

SUMMARY
Monocarboxylate Transporter 1 (MCT1) inhibition is thought to block tumor growth through disruption of lactate transport and glycolysis. Here we show MCT1 inhibition impairs proliferation of glycolytic breast cancer cells co-expressing MCT1 and MCT4 via disruption of pyruvate rather than lactate export. MCT1 expression is elevated in glycolytic breast tumors, and high MCT1 expression predicts poor prognosis in breast and lung cancer patients. Acute MCT1 inhibition reduces pyruvate export but does not consistently alter lactate transport or glycolytic flux in breast cancer cells that co-express MCT1 and MCT4. Despite the lack of glycolysis impairment, MCT1 loss-of-function decreases breast cancer cell proliferation and blocks growth of mammary fat pad xenograft tumors. Our data suggest MCT1 expression is elevated in glycolytic cancers to promote pyruvate export, which when inhibited enhances oxidative metabolism and reduces proliferation. This study presents an alternative molecular consequence of MCT1 inhibitors further supporting their use as anti-cancer therapeutics.
INTRODUCTION
Since glycolytic metabolism contributes to tumor growth in many cancers, efforts have been made to block tumor glycolysis by inhibiting the monocarboxylate transporters (MCTs) that regulate cancer cell lactate export. The MCT family includes 14 members, but only MCT1-4 have been demonstrated to mediate proton-linked bi-directional transport of monocarboxylates such as lactate, pyruvate, and ketone bodies across the plasma membrane (Halestrap and Meredith, 2004). Tumor lactate export is thought to be primarily mediated by MCT1 and MCT4, since these are the family members most commonly upregulated in cancers (Halestrap and Meredith, 2004; Halestrap and Wilson, 2012). SLC16A1, the gene that encodes MCT1, was recently reported to be a MYC transcriptional target essential for lactate transport and glycolytic flux of certain cancer cell lines (Doherty et al., 2014). MCT1 inhibition induces cell death in Burkitt lymphoma cells and MCF7 breast cancer cells through disruption of lactate export, glycolysis and glutathione synthesis (Doherty et al., 2014). Consistently, small molecule inhibitors of MCT1 block activation of T cells reliant on increased glycolysis for proliferation through abrogation of lactate export (Guile et al., 2006; Murray et al., 2005). AZD3965 is a MCT1 inhibitor that is currently undergoing phase I evaluation in the United Kingdom for patients with solid tumors, prostate cancer, gastric cancer, and diffuse large cell B lymphoma (Polanski et al., 2014). Multiple studies, including one using AZD3965, show that MCT4 expression can portend resistance to MCT1 inhibition. Consistent with previous studies, here we show that MCT1 expression correlates with breast cancer glycolytic phenotype and aggressiveness. However, we also find that MCT1 loss of function reduces pyruvate, but not lactate export in glycolytic breast cancer cells that co-express MCT1 and MCT4, which leads to enhanced oxidative metabolism and decreased proliferation, thus presenting an alternative mode of action of MCT1 inhibitors.
RESULTS
Unbiased gene expression analysis finds that MCT1 mRNA levels correlate with glycolytic metabolism in breast cancer cells
To identify specific transcriptional events that correlate with glycolytic phenotype in breast cancer, we analyzed gene expression profiles from eleven patient breast tumors stratified by FDG uptake and thirty-one breast cancer cell lines that we stratified based on glycolytic versus oxidative phenotype (nmol lactate produced/nmol oxygen consumed) (Figure S1a,b) (Neve et al., 2006; Palaskas et al., 2011). As shown in Fig. 1a, tumors with high FDG uptake exhibit a distinct transcriptional signature from those with low FDG uptake. Gene Set Enrichment Analysis confirmed that MYC-regulated gene sets are significantly enriched in the glycolytic breast tumors and cell lines (Figure S1c, Table S1) (Palaskas et al., 2011). Additionally, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways involved in nucleotide metabolism and glycolysis are also enriched in the glycolytic tumors and cell lines (Fig. 1a, Table S2) (Kanehisa et al., 2014). Consistent with previous findings (Palaskas et al., 2011), the glycolytic tumor and cell line gene expression signature significantly correlates with the basal gene expression signature in breast cancer (Chang et al., 2005) (Figure S1d,e). Mapping the glycolytic gene expression signature to the KEGG glycolysis pathway demonstrates coordinated upregulation of glycolytic genes including HK2, PFKP, BPGM, ENO3 and LDHB (Fig. 1b, Figure S1f,g). Together, these data demonstrate that glycolytic tumors and cell lines exhibit a gene expression signature consistent with the Warburg effect.
Figure 1. Unbiased gene expression analysis finds that MCT1 correlates with glycolytic phenotype in breast cancer.

a, Breast tumors with high and low FDG uptake have distinct gene expression signatures. Transcript levels from 11 human breast cancers were ranked by the average correlation with tumor FDG maximum standardized uptake value (SUVmax) and cell line glycolytic phenotype (nmol lactate produced/nmol oxygen consumed) and arranged from left to right in order of increasing FDG uptake. Red and green denote high and low average correlation coefficients, respectively. MCT1 is the top-ranked of 13,374 genes. The inset table lists metabolic pathways enriched in highly glycolytic tumors and cell lines (PPP, pentose phosphate pathway; Glyc/Gluc, glycolysis/gluconeogenesis). b, Highly glycolytic tumors and cell lines demonstrate coordinate upregulation of glycolysis genes and MCT1. Genes within the glycolysis pathway are colored red or green to denote high and low correlation coefficients with glycolytic phenotype, respectively. c, Levels of MCT1, but not other MCT family members, are highly correlated with glycolytic phenotype in breast tumors and cell lines. The Pearson correlation coefficient with FDG uptake in human breast tumors (left) and glycolytic phenotype in human breast cancer cell lines (right) is depicted for microarray probes recognizing MCT1-4 family members. d, Scatter plots of MCT1 and MCT4 expression demonstrate that MCT1 but not MCT4 is highly correlated with glycolytic phenotypes. Transcript levels are plotted versus FDG uptake for human breast tumors (left) and glycolytic phenotype for human breast cancer cell lines (right). P-values are the two-tailed significance of the Pearson correlation coefficient. See also Figure S1.
Notably, the top ranked transcript correlating with glycolytic phenotype in breast tumors and cell lines is Solute Carrier 16A1 (SLC16A1), encoding MCT1 (Fig. 1a, b). Since MCT1-4 mediate monocarboxylate transport in cells, we analyzed mRNA expression patterns of the corresponding genes in breast tumors and cell lines (Fig. 1b, c). Only MCT1 mRNA expression yields consistently strong correlation coefficients with glycolytic phenotype in both breast tumors and cell lines (Fig. 1b–d). In contrast, MCT4 mRNA expression is less correlated with glycolytic phenotype (Fig. 1b–d). However, MCT1-4 mRNA levels in cancer cells may not reflect protein levels or transporter activity, especially since MCT4 mRNA levels have been shown to correlate poorly with protein levels in muscle (Bonen et al., 2000).
