41Georgieva JV, Hoekstra D, Zuhorn IS. Smuggling drugs into the brain: an overview of ligands targeting transcytosis for drug delivery across the blood-brain barrier. Pharmaceutics 2014; 6: 557–83. [DOI] [PMC free article] [PubMed] [Google Scholar]