

SUMMARY
Induction of compensatory mechanisms and ERK reactivation has limited the effectiveness of Raf and MEK inhibitors in RAS-mutant cancers. We determined that direct pharmacologic inhibition of ERK suppressed the growth of a subset of KRAS-mutant pancreatic cancer cell lines and that concurrent PI3K inhibition caused synergistic cell death. Additional combinations that enhanced ERK inhibitor action were also identified. Unexpectedly, long-term treatment of sensitive cell lines caused senescence, mediated in part by MYC degradation and p16 reactivation. Enhanced basal PI3K-AKT-mTOR signaling was associated with de novo resistance to ERK inhibitor, as were other protein kinases identified by kinome-wide siRNA screening and a genetic gain-of-function screen. Our findings reveal distinct consequences of inhibiting this kinase cascade at the level of ERK.
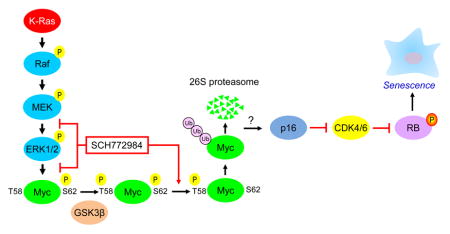
Graphical Abstract
INTRODUCTION
Mutational activation of KRAS is found in >95% of pancreatic ductal adenocarcinomas (PDAC) (Bryant et al., 2014). With strong and compelling evidence that the continued function of mutant KRAS is essential for PDAC maintenance, there has been intense effort in developing pharmacologic approaches to block mutationally activated KRAS for cancer treatment (Cox et al., 2014; Stephen et al., 2014). Currently the most promising strategy involves inhibitors of RAS effector signaling, in particular the RAF serine/threonine kinases (Cox et al., 2014; Stephen et al., 2014). Activated RAS binds to RAF and promotes its activation. RAF then phosphorylates and activates the MEK1 and MEK2 dual specificity protein kinases, which in turn phosphorylate the related ERK1 and ERK2 MAPKs. Activated ERK1/2 then phosphorylate more than 200 substrates (Yoon and Seger, 2006).
The limited substrates of RAF and MEK prompted earlier assumptions that pharmacologic inhibitors of either kinase would be equivalent and equally effective in blocking ERK activation. This led to the development and evaluation of small molecule inhibitors of RAF or MEK, with at least 27 under clinical evaluation (ClinicalTrials.gov). However, RAF kinase inhibitors have been ineffective in RAS-mutant cancers as a consequence of paradoxical induction of Ras-dependent RAF dimerization and activation, with subsequent enhanced ERK activation (Lito et al., 2013). MEK inhibitors have shown limited to no activity in RAS-mutant cancers, most commonly attributed to loss of ERK feedback inhibition and compensatory mechanisms that cause reactivation of ERK (Samatar and Poulikakos, 2014).
Since reactivation of ERK is a major mechanism overcoming RAF or MEK inhibitor efficacy, we hypothesized that direct inhibition of ERK may overcome these limitations. In support of this, we recently described the development of a ERK1/2-selective pharmacologic inhibitor (SCH772984) and showed that BRAF-mutant melanomas with acquired resistance to RAF and/or to MEK inhibitors were still sensitive to ERK1/2-selective pharmacologic inhibitors (Hatzivassiliou et al., 2012; Morris et al., 2013). ERK inhibition suppressed the growth of ~50% of RAS-mutant human tumor cell lines in vitro. However, the mechanisms behind ERK inhibitor susceptibility versus resistance of subsets of RAS-mutant cancers remain unresolved (Hatzivassiliou et al., 2012). In the present study we assessed the mechanistic basis of ERK inhibitor sensitivity in KRAS-mutant PDAC, and applied unbiased chemical and genetic library screens to identify combination approaches to enhance anti-ERK therapies.