MCT1 protein levels are elevated in malignant breast and lung cancer lesions
To determine whether MCT1 protein expression is elevated in primary cancers, we analyzed normal and malignant breast and lung tissues by immunohistochemistry using high-density tissue microarrays (TMAs). MCT1 protein expression is significantly increased in malignant breast and lung tissues compared to adjacent non-malignant tissues (Fig. 2a–c). Later stage lung cancers (Stage II–IV) have greater MCT1 expression than those in early stages (Stage I) (integrated intensity = 0.44 ± 0.08 in Stage II–IV (n = 174) versus integrated intensity = 0.29 ± 0.03 in Stage I (n=216), p < 0.01). Additionally, high MCT1 expression is associated with worse prognosis in breast and lung cancer patients (Fig. 2d, e). These findings corroborate published results showing MCT1 elevation in basal-like breast carcinoma (Pinheiro et al., 2010) and colorectal carcinomas (Pinheiro et al., 2008) as well as studies showing association of MCT1 expression with poor prognosis in epithelial ovarian cancer (Chen et al., 2010) and gastric cancer (Pinheiro et al., 2009). These results are inconsistent with a previous report that did not find elevated MCT1 expression by immunohistochemistry in lung adenocarcinomas (McCleland et al., 2013). One potential explanation for this discrepancy is the use of more lung adenocarcinoma patient samples in our study (715 versus 226). Additionally, while we found significantly elevated MCT1 expression in lung adenocarcinomas compared to adjacent nonmalignant tissue, we found an even greater increase in MCT1 expression in squamous cell and large cell carcinoma tissues. Furthermore, we found that serum lactate and pyruvate concentrations are significantly elevated in Stage IV versus Stage I lung cancer patients (Fig. 2f), consistent with MCT1 modulation of tumor cell lactate and pyruvate export in vivo. However, no significant difference in lactate or pyruvate levels was observed in serum from Stage IV versus Stage I breast cancer patients (data not shown).
Figure 2. Elevated MCT1 levels are indicative of tumor malignancy and poor breast and lung cancer patient survival.
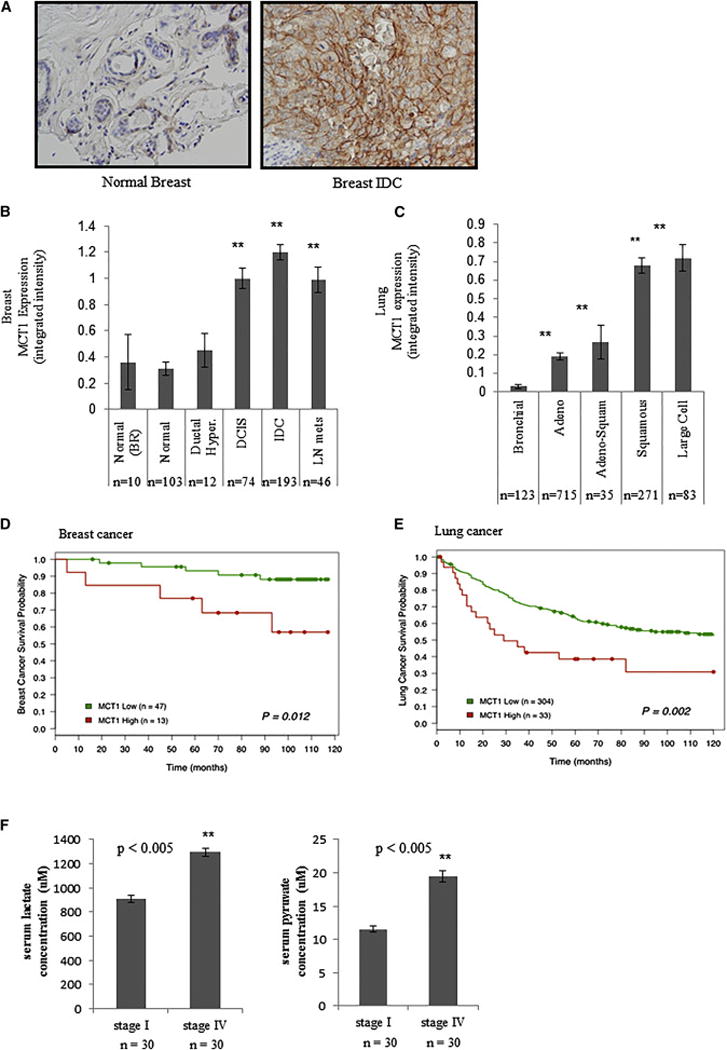
a, Immunohistochemistry of human normal breast and breast invasive ductal carcinoma (IDC) with an antibody towards MCT1 indicates higher MCT1 expression in the malignant tissue. Images are shown at X 100 magnification. b, The mean integrated MCT1 expression as determined by immunohistochemistry on a breast tissue microarray is compared across breast histologies and histopathologies. MCT1 expression is significantly increased in ductal carcinoma in situ (DCIS, P = 0.003), invasive ductal carcinoma (IDC, P < 0.001), and lymph node metastatic lesions (LN mets, P = 0.004) compared to adjacent non-malignant glandular epithelium (“normal”) or epithelium from voluntary breast reductions (BR). There is no significant difference between normal breast glandular epithelium and ductal hyperplasia (Ductal Hyper.) lesions. c, The mean integrated MCT1 expression as determined by immunohistochemistry on a lung tissue microarrary is compared across lung histologies and histopathologies. MCT1 expression is significantly increased in adenocarcinoma (Adeno, P < 0.001), adeno-squamous (Adeno-Squam, P < 0.001), squamous cell (P < 0.001) and large cell (P < 0.001) compared to adjacent non-malignant bronchial epithelium. Expression of MCT1 is significantly elevated in squamous and large cell tumors compared to adenocarcinomas and adeno-squamous tumors (P < 0.001 for both). For b, c, error bars denote standard error of the mean, and n = number of tissue array spots analyzed. d, Higher MCT1 expression levels predicts poorer survival in women with invasive ductal carcinoma of the breast. Kaplan-Meier survival plot shows patients with lower MCT1 expression (< 2.0 mean integrated intensity) depicted as a green line, and higher MCT1 expression (≥ 2.0 mean integrated intensity) depicted as a red line. e, Higher MCT1 expression levels predict poorer survival in individuals with NSCLC. Kaplan-Meier survival plot shows patients with lower MCT1 expression (≤ 1.0 mean integrated intensity) depicted as a green line and higher MCT1 expression (> 1.0 mean integrated intensity) depicted as a red line. f, Serum lactate (left) and pyruvate (right) concentrations from Stage I versus Stage IV lung cancer patients. For d, e, f, n = number of individuals in each category.