RESULTS
MEK Inhibitor-Resistant PDAC Cell Lines Are Sensitive to ERK Inhibitor
Our recent analyses showed that the ERK1/2-selective inhibitor SCH772984 potently suppressed the growth of ~50% of RAS-mutant human tumor cell lines in vitro (Morris et al., 2013). However, the mechanistic bases for ERK inhibitor sensitivity and de novo resistance were not addressed. Since PDAC is the most KRAS-addicted cancer, we first focused on evaluating SCH772984 in a panel of 11 KRAS-mutant PDAC cell lines (Table S1). Anchorage-dependent proliferation was monitored for 72 hr (Figure 1A). We found that five cell lines were sensitive to SCH772984 (GI50 <4 μM), whereas six exhibited de novo resistance (GI50 >4 μM). Surprisingly, three out of four SCH772984-sensitive cell lines were resistant to the MEK inhibitor selumetinib (Figure 1B), independent of the degree of suppression of ERK phosphorylation (Figure S1C). Thus, inhibition of the pathway at the level of ERK has distinct consequences from inhibition at the level of MEK.
Figure 1. PDAC Cell Line Sensitivity to the ERK-Selective Inhibitor SCH772984 Is Not Associated with KRAS Dependency.

(A) KRAS-mutant PDAC cell lines were maintained on plastic in growth medium with DMSO vehicle or SCH772984 (3.9 nM – 4 μM). Proliferation was monitored by MTT assay to assess growth inhibition after 72 hr treatment. GI50 values were determined using CalcuSyn. Data are representative of three independent experiments. Bars indicate standard deviation from triplicate samples for each cell line.
(B) SCH772984-sensitive and -resistant PDAC cell lines were maintained on plastic in growth medium with vehicle or the MEK inhibitor selumetinib (3.9 nM – 4 μM). MTT assays were performed and GI50 values were determined as in (A).
(C) Cells were transfected with scrambled (NS) or one of two individual siRNAs targeting KRAS (designated KRAS1 or KRAS2) for 48 hr, followed by western blot for total K-Ras4B, ERK1/2 (ERK), AKT1–3 (AKT) and for vinculin to verify equivalent loading of total protein. Phospho-specific antibodies were used to monitor phosphorylation and activation of ERK (T202/Y204; pERK) and AKT (S473; pAKT). Data are representative of two independent experiments.
(D) Cells transfected with NS or KRAS siRNAs were monitored for proliferation on plastic at 6 days post-transfection by MTT assay. Error bars represent the standard error of the mean. Data are representative of three independent experiments. Asterisks represent statistical significance using one-way ANOVA analysis, where * = p < 0.05, ** = p < 0.001, and ns = not significant.
(E) Cells transfected with NS or KRAS siRNAs were plated at low density and clonogenic growth was monitored at 9–12 days post-transfection. Error bars represent standard error of the mean. Data are representative of three independent experiments. Asterisks represent statistical significance using one-way ANOVA analysis, where * = p < 0.05, ** = p < 0.001, and ns = not significant.
See also Figure S1.
We next expanded our panel of KRAS-mutant pancreatic cancer cell lines to include those derived from PDAC patient-derived xenografts (PDX cell lines, Table S2). Interestingly, we found a similar pattern of sensitivity to SCH772984, where some cell lines were sensitive and others were resistant (Figure S1A). We observed that treatment with a chemically and mechanistically distinct ERK inhibitor, BVD-523 (ulixertinib), a current clinical candidate (NCT01781429) (Germann, 2015), resulted in a similar pattern of sensitivity in both established and PDX cell lines (Figure S1A and S1B).
ERK Inhibitor Sensitivity Is Not Associated with KRAS Dependency or with K-Ras-dependent Effector Signaling
Previous studies showed that only a subset of KRAS-mutant PDAC cell lines exhibited KRAS dependency (Singh et al., 2009). To determine if KRAS dependency correlates with sensitivity to SCH772984, we evaluated the consequence of transient siRNA-mediated suppression of KRAS expression in our cell lines (Figure 1C). KRAS knockdown resulted in ~50% reduction in anchorage-dependent viability (Figures 1D and S1D) and 50% or greater reduction in clonogenic growth (Figures 1E and S1E). Using the same shRNA KRAS vectors used in the previous study (Singh et al., 2009), we established mass populations of stably infected cells displaying >80% reduction in K-Ras4B protein (Figure S1F). We found >50% reduction in both anchorage-dependent and anchorage-independent growth in all cell lines (Figures S1G and S1H). We conclude that KRAS-dependent growth is not a predictor of ERK inhibitor sensitivity.