MCT1 inhibition reduces pyruvate but not lactate export, and enhances oxidative metabolism in glycolytic breast cancer cells
Since MCT1 levels are elevated in glycolytic and malignant breast tumors, we hypothesized that MCT1 may contribute to the Warburg effect metabolic phenotype. To test this hypothesis, we generated breast cancer cell lines with short hairpin (sh)RNA-mediated stable knockdown of MCT1 (Figure S2a). The cell lines used – HS578T, SUM149PT, and SUM159PT – are among the most glycolytic in our panel of 31 breast cancer cell lines (Figure S1b). Using a threshold-free comparison of genome-wide expression patterns (Plaisier et al., 2010), we found that MCT1 knockdown in SUM149PT cells abrogated the similarity of these glycolytic cells to tumors with high FDG uptake (Fig. 3a). Additionally, MCT1 knockdown results in altered expression of several nodes associated with highly glycolytic phenotypes, including HK1, PFKM. BPGM, and ENO1 (Figure S2b). Treatment of HS578T cells for 24 hours with an MCT1 inhibitor (AZD3965) induced a gene expression signature that strongly resembles that of SUM149PT cells with stable MCT1 knockdown (Figure S2c). Treatment of SUM149PT and SUM159PT cells with the MCT1 inhibitor abrogated the similarity to SUM149PT cells expressing scrambled shRNA. Additionally, MCT1 inhibition decreased the similarity of these glycolytic cells to tumors with high FDG uptake (Figure S2d). Gene set enrichment analysis (GSEA) of MCT1 knockdown SUM149PT cells, as well as MCT1 inhibitor (AZD3965)-treated SUM149PT cells, showed enrichment in genes involved in oxidative phosphorylation, pyruvate metabolism, the TCA cycle, and, surprisingly, glycolysis (Fig. 3b, Figure S2e). Enriched expression of oxidative phosphorylation genes was also observed upon MCT1 inhibition for 24 hours in additional breast cancer cell lines (Figure S2f). Consistently, MCT1 inhibition results in increased oxygen consumption rates in multiple breast cancer cell lines after 24 hours treatment, but not after 0.5 or 4 hours AZD3965 treatment (Fig. 3c, Figure S3a,b,c). Notably however, lactate export rates are not consistently altered upon MCT1 inhibition (Fig. 3d, Figure S3d,e) or MCT1 knockdown (Figure S3f). Additionally, glycolytic flux as determined by conversion of 1,2-13C-glucose to 1,2-13C-lactate is also not consistently altered by MCT1 inhibition in breast cancer cell lines (Fig. 3e, Figure S3g). These data suggest that MCT1 inhibition leads to enhanced oxidative metabolism in breast cancer cells through an alternative mechanism than reduced lactate export and disrupted glycolysis.
Figure 3. MCT1 is critical for pyruvate export in breast cancer cell lines.
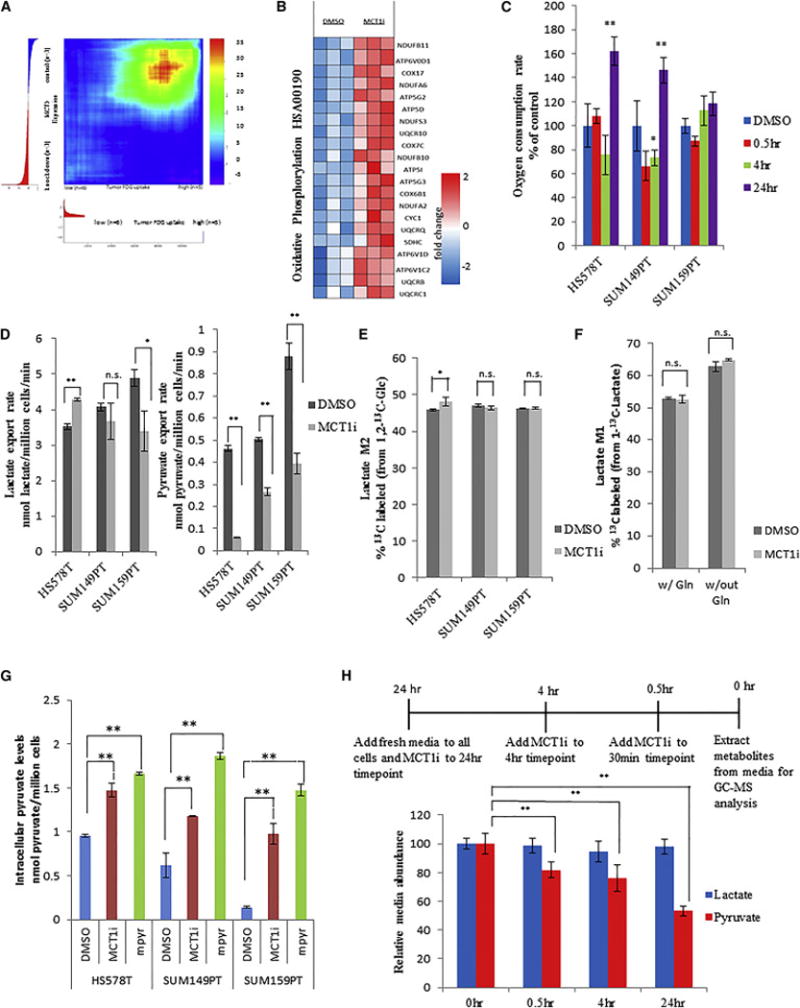
a, Gene expression profiles from SUM149PT cells expressing scrambled shRNA (control) and shRNA towards MCT1 (knockdown) were used to generate a ranked list of transcripts that are differentially expressed upon MCT1 knockdown. This ranked list was compared to a ranked list of transcripts that correlate with high FDG uptake in breast tumors from patients using the rank-rank hypergeometric overlap (RRHO) algorithm. The resulting overlap from the ranked lists, represented as a hypergeometric heat map, indicates that MCT1 knockdown renders glycolytic SUM149PT breast cancer cells less similar to tumors with high FDG uptake. The direction-signed log10-transformed hypergeometric p-values are indicated in the accompanying color scale. b, Heatmap indicating fold changes in expression levels of genes in the oxidative phosphorylation gene set from SUM149PT cells treated with DMSO or AZD3965 (MCT1i) for 24 hr. c, Cellular oxygen consumption rates 24 hrs post treatment with DMSO versus 250 nM AZD3965 (MCT1i). d, Lactate and pyruvate export rates 4 hours post treatment of the indicated cell lines with DMSO or 250 nM AZD3965 (MCT1i). e, Percentage of the 13C-labeled M1 lactate isotopomer from cells 24 hr post labeling with 1-13C-lactate and treatment with DMSO or 250 nM AZD3965 (MCT1i) in the presence or absence of 4 mM glutamine as determined by LC-MS/MS. f, Percentage of the 13C-labeled M2 lactate isotopomer from cells 24 hr post labeling with 1,2-13C-glucose and treatment with DMSO or 250 nM AZD3965 (MCT1i) as determined by LC-MS/MS. g, Intracellular pyruvate levels from the indicated cell lines treated with DMSO, 250 nM MCT1i, or 5 mM methyl-pyruvate for 30min. h, Relative abundance of media lactate and pyruvate post treatment of SUM149PT cells with 250 nM AZD3965 (MCT1i) for the indicated times as determined by GC-MS. Error bars in c denote standard error of mean (n=5). Error bars in d–g denote standard deviation (n=3). * denotes p < 0.05; ** denotes p < 0.01. See also Figure S2 and S3
Since MCT1 has been shown to regulate lactate uptake in cultured endothelial cells and certain cancer cell lines (Sonveaux et al., 2008; Vegran et al., 2011), we next measured the effect of MCT1 inhibition on lactate uptake in glycolytic breast cancer cells. To assess lactate uptake, 11 mM 1-13C-lactate was added to the normal culture medium of SUM149PT cells along with 250 nM AZD3965 or DMSO, and 24 hours later, metabolites were extracted from the cells and media and analyzed by LC-MS/MS. AZD3965 treatment of SUM149PT cells had no effect on intracellular lactate levels, extracellular lactate levels, or percentage of 13C-labeled intracellular lactate (Fig. 3f and Figure S3h,i,j). The same experiment was conducted on SUM149PT cells cultured in media lacking glutamine, and while the percentage of 13C-labeled intracellular lactate increased in the glutamine-deprived cells, MCT1 inhibition again had no effect on lactate levels or labeling (Fig. 3f and Figure S3h,i,j). These results suggest that MCT1 does not impact lactate uptake in glycolytic breast cancer cells.
Since the published Km values for MCT1 interaction with pyruvate are lower than those for other monocarboxylates including lactate and acetate (Km pyruvate = 0.6 – 1.0 mM versus Km lactate = 2.2 – 4.5 mM, Km acetate = 3.7)(Halestrap and Wilson, 2012), we next measured the effect of MCT1 inhibition on pyruvate export rates. As shown in Fig. 3d, all cell lines tested show decreased pyruvate export rates upon MCT1 inhibition with AZD3965, as well as upon stable MCT1 knockdown (Figure S3k). Reduced pyruvate export rates were also observed using an alternative shRNA sequence that knocks down MCT1 expression (Figure S3b,l,m), and expression of shRNA-resistant MCT1 cDNA rescued the reduced pyruvate export rate caused by MCT1 knockdown (Figure S2n,o). Consistent with a reduction in pyruvate export, MCT1 inhibition with AZD3965 leads to an increase in intracellular pyruvate levels (Fig.3g, Figure S3p). Together these data confirm that MCT1 loss of function reduces cellular pyruvate export.
To better understand the role of MCT1 in regulating lactate versus pyruvate transport in breast cancer cells, we also measured lactate and pyruvate export rates over a time course post AZD3965-mediated MCT1 inhibition by analyzing metabolites extracted from the media via gas chromatography – mass spectrometry (GC-MS). We found that pyruvate media levels are reduced by acute MCT1 inhibition in SUM149PT cells, whereas lactate media levels are unchanged (Fig. 3h). Other breast cancer cell lines showed similar results (data not shown). These data suggest that a primary function of MCT1 in glycolytic breast cancer cells may be to mediate pyruvate export.
MCT1 loss of function decreases breast cancer cell proliferation in vitro and tumor growth in vivo
To examine whether MCT1 function affects breast cancer cell proliferation and survival, we measured the proliferation rates and viability of breast cancer cell lines stably expressing MCT1 shRNA or treated with AZD3965. MCT1 knockdown reduces glycolytic breast cancer cell proliferation (Fig. 4a), and this proliferative defect is rescued by expression of shRNA-resistant MCT1 cDNA (Figure S3b,n,4a,b). AZD3965 treatment also reduces proliferation rates of glycolytic breast cancer cell lines (Fig. 4a), and dose-response curves for AZD3965 indicate that the reduction in proliferation in glycolytic breast cancer cells tracks with the reduction in pyruvate export rate caused by MCT1 inhibition (Figures S4c,d). Since MCT1 inhibition leads to increased intracellular pyruvate levels (Fig.3g), we wondered whether increased intracellular pyruvate levels could reduce proliferation of glycolytic breast cancer cells. Consistent with this notion, treatment with 5mM methyl-pyruvate results in elevated intracellular pyruvate levels and significantly reduced proliferation in glycolytic breast cancer cells (Figures Fig.4b). MCT1 knockdown increases the percentage of cells in the G0/G1 phase of the cell cycle (Fig. 4c). However, apoptosis-mediated cell death as measured by Annexin A5 staining is not affected by MCT1 knockdown in the breast cancer cell lines tested (Figure S4e,f). These results suggest that increased intracellular pyruvate levels as a result of MCT1 loss-of-function reduces proliferation of glycolytic breast cancer cells without enhancing apoptosis.
Figure 4. MCT1 loss-of-function reduces breast cancer cell proliferation and tumor growth.