We next determined whether ERK inhibitor sensitivity correlates with K-Ras-dependent ERK activation. Neither transient nor sustained KRAS suppression reproducibly suppressed pERK in any cell line (Figures 1C and S1F). Transient KRAS suppression significantly reduced pAKT in 3 of 9 cell lines, whereas stable suppression did not. Thus, SCH772984 sensitivity was not associated with K-Ras-dependent ERK or AKT activation.
Short-term Treatment with SCH772984 Enhances Apoptosis and Alters Cell Cycle Regulation
Next, we investigated the mechanism of SCH772984-induced growth suppression. After 72 hr treatment, we observed a significant fraction of non-adherent cells in the sensitive cell lines. Enhanced caspase-3 cleavage was detected in both non-adherent (Figure 2A) and adherent (Figure S2A) cell populations.
Figure 2. Short-term SCH772984 Treatment Induces Apoptosis and Altered Cell Cycle Progression.

(A) SCH772984-sensitive or -resistant cell lines were treated for 72 hr with DMSO vehicle or SCH772984. Non-adherent cells were collected and monitored for apoptosis by western blot for cleaved caspase-3. Data are representative of three independent experiments.
(B) Cells treated as above were stained with propidium iodide followed by flow cytometry. Error bars represent standard error of the mean. Asterisks represent statistical significance using one-way ANOVA analysis, where * = p < 0.05.
(C) Cells treated as above were collected for western blot for total cyclin B1, cyclin D1 and p21, and of phosphorylated, inactivated RB (S807/811; pRB). Western blot for pERK was done to verify SCH772984 inhibition; β-actin was the loading control.
See also Figure S2.
We then determined if ERK inhibition perturbed cell cycle progression. Using flow cytometry, we observed that three of four sensitive cell lines showed a significant treatment-induced increase in cells in G0/G1 and a concomitant decrease in cells in S and G2/M (Figure 2B). Treated cell lines also exhibited reduced levels of cyclin D1 and B1, regulators of progression through G1 and M, respectively, as well as hypophosphorylation and activation of RB, and reduced p21 protein levels (Figure 2C). Additionally, we found that sensitive cell lines exhibited increased sensitivity to SCH772984 over time as measured by changes in GI50 values (Figure S2B). We conclude that short-term treatment with SCH772984 suppresses PDAC tumor cell growth by enhancing apoptosis and/or by impairing progression through G1 and mitosis.
ERK Inhibitor Induction of AKT Activation Is a Marker of Sensitivity
Rapid ERK inhibitor-induced kinome reprogramming may cause dynamic changes in the activities of signaling components that distinguish sensitive from resistant cell lines (Duncan et al., 2012). Therefore, we evaluated signaling changes caused by SCH772984 treatment at 4, 24 and 72 hr. We monitored the consequences of both mechanisms of SCH772984 inhibition of ERK (Morris et al., 2013): inhibition of MEK1/2 binding and phosphorylation of ERK1/2, and inhibition of ATP binding and ERK1/2 phosphorylation of their cytoplasmic substrate, p90 RSK. After 4 hr, pERK and pRSK levels were reduced in both sensitive and resistant cell lines (Figure 3A). After 24 hr, pERK but not pRSK levels rebounded over vehicle control levels in both sensitive and resistant cell lines. However, by 72 hr, pERK increased further in a dose-dependent manner, whereas pRSK levels remained reduced even at 72 hr (Figures S3A and S3C). Thus, kinome reprogramming overcomes SCH772984 inhibition of ERK phosphorylation by MEK, but not ERK signaling, in both sensitive and resistant cell lines.
Figure 3. SCH772984 Sensitivity Is Associated with Treatment-Induced AKT Phosphorylation.