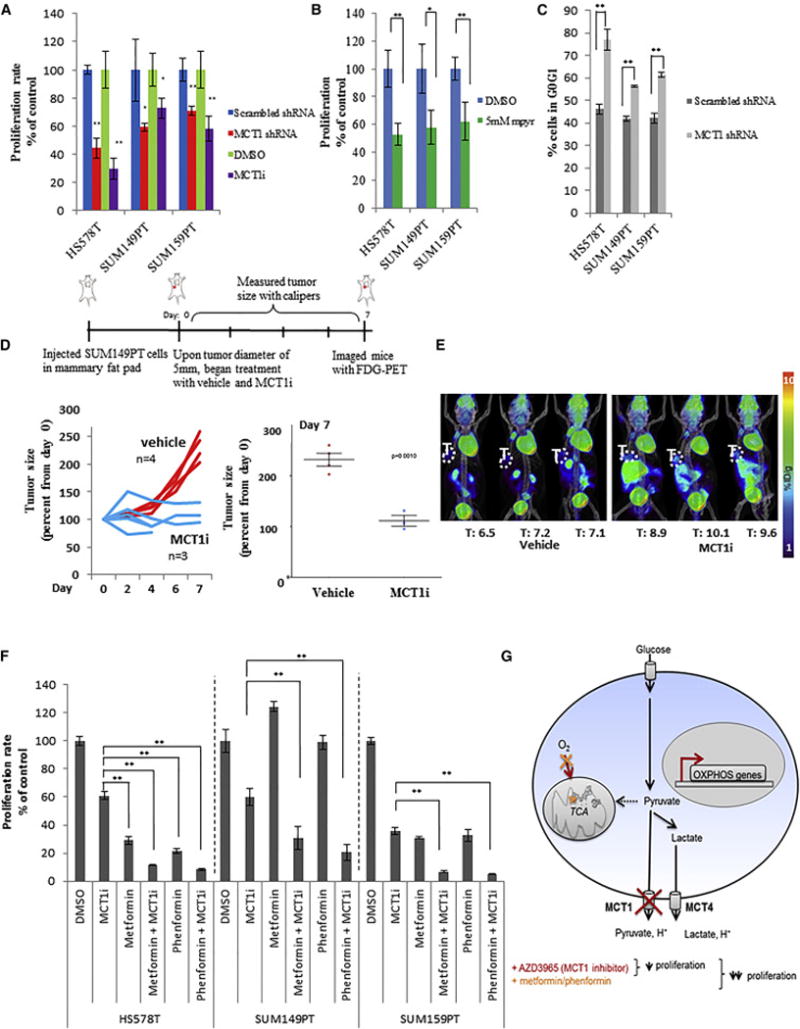
a, Proliferation rates of the indicated breast cancer cell lines stably expressing shRNA that knocks down MCT1 expression (MCT1 shRNA) versus control scrambled shRNA (scramble shRNA), and vehicle (DMSO) treated cells versus 250 nM AZD3965 (MCT1i). b, Proliferation rates of the indicated cell lines treated for 4 days with DMSO or 5mM methyl-pyruvate. c, Percentage of MCT1 knockdown cells (MCT1 shRNA) versus control cells (scramble shRNA) in the G0/G1 phase of the cell cycle. d and e, Relative tumor volumes (d) and FDG-PET/CT images (e) from NSG mice with mammary fat pad xenograft tumors derived from SUM149PT cells treated by oral gavage twice daily with either 0.5% hydroxypropyl methyl cellulose/0.1%tween (vehicle) or 0.1ml/10g AZD3965 (MCT1i) for seven days. In (e) T indicates tumor, and values shown represent mean injected dose per gram (%ID/g) calculated from tumor regions of interest. f, Proliferation rates of indicated breast cancer cell lines after five days of treatment with DMSO, MCT1i (IC50), metformin (IC50), metformin + MCT1i (IC50 for both), or phenformin (IC50), phenformin + MCT1i (IC50 for both). MCT1i was replenished every day. g, Schematic representation of the impact of MCT1 inhibition on breast cancer cell metabolism and proliferation. AZD3965 consistently reduces pyruvate export, increases oxygen consumption, and reduces proliferation rates. Dual treatment of AZD3965 with metformin or phenformin further reduces proliferation rates, presumably by blocking the compensatory switch to oxidative metabolism caused by MCT1 inhibition to sustain proliferation. Error bars in a–c denote standard deviation n=3). Error bars in e denote standard error of mean. For a–c and f, * denotes p < 0.05, and ** denotes p < 0.01. See Figure S4.
The consistent reduction in proliferation rate upon MCT1 knockdown and AZD3965 treatment is surprising given that these cells express other MCTs (Figure S4g), and MCT4 expression has been associated with resistance to MCT1 inhibition (Doherty et al., 2014; Le Floch et al., 2011; Polanski et al., 2014). MCT4 is expressed in the breast cancer cell lines tested with relatively high expression in SUM149PT and SUM159PT cells (Figure S4h). The MCT1 shRNA sequences used in this study are not cognate to MCT2-4 mRNAs, and in fact SUM149PT cells with stable expression of MCT1 shRNA exhibit increased MCT2-4 transcript levels (Figure S2b). However, MCT4 protein levels are not grossly altered in the context of MCT1 knockdown or inhibition in the breast cancer cell lines tested (Figure S4i). Additionally, protein levels of the MCT chaperone protein CD147 are not altered in the context of MCT1 knockdown or inhibition in the breast cancer cell lines tested (Figure S15). Since we found that MCT1 loss-of-function reduces proliferation of glycolytic breast cancer cells that express other MCTs, including MCT4, our results suggest an important and specific role for MCT1 function in breast cancer cell proliferation.
To examine whether MCT1 inhibition impacts tumor growth and glycolysis in vivo, we generated mammary fat pad xenograft tumors from SUM149PT cells in NOD scid gamma (NSG) mice, and began AZD3965 treatment after the tumors reached 5mm in diameter. AZD3965 treatment robustly blocked growth of the mammary fat pad xenograft tumors, with significant differences in tumor growth between the vehicle-treated and AZD3965-treated cohorts after only one week of treatment (Fig. 4d). However, despite the reduced tumor growth, tumor FDG uptake as measured by PET was not decreased in the AZD3965-treated mice, and instead was slightly but significantly increased (Fig. 4e). These data are consistent with our in vitro results showing that AZD3965 treatment reduces proliferation but not glycolytic flux of breast cancer cells.
Since we found that breast cancer cells adapt to MCT1 inhibition by enhancing oxidative metabolism (Fig. 3c), we reasoned that co-treatment of AZD3965 along with an oxidative phosphorylation inhibitor may further reduce proliferation rates of glycolytic breast cancer cells treated with AZD3965 by blocking the compensatory switch to increased oxidative metabolism. We therefore tested the effects of the biguanides, metformin and phenformin, known inhibitors of mitochondrial complex I, on glycolytic breast cancer cell proliferation in combination with AZD3965 treatment. Consistent with our hypothesis, dual treatment of AZD3965 with metformin or phenformin further lowered proliferation rates, beyond those seen with AZD3965 treatment alone, in an additive to synergistic fashion in multiple breast cancer cell lines (Fig. 4f). Similar synergistic effects between MCT1 inhibition and metformin treatment were recently reported in mouse xenograft models of cancer (Doherty et al., 2014). Collectively, these additive to synergistic effects of metformin or phenformin dual treatment with AZD3965 in cancer cell lines and tumors suggest a promising combination treatment strategy for patients with glycolytic tumors.