(A) Cells were treated for 4 or 24 hr with DMSO vehicle or SCH772984, then evaluated by western blot with phospho-specific antibodies for RSK (T395/S363; pRSK), MEK1/2 (S217/221; pMEK), AKT (S473; pAKT), and ERK (T202/Y204; pERK). Total RSK, ERK, AKT, MEK and β-actin were also analyzed. Data are representative of three independent experiments.
(B) Cells were treated as in (A) for 72 hr and evaluated by western blot for DUSP1, DUSP4, DUSP6 and β-actin.
(C) SCH772984-sensitive (Panc10.05) and -resistant (CFPAC-1) cell lines were treated concurrently with selumetinib (5 μM) and either vehicle or the indicated concentrations of SCH772984. Phosphorylated and total RSK, MEK, ERK and vinculin were evaluated by western blot.
(D) Cells were treated as above and collected for western blot for pCRAF (S338), pCRAF (S289/296/301), total CRAF and β-actin.
(E) Cells were maintained in growth medium with vehicle or SCH772984 (3.9 nM – 4 μM), with or without the PI3K inhibitor AZD8186 (1 μM). MTT was used to assess growth inhibition after 72 hr. Data are representative of three independent experiments. Bars indicate standard deviation from triplicate samples for each cell line.
(F) Cells were co-treated with SCH772984 and AZD8186. Western blots were performed for pAKT (S473) and total AKT, caspase-3 and β-actin.
See also Figure S3.
We next determined a basis for the transient and reversible suppression of ERK1/2 phosphorylation by SCH772984. Treatment with SCH772984 for 72 hr caused suppression of the ERK phosphatase DUSP4 in 5 of 6 cell lines, which could contribute to pERK restoration (Figures 3B and S3E). However, an alternative mechanism was suggested by the increased levels of pMEK1/2 observed at 4 and 24 hr (Figure 3A and S3B), indicating potential loss of ERK-mediated negative feedback inhibition of KRAS-RAF-MEK signaling (Dougherty et al., 2005). Consistent with this possibility, concurrent treatment with the MEK inhibitor selumetinib prevented the increase in pERK1/2 (Figure 3C). Furthermore, SCH772984 both reduced ERK-mediated phosphorylation of CRAF at S289/296/301 and increased phosphorylation at S338, a marker of CRAF activation (Figures 3D and S3D). Thus, SCH772984-initiated loss of ERK-dependent negative feedback leads to increased CRAF and MEK activation in both sensitive and resistant cell lines without inducing the restoration of active ERK signaling as measured by pRSK.
MEK inhibitor treatment has been linked to activation of phosphatidylinositol-3 kinase (PI3K)-AKT serine/threonine kinase signaling to re-sensitize BRAF inhibitor-resistant melanoma cell lines (Villanueva et al., 2010). Therefore, we determined whether phosphorylation of the PI3K substrate AKT was also elevated as a consequence of SCH772984 treatment in our sensitive cell lines. Consistent with that previous study (Villanueva et al., 2010), we observed a dose-dependent increase in pAKT at 72 hr in all sensitive but not resistant cell lines (Figure S3B and S3D). Next, we expanded our analysis to the PDX cell lines. We found that 66% of sensitive cell lines exhibited increased levels of pAKT (Figure S3G). Thus, treatment-induced pAKT elevation provided a biomarker for ERK inhibitor sensitivity.
To assess whether increased AKT activation is a compensatory protective response to overcome ERK inhibition-induced growth suppression, we co-treated cells with both SCH772984 and the PI3K inhibitor AZD8186 (Hancox et al., 2015) to block the ERK inhibitor-associated increases in pAKT (Figures 3F and S3F). Concurrent PI3K inhibition increased growth inhibition in sensitive cell lines, associated with caspase-3 cleavage, but surprisingly not in resistant cell lines (Figure 3E). SCH772984-sensitive PDX cell lines also exhibited synergy when exposed to the combination of SCH772984 and AZD8186 (Figure S3H). Similarly, in sensitive cell lines we also observed synergy with BVD-523 in combination with PI3K inhibition (Figure S3I–J). Together, our data suggest ERK and PI3K inhibitors as a potential clinically relevant combination therapy in KRAS-mutant PDAC.