DISCUSSION
Here we show that MCT1 is critical for pyruvate export and proliferation of glycolytic breast cancer cells (schematic representation in Figure 4g). MCT1 loss of function consistently reduces pyruvate export, increases oxygen consumption, and reduces proliferation rates. Dual treatment of AZD3965 with metformin or phenformin further reduces proliferation rates, presumably by blocking the compensatory switch to oxidative metabolism caused by MCT1 inhibition to sustain proliferation. Our results imply an important role for pyruvate export in promoting proliferation and reducing oxidative metabolism in glycolytic breast cancer cell lines. However, the mechanism by which blocking pyruvate export may impact proliferation remains unclear – modulation of cellular redox status, intracellular pH, and/or ATP levels could be involved. Additionally, the mechanism by which MCT1 inhibition leads to increased cellular respiration requires further investigation. Pyruvate export inhibition may lead to increased entry of pyruvate carbons into the mitochondria to provide more substrate for oxidative phosphorylation. However, our preliminary studies tracing the fate of 13C-glucose metabolites, and specifically pyruvate, in MCT1-inhibited cells have yielded varying results across the breast cancer cell lines tested. Also, elevated oxygen consumption rates are observed at 24 hours post AZD3965 treatment, but not at 30 minutes or 4 hours post treatment (Fig. 3c), suggesting that the effect on respiration is not direct but rather through programmed changes in transcription. Consistent with this notion, genes involved in oxidative phosphorylation are enriched in breast cancer cells after 24 hours AZD3965 treatment (Figure 3b and Figure S2f). These changes in gene expression may occur through an unknown mechanism downstream of MCT1 inhibition and reduction of pyruvate export, or may simply be a cellular adaptation to survive MCT1 inhibition. It will be interesting to determine whether similar changes in gene expression also happen in tumors of breast cancer patients treated with AZD3965.
Doherty et al. recently found that MCT1 inhibition blocks lactate transport and decreases glycolysis to ultimately trigger cancer cell death in lymphoma and breast cancer cells (Doherty et al., 2014), however we did not observe reduced lactate export, decreased glycolysis, or increased cell death upon MCT1 inhibition in glycolytic breast cancer cell lines. One potential explanation for these discrepancies is the varying levels of MCT4 in the different cell lines used. The Burkitt lymphoma cell lines and MCF7 and T47D breast cancer cell lines used in the study by Doherty et al. expressed very little, if any, MCT4. In contrast, the relatively more glycolytic breast cancer cell lines used in our study - SUM149PT, SUM159PT, and HS578T (Figure S1b) - expressed measurable amounts of MCT4 (Figure S4g,h). MCT4 expression is likely responsible for continued lactate transport which likely sustains the glycolytic flux and survival of glycolytic breast cancer cell lines in the context of MCT1 inhibition. Consistent with an important role for lactate export in promoting cancer cell proliferation, and in agreement with results shown by Doherty et al., we found that supplementation of complete medium with 11 mM lactate reduced proliferation of glycolytic breast cancer cells that co-express MCT1 and MCT4 (data not shown).
Previous reports have found that MCT1 and MCT4 are commonly co-expressed in tumors (Choi et al., 2014; Kim et al., 2015). In support of this notion, we have found that MCT1 and MCT4 are often coexpressed at the mRNA level in patient breast and lung tumors (Figure S4l,m). Examination of MCT1 and MCT4 mRNA levels across cancer cell lines shows a high number of cancer cell lines with dual expression of MCT1 and MCT4, and another population with high expression of MCT1 only (Figure S4n). While several previous studies have found that MCT1 modulates cancer cell lactate transport in the absence of MCT4 expression, our results support a lactate-transport independent role for MCT1 in promoting proliferation of cancer cells with naturally-derived dual MCT1/MCT4 expression. One potential reason why cancer cells may upregulate MCT1 expression even in the context of MCT4 expression, is the ability of MCT1, but not MCT4, to transport both lactate and pyruvate to ensure cytosolic redox equilibration between cells and tissues (Halestrap and Wilson, 2012). Future experiments on patient-derived primary breast cancers that exclusively express MCT1, MCT4, or both together, are needed to more clearly delineate the roles of MCT1 versus MCT4 in tumor metabolism and growth.
AZD3965 is thought to kill tumor cells reliant on glycolysis through inhibition of lactate transport (Doherty et al., 2014; Polanski et al., 2014). However, our data suggests that AZD3965 does not reduce lactate export, glycolytic flux, or survival of glycolytic breast cancer cell lines coexpressing MCT1 and MCT4 in vitro, but still impacts proliferation through an alternative mechanism, potentially via reduction of pyruvate export and/or induction of oxidative metabolism. In our mammary fat pad xenograft model, AZD3965 treatment blocked tumor growth despite causing a slight increase in tumor FDG uptake as measured by PET. These data support an alternative mode of action for AZD3965 than impaired glycolysis in blocking tumor growth and suggest that loss of tumor FDG uptake by PET is not a good biomarker of response to AZD3965 in breast cancer patients.
Pyruvate is a commonly secreted metabolite from cancer cell lines in vitro (Jain et al., 2012), however whether pyruvate is secreted from tumors in vivo remains unknown. Our finding that serum samples from Stage IV lung cancer patients have elevated pyruvate levels compared to serum from Stage I lung cancer patients is consistent with MCT1 modulation of tumor pyruvate export in vivo (Figure 2e). However, further studies are necessary to confirm MCT1-mediated pyruvate export from cancer cells within tumor tissues, and to determine whether and how pyruvate export impacts tumor growth. Our results expand upon a growing literature that MCT1 is necessary for optimal cancer cell proliferation and provide further support for use of MCT1 inhibitors as anti-cancer therapeutics.
EXPERIMENTAL PROCEDURES
Cell culture conditions
HS578T cells were grown in DMEM media with 1% Penstrep, and 10%FBS. SUM149PT and SUM159PT cells were grown in F12 media with 1%Penstrep, 10% FBS, 10ug/ml insulin, 10ug/ml hydrocortisone, 10ng/ml EGF, and 0.5mg/ml of gentamicin.
Enrichment Analysis
Gene expression data was analyzed using Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) and Rank-rank hypergeometric overlap (RRHO) (Plaisier et al., 2010) algorithms as described previously (Palaskas et al., 2011). Pathway annotations for GSEA were from i) KEGG metabolic pathways (Kanehisa et al., 2010) or ii) the MSigDB curated gene sets (Subramanian et al., 2005). To visualize gene expression data within the context of metabolic pathway structure, we used Cytoscape (Smoot et al.) to color code the KEGG glycolysis pathway genes (hsa00010) on a green-to-red scale according to the average Pearson correlation with glycolytic phenotype. Microarray data accession number GSE76675.
Immunohistochemistry
Paraffin-embedded breast and lung cancer tissue microarray blocks were cut into 4 μm sections immediately prior to immunohistochemistry with polyclonal MCT1 antibody, and staining, blinded scoring, and statistical analyses were carried out as described previously (Mah et al., 2011; Mah et al., 2007; Yoon et al., 2010; Yoon et al., 2011). Comparisons of MCT1 expression across lung and breast histopathological categories were performed using Kruskal-Wallis tests, and the Cox proportional hazards model was used to determine prognostic values of survival.
Measurement of Glucose Consumption Rates and Pyruvate/Lactate Export Rates
Glucose consumption and lactate export rates were measured using a NOVA Bioanalyzer, and oxygen consumption rates were measured using a Seahorse XF-3 analyzer. Serum pyruvate/lactate levels and pyruvate/lactate export rates were measured using an absorbance-based assay kit (BioVision) and GC-MS. Following MCT1 inhibition accomplished by treating cells with 250 nM AZD3965, pyruvate/lactate export rates were measured using colorimetric-based assay kits (BioVision).
Intracellular Metabolite Analysis Using LC-MS
Cells were carefully scraped off in 800 μL of 50% ice cold methanol. An internal standard of 10 nmol norvaline was added to the cell suspension, followed by 400 μL of cold chloroform. After vortexing for 15 min, the aqueous layer was transferred to a glass vial and the metabolites dried under vacuum. Metabolites were resuspended in 100 μL 70% acetonitrile (ACN) and 5 μL of this solution used for the mass spectrometer-based analysis. The analysis was performed on a Q Exactive (Thermo Scientific) in polarity-switching mode with positive voltage 3.0 kV and negative voltage 2.25 kV. The mass spectrometer was coupled to an UltiMate 3000RSLC (Thermo Scientific) UHPLC system. Mobile phase A was 5 mM NH4AcO, pH 9.9, B) was ACN, and the separation achieved on a Luna 3μm NH2 100A (150 × 2.0 mm) (Phenomenex) column. The flow was 300 μL/min, and the gradient ran from 15% A to 95% A in 18 min, followed by an isocratic step for 9 min and re-equilibration for 7 min. Metabolites were detected and quantified as area under the curve (AUC) based on retention time and accurate mass (≤ 3 ppm) using the TraceFinder 3.1 (Thermo Scientific) software. Relative amounts of metabolites between various conditions, as well as percentage of labeling was calculated, corrected for naturally occurring 13C abundance as described (Yuan et al., 2008), and depicted in bar graphs.
Cell Culture Medium Metabolite Analysis Using GC-MS
A total of 2.5 × 105 cells of each breast cancer cell line were seeded onto 6-well plates, medium was replaced after 24 hours and inhibitor added at appropriate times. Twenty microliters of cell-free medium samples were taken 24 hours thereafter. Metabolites were extracted by adding 300 μl 80% methanol to the medium samples, followed by vortexing 3X, then centrifugation for 10 minutes at 13k rpm at 4°C. The supernatant was transferred to a fresh tube, and the solvent was evaporated using a SpeedVac. Metabolites were derivatized by adding 20 μl of 2% methoxyamine hydrochloride in pyridine (Pierce) for 1.5h at 37 °C followed by 30 μl N-methyl-N-(tert-butyldimethylsilyl)trifluoroacetamide (Pierce) for 1h at 55 °C. Samples were run on an Agilent 5975C MSD coupled to an Agilent 7890A GC as described (Metallo et al.). Data extraction was done with Agilent MSD ChemStation software and analysis performed with Microsoft Excel. Metabolite isotopomers were not corrected for naturally occuring 13C.
Xenograft Models and MicroPET/CT Imaging
Breast cancer tumor xenografts were generated by injecting 1 × 106 SUM149PT cells into female NSG mice. Half of the mice were treated with MCT1i (AZD3965) once tumors reached a size of 5mm in diameter, and were treated twice daily by oral gauvage. Half of the mice were treated as control micewith an equal volume of vehicle. Tumor size was monitored every other day with calipers. For microPET imaging, animals were anesthetized with 1.5% isoflurane, USP (Phoenix Pharmaceutical Inc.) and injected intravenously with 200 uCi 18F-FDG. PET imaging was conducted on a Focus 220 microPET scanner (Siemens) and, subsequently, CT recorded using a MicroCAT II CT instrument (Siemens). Data was analyzed by drawing 3-dimensional ROIs using AMIDE software (Loening and Gambhir, 2003).
Supplementary Material
Acknowledgments
We kindly thank Frank McCormick and Joe Gray for the breast cancer cell lines and early advice on study design. We thank Adrian Garcia for technical assistance, and Steve Bensinger, Caius Radu, and members of the Christofk laboratory for helpful discussions. Nicholas A. Graham is a postdoctoral trainee supported by the UCLA Scholars in Oncologic Molecular Imaging (SOMI) program, NIH grant R25T CA098010. This work was also supported by the Early Detection Research Network NCI CA86366 awarded to Lee Goodglick and David Chia. The collection and use of the lung cancer serum samples was supported by an Early Detection Research Network NCI CA152751 grant awarded to Steven M. Dubinett, and aspects of this study were supported by the NIH NCATS, UCLA CTSI UL1TR000124 grant. Thomas G. Graeber is supported (in part) by the National Cancer Institute/NIH (P01 CA168585), an American Cancer Society Research Scholar Award (RSG-12-257-01-TBE), a Melanoma Research Alliance Established Investigator Award (20120279), and the UCLA Jonsson Cancer Center Foundation. Heather R. Christofk is a Damon Runyon-Rachleff Innovation Awardee supported (in part) by the Damon Runyon Cancer Research Foundation, the Searle Scholars Program, the NIH Director’s New Innovator Award (DP2 OD008454-01), and the Caltech/UCLA Nanosystems Biology Cancer Center (NCI U54 CA151819).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This manuscript is accompanied by supplementary methods, tables, and figures.
AUTHOR CONTRIBUTIONS
C.S.H. performed all cell culture experiments, collected and analyzed experimental data, and prepared the manuscript. N.A.G. performed and analyzed all computational experiments and prepared the manuscript. D.B. performed the metabolomics experiments and the tumor growth and imaging experiment. W.G. assisted with cell culture experiments and analysis. C.E.C. assisted with cell culture experiments and experiments involving breast cancer patient serum samples. V.M, E.L.M, M.A., C.B., D.C., and S.H. conducted the tissue microarray experiments and analysis. P.A.L.K. assisted with computational experiments. B.K.G. and S.M.D. contributed and analyzed experiments involving lung cancer patient serum samples. S.A.H. collected breast cancer patient serum samples. I.M. generated the breast cancer gene expression datasets. S.E.C. and S.K.K. provided valuable experimental feedback and prepared the manuscript. L.G. conceived the tissue microarray experiments, reviewed experimental data involving the tissue microarrays, and prepared the manuscript. T.G.G. conceived the computational experiments, reviewed experimental data involving computational methods, and prepared the manuscript. H.R.C. designed the study, conceived all experiments, reviewed all experimental data, and prepared the manuscript.
The authors declare no competing financial interests.