Mechanistically Diverse Inhibitor Combinations Synergistically Enhance the Growth Inhibitory Activity of ERK Inhibitor versus MEK Inhibitor
Since we determined that concurrent inhibition of PI3K synergistically enhanced ERK inhibitor-induced anti-proliferative activity, we next performed an unbiased chemical library screen to identify additional combinations for SCH772984-based therapy. We applied a chemical library, comprised of 304 approved cytotoxic chemotherapeutic drugs and clinically available molecularly targeted drugs (Pemovska et al., 2013), to two SCH772984-sensitive cell lines, and we additionally screened for combinations with selumetinib. Synergistic and antagonistic interactions were assessed by comparing the response to each drug in the drug collection in the presence or absence of SCH772984 or selumetinib (deltaDSS score). DeltaDSS scores greater than 5 or less than −5 were cutoffs for likely synergistic or antagonistic interactions, respectively. Complementing our findings with the PI3K inhibitor AZD8186, additional inhibitors of PI3K signaling (mTOR, AKT and S6 kinase inhibitors) also enhanced SCH772984 activity in both cell lines (Figure 4). Interestingly, the drug interactions of SCH772984 and selumetinib were strikingly different. For example, in HPAC cells, SCH772984, but not selumetinib, was synergistic with mTOR and AKT inhibitors, as well as with a bromodomain inhibitor (Figure 4, upper right panels). The latter results indicate distinct mechanisms of resistance to inhibitors of these two nodes of the ERK-MAPK cascade.
Figure 4. Distinct Patterns of Drug Synergies in ERK versus MEK Inhibitor Combinations.

HPAC and Panc10.05 cells were exposed to dose-dependent drug sensitivity testing (DSRT) against 309 oncology-related compounds in the presence or absence of the ERK inhibitor SCH772984 (2 μM) or the MEK inhibitor AZD6244/selumetinib (1 μM). Cell viability was measured using CellTiter-Glo and drug responses were calculated as drug sensitivity scores (DSS). Plotted in the heatmap are the deltaDSS values (DSS in the presence of ERK/MEK inhibitor – DSS in the absence of overlaid inhibitor) for each condition, where red signifies potential synergies and blue indicates negative interactions between the two drugs. Drugs where the deltaDSS remained between −5 and 5 for all four conditions were excluded from the heatmap.
Long-term ERK Inhibition Causes a Senescence-like Phenotype
Since acquired resistance is a limitation of essentially all protein kinase inhibitor-based therapies, we expect that ERK inhibitor treatment will be similarly limited. To investigate likely mechanisms of acquired resistance, we first attempted to select resistant subpopulations by long-term high-dose inhibitor treatment of sensitive cells, an approach used widely with inhibitors of RAF and MEK (Samatar and Poulikakos, 2014). However, even after 2–3 months of SCH772984 treatment, we did not observe outgrowth of ERK inhibitor-resistant cells. Instead, treated cells remained quiescent and displayed a flattened cellular morphology characteristic of senescent cells (Campisi, 2013) (Figure 5A). To investigate whether they were indeed senescent, we determined if the treated cells exhibited elevated β-galactosidase activity, the only reliable marker of oncogene-induced senescence in pancreatic cancer (Caldwell et al., 2012). We found that treatment of sensitive cell lines with SCH772984 at 1x or 2x GI50 induced increased β-galactosidase activity in ~60% of cells after 7 to 14 days (Figures 5A, 5B and S4E) or even 72 hr (e.g., HPAF-II cells, Figure S4A and S4B).
Figure 5. Long-term SCH772984 Treatment Induces Markers of Senescence and Ubiquitination in Sensitive but Not Resistant Cell Lines.

(A and B) Cells were treated with SCH772984 at GI50 or at 2x GI50 concentrations for 14 days and stained for β-galactosidase. Representative images (scale bar, 50 μm) are shown in the left panel, with quantitation in the right panel. Error bars represent standard error of the mean. Data are representative of three independent experiments. Asterisks represent statistical significance using one-way ANOVA analysis, where * = p < 0.05 and ** = p < 0.001.
(C) Cells were treated with SCH772984 for 14 days on plastic, followed by western blot for pRB, pRSK and pERK, and for total p16, RSK and ERK; β-actin was a loading control.
(D and E) Cells were treated with vehicle or SCH772984 for 7 days, and then immunoblotted for ubiquitin or for β-actin.
See also Figure S4.
Mutant RAS-induced senescence can be prevented through loss of one of two tumor suppressor pathways: p53 and its target p21, or p16-mediated dephosphorylation and activation of RB (Campisi, 2013). We found that the p53-p21 pathway was not induced in either sensitive or resistant cell lines after 7 or 14 days of continuous exposure to SCH772984 (Figure S4C, S4F, S4H, and S4K). However, sensitive cell lines displayed both induction of p16 and loss of phospho-RB (Figures 5C, S4C, and S4G). Furthermore, we observed onset of senescence in the SCH772984-sensitive PDX cell lines (Figure S4M and S4N). Thus, consistent with previous studies where loss of p16 was required to prevent KRAS-induced senescence in pancreatic cancer (Bardeesy et al., 2006; Eser et al., 2013), we suggest that restoration of p16 function upon ERK inhibitor treatment in turn unmasked KRAS-induced senescence activity in sensitive PDAC cell lines.
Senescence is conventionally an irreversible process. We found that removal of SCH772984 for as long as 14 days reversed neither the sustained β-galactosidase activity nor the reduced cell growth induced by 28 days of treatment (Figure S4O–R). Additionally, p16 protein expression remained high whereas Myc and pRB protein expression remained low after SCH772984 removal (Figure S4S and S4T). Thus, ERK inhibition induced an irreversible senescence-like phenotype.
ERK inhibition-induced Degradation of MYC Is Necessary and Sufficient for Induction of Senescence
Next, we determined the signaling mechanism whereby ERK inhibition induced this phenotype. Recently, one study showed that Ras activation-induced senescence was characterized by both high ERK activity and increased degradation of selected proteins (Deschenes-Simard et al., 2013). In contrast, we found that long-term ERK inhibitor treatment (7 or 14 days) increased widespread protein ubiquitination in sensitive cell lines (Figures 5D, 5E, S4D, S4I, S4J, and S4L). Thus, ERK inhibition may cause the degradation and loss of one or more specific ERK substrate(s) to induce senescence-like growth suppression.
We next determined if long-term SCH772984 treatment caused loss of specific ERK substrates or gene targets with known roles in senescence. The MYC transcription factor is an ERK substrate, and loss of MYC induced tumor cell senescence by an RB- or p16-dependent mechanism (Ohtani et al., 2001; Wu et al., 2007). Altered expression of ERK-regulated ETS family transcription factors (Ets-1 and Ets-2) stimulated CDKN2A (encoding p16) gene expression in senescent fibroblasts (Ohtani et al., 2001; Wu et al., 2007). In BRAF-mutant melanoma, ERK stimulated AURKB gene transcription, and suppression of AURKB expression induced senescence (Bonet et al., 2012). We therefore determined if long-term treatment with SCH772984 altered the levels of ETS1, ETS2, MYC or Aurora B proteins. We found that 7-day treatment resulted in decreased their levels in sensitive cell lines (Figure 6A).
Figure 6. Induction of Senescence Is Dependent on Proteasomal Degradation of cMYC.

(A) Cells were treated with vehicle or SCH772984 for 7 days, then immunoblotted for Aurora B, MYC, ETS1 or ETS2, and β-actin. Data are representative of three independent experiments.
(B) Sensitive cell lines were treated with vehicle or the indicated concentrations of SCH772984 for 7 days, and then co-treated with either vehicle (−) or MG132 (10 μM, +) for an additional 8 hr, followed by western blots for Aurora-B, MYC, ETS-1, and ETS-2. β-actin is the loading control.
(C) Densitometry analysis of MYC protein levels from (B).
(D) Sensitive cell lines were treated with vehicle or SCH772984 for 7 days, and then immunoblotted with phospho-specific antibodies for Myc residues T58 or S62 (pMYC), or for total MYC and β-actin.
(E) Cells were treated as in (C) and then MG132 (10 μM) was added for 8 hr.
(F) Sensitive Panc10.05 cells maintained on plastic were treated with either vehicle (−) or SCH772984 (1,000 nM, +) for 7 days, then co-treated with either vehicle (−) or MG132 (10 μM, +) for an additional 8 hr. Whole cell lysates (WCL) were then subjected to control normal serum (mock) or to anti-MYC immunoprecipitation (IP), resolved by SDS-PAGE and then immunoblotted for ubiquitin or for MYC, and for vinculin to verify equivalent loading of total cellular protein.
(G) Sensitive Panc10.05 and HPAC cells stably infected with either the empty pMSCV retrovirus vector (EV) or pMSCV encoding a FLAG epitope-tagged ubiquitination-deficient MYC T58A mutant were treated with vehicle or SCH772984 for 10 or 14 days. Western blot was performed to determine levels of Aurora-B, MYC, and FLAG. Asterisk denotes band of interest. β-actin is the loading control.
(H) Panc10.05 and HPAC cells expressing either EV or MYC T58A were treated with SCH772984 for 10 or 14 days, then stained for β-galactosidase. The percentage of β-galactosidase-positive cells was determined. Error bars represent standard error of the mean. Asterisks represent statistical significance using an unpaired t-test, where ** = p < 0.01 and * = p < 0.05.
(I) Images (scale bar, 100 μm) of β-galactocidase-positive cell staining in Panc10.05 and HPAC cells, stably infected with the empty pMSCV puro retrovirus vector (EV) or pMSCV encoding MYC T58A, after 10 or 14 days of SCH772984 treatment.
See also Figure S5.
Next, we investigated the mechanisms whereby SCH772984 modulates Aurora B, MYC, ETS1, and ETS2 protein levels. First, RT-PCR analyses revealed no reduction in mRNA levels of any of the four proteins after 7 days of treatment, suggesting posttranscriptional mechanisms (Figure S5A). ERK phosphorylation of MYC is known to prevent its degradation (Farrell and Sears, 2014), implying that ERK inhibition would enhance its degradation. Consistent with this possibility, the proteasome inhibitor MG132 restored the protein levels of ETS1, ETS2, and MYC in SCH772984-treated cells (Figures 6B, 6C and S5B).
MYC protein stability can be controlled in a multi-step process by RAS effector signaling (Farrell and Sears, 2014). ERK phosphorylation of MYC at residue S62 facilitates its subsequent phosphorylation at T58 by GSK3β, leading to proteasomal degradation. We found that SCH772984 induced loss of the majority of MYC protein. Consistent with the current model, each sensitive cell line displayed hypophosphorylation at T58 in the remaining MYC protein (Figures 6D, 6E and S5E). Furthermore, MYC ubiquitination was increased in sensitive cells after SCH772984 treatment (Figures 6F, S5C and S5D). Interestingly, the remaining low level of MYC was hyperphosphorylated at S62 in HPAF-II cells, suggesting ERK-independent phosphorylation at S62 in these cells.
We then determined whether loss of MYC is a driver of the SCH772984-induced senescence-like phenotype, using a well-characterized MYC variant in which the T58 phosphorylation site is substituted with an alanine residue (T58A) to prevent degradation by the proteasome (Yeh et al., 2004). We found that Panc10.05 and HPAC cells ectopically expressing MYC T58A partially retained MYC expression upon SCH772984 treatment (Figure 6G), and that induction of β-galactosidase activity was significantly inhibited (Figure 6H and 6I). Conversely, depletion of MYC with siRNA in both sensitive and resistant cell lines was associated with increased β-galactosidase activity (Figure S5F–I). Thus, loss of MYC is necessary and sufficient to cause senescence-associated increased β-galactosidase activity.
Since we noted that MYC T58A protein stability unexpectedly decreased upon ERK inhibition, we assessed the requirements for phosphorylation at S62 and T58 to modulate SCH772984-mediated loss of MYC protein. Our analyses indicated that the regulation of MYC protein stability by ERK signaling is more complex than currently understood and must involve additional phosphorylation events and sites (Figure S5J–W).
SCH772984 Treatment Reduces Tumor Xenograft Growth and MYC Protein Levels in vivo
We next determined if loss of MYC also correlated with sensitivity to SCH772984 in vivo. We first determined the ability of SCH772984 to inhibit tumor xenograft growth of two PDAC cell lines that were sensitive to SCH772984 in vitro. We found that SCH772984 treatment of established tumors caused tumor regression (HPAC) or impaired progression (HPAF-II) (Figure 7A). No weight loss was seen, indicating that the treatment was well tolerated (Figure S6A). These anti-tumor activities were associated with reductions in pRSK in the tumor tissues (Figure S6B, S6E and S6F), consistent with ERK inhibition at the tumor sites, and with reduced total protein levels of MYC and Aurora B (Figure 7B). We did not observe depleted levels of MYC mRNA, supporting a mechanism whereby loss of MYC in vivo was due to protein degradation (Figure 7C). Furthermore, two in vitro-resistant cell lines were similarly not sensitive to SCH772984 in vivo (Figure S6C and S6D).
Figure 7. SCH772984 Inhibition of Tumor Growth Is Associated with Suppression of Myc and Aurora B Abundance.

(A) HPAC or HPAF-II cells were injected subcutaneously into the flanks of nude mice. Tumors were allowed to reach 300–450 mm3, then mice were treated i.p. daily with vehicle (20% hydroxypropyl beta-cyclodextrin or HPBCD) or with SCH772984 at 75 or 90 mpk for 21 or 14 days, respectively. Error bars represent standard error of the mean (n=10).
(B) Tumors from mice treated as in (A) were harvested after 21 days and lysates were analyzed by blotting for Aurora B and MYC protein.
(C) Tumors from mice treated as in (A) were harvested after 21 days and analyzed by RT-PCR for AURKB and MYC mRNA.
(D) KRAS-mutant pancreatic cancer patient-derived xenograft (PDX) cell lines (Panc354, Panc215, Panc185, and Panc374) were injected subcutaneously into the flanks of nude mice. Tumors were allowed to reach 200 mm3, then mice were treated with vehicle (10% HPBCD) or SCH772984 at 50 mpk b.i.d. for 14 days. Error bars represent standard error of the mean (n=10). Asterisks represent statistical significance using two-way ANOVA, where * = p < 0.05.
(E) Panc215 xenograft tumors treated as in (B) were harvested after 14 days and lysates were analyzed by blotting for Aurora B, MYC, pRB, cyclin B1, cyclin D1, caspase-3 and vinculin.
(F) Panc215 xenograft tumors treated as in (C) were harvested after 14 days and lysates were analyzed by RT-PCR for AURKB and MYC mRNA.
(G) Panc253 tumors were orthotopically implanted in nude mice and allowed to grow to a mean size of 200 mm3, as measured by ultrasound. Mice were treated i.p. with SCH772984 at 25 mpk three times per week or with dinaciclib at 20 mpk twice weekly. After 25 days, tumors were harvested and weighed. All data represent mean ± SE (n= 7); ***, p < 0.001, each arm versus vehicle control.
(H) Tumors harvested from mice treated as in (B) were analyzed by western blot for phospho-RSK and -ERK, and total RSK, ERK, and MYC. Vinculin is a loading control.
(I) Images of Panc253 tumors treated as described in (G).
See also Figure S6.
Since PDAC PDX mouse models may more accurately model drug response in cancer patients (Hidalgo et al., 2014), we next evaluated SCH772984 sensitivity in four different serially passaged KRAS-mutant PDX tumors surgically implanted into the flanks of nude mice. SCH772984 significantly reduced (70–90%) tumor growth in all four PDX models (Figure 7D), and this was associated with decreases in protein but not RNA levels of MYC and Aurora B (Figures 7E, 7F, S6G, and data not shown). We also observed loss of cyclin B1, cyclin D1, and pRB, but no induction of cleaved caspase-3, suggesting halted cell growth rather than induction of apoptosis (Figures 7E and S6G). Together with our in vitro observations, these results in both cell line- and patient-derived tumor xenografts indicate that loss of MYC and Aurora B proteins