References
- Bonen A, Miskovic D, Tonouchi M, Lemieux K, Wilson MC, Marette A, Halestrap AP. Abundance and subcellular distribution of MCT1 and MCT4 in heart and fast-twitch skeletal muscles. American journal of physiology. Endocrinology and metabolism. 2000;278:E1067–1077. doi: 10.1152/ajpendo.2000.278.6.E1067. [DOI] [PubMed] [Google Scholar]
- Chang HY, Nuyten DS, Sneddon JB, Hastie T, Tibshirani R, Sorlie T, Dai H, He YD, van’t Veer LJ, Bartelink H, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci U S A. 2005;102:3738–3743. doi: 10.1073/pnas.0409462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Wang L, Beretov J, Hao J, Xiao W, Li Y. Co-expression of CD147/EMMPRIN with monocarboxylate transporters and multiple drug resistance proteins is associated with epithelial ovarian cancer progression. Clin Exp Metastasis. 2010;27:557–569. doi: 10.1007/s10585-010-9345-9. [DOI] [PubMed] [Google Scholar]
- Choi JW, Kim Y, Lee JH, Kim YS. Prognostic significance of lactate/proton symporters MCT1, MCT4, and their chaperone CD147 expressions in urothelial carcinoma of the bladder. Urology. 2014;84:245 e249–215. doi: 10.1016/j.urology.2014.03.031. [DOI] [PubMed] [Google Scholar]
- Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, Hall MA, Amelio AL, Mishra JK, Li F, et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014;74:908–920. doi: 10.1158/0008-5472.CAN-13-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guile SD, Bantick JR, Cheshire DR, Cooper ME, Davis AM, Donald DK, Evans R, Eyssade C, Ferguson DD, Hill S, et al. Potent blockers of the monocarboxylate transporter MCT1: novel immunomodulatory compounds. Bioorg Med Chem Lett. 2006;16:2260–2265. doi: 10.1016/j.bmcl.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Meredith D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004;447:619–628. doi: 10.1007/s00424-003-1067-2. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Wilson MC. The monocarboxylate transporter family–role and regulation. IUBMB Life. 2012;64:109–119. doi: 10.1002/iub.572. [DOI] [PubMed] [Google Scholar]
- Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Choi JW, Lee JH, Kim YS. Expression of lactate/H(+) symporters MCT1 and MCT4 and their chaperone CD147 predicts tumor progression in clear cell renal cell carcinoma: immunohistochemical and The Cancer Genome Atlas data analyses. Hum Pathol. 2015;46:104–112. doi: 10.1016/j.humpath.2014.09.013. [DOI] [PubMed] [Google Scholar]
- Le Floch R, Chiche J, Marchiq I, Naiken T, Ilc K, Murray CM, Critchlow SE, Roux D, Simon MP, Pouyssegur J. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc Natl Acad Sci U S A. 2011;108:16663–16668. doi: 10.1073/pnas.1106123108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loening AM, Gambhir SS. AMIDE: a free software tool for multimodality medical image analysis. Mol Imaging. 2003;2:131–137. doi: 10.1162/15353500200303133. [DOI] [PubMed] [Google Scholar]
- Mah V, Marquez D, Alavi M, Maresh EL, Zhang L, Yoon N, Horvath S, Bagryanova L, Fishbein MC, Chia D, et al. Expression levels of estrogen receptor beta in conjunction with aromatase predict survival in non-small cell lung cancer. Lung Cancer. 2011;74:318–325. doi: 10.1016/j.lungcan.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah V, Seligson DB, Li A, Marquez DC, Wistuba II, Elshimali Y, Fishbein MC, Chia D, Pietras RJ, Goodglick L. Aromatase expression predicts survival in women with early-stage non small cell lung cancer. Cancer Res. 2007;67:10484–10490. doi: 10.1158/0008-5472.CAN-07-2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleland ML, Adler AS, Deming L, Cosino E, Lee L, Blackwood EM, Solon M, Tao J, Li L, Shames D, et al. Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin Cancer Res. 2013;19:773–784. doi: 10.1158/1078-0432.CCR-12-2638. [DOI] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CM, Hutchinson R, Bantick JR, Belfield GP, Benjamin AD, Brazma D, Bundick RV, Cook ID, Craggs RI, Edwards S, et al. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat Chem Biol. 2005;1:371–376. doi: 10.1038/nchembio744. [DOI] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaskas N, Larson SM, Schultz N, Komisopoulou E, Wong J, Rohle D, Campos C, Yannuzzi N, Osborne JR, Linkov I, et al. 18F-fluorodeoxy-glucose positron emission tomography marks MYC-overexpressing human basal-like breast cancers. Cancer Res. 2011;71:5164–5174. doi: 10.1158/0008-5472.CAN-10-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro C, Albergaria A, Paredes J, Sousa B, Dufloth R, Vieira D, Schmitt F, Baltazar F. Monocarboxylate transporter 1 is up-regulated in basal-like breast carcinoma. Histopathology. 2010;56:860–867. doi: 10.1111/j.1365-2559.2010.03560.x. [DOI] [PubMed] [Google Scholar]
- Pinheiro C, Longatto-Filho A, Scapulatempo C, Ferreira L, Martins S, Pellerin L, Rodrigues M, Alves VA, Schmitt F, Baltazar F. Increased expression of monocarboxylate transporters 1, 2, and 4 in colorectal carcinomas. Virchows Arch. 2008;452:139–146. doi: 10.1007/s00428-007-0558-5. [DOI] [PubMed] [Google Scholar]
- Pinheiro C, Longatto-Filho A, Simoes K, Jacob CE, Bresciani CJ, Zilberstein B, Cecconello I, Alves VA, Schmitt F, Baltazar F. The prognostic value of CD147/EMMPRIN is associated with monocarboxylate transporter 1 co-expression in gastric cancer. Eur J Cancer. 2009;45:2418–2424. doi: 10.1016/j.ejca.2009.06.018. [DOI] [PubMed] [Google Scholar]
- Plaisier SB, Taschereau R, Wong JA, Graeber TG. Rank-rank hypergeometric overlap: identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res. 2010;38:e169. doi: 10.1093/nar/gkq636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanski R, Hodgkinson CL, Fusi A, Nonaka D, Priest L, Kelly P, Trapani F, Bishop PW, White A, Critchlow SE, et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin Cancer Res. 2014;20:926–937. doi: 10.1158/1078-0432.CCR-13-2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71:2550–2560. doi: 10.1158/0008-5472.CAN-10-2828. [DOI] [PubMed] [Google Scholar]
- Yoon NK, Maresh EL, Elshimali Y, Li A, Horvath S, Seligson DB, Chia D, Goodglick L. Elevated MED28 expression predicts poor outcome in women with breast cancer. BMC Cancer. 2010;10:335. doi: 10.1186/1471-2407-10-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon NK, Maresh EL, Shen D, Elshimali Y, Apple S, Horvath S, Mah V, Bose S, Chia D, Chang HR, et al. Higher levels of GATA3 predict better survival in women with breast cancer. Hum Pathol. 2011;41:1794–1801. doi: 10.1016/j.humpath.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Bennett BD, Rabinowitz JD. Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat Protoc. 2008;3:1328–1340. doi: 10.1038/nprot.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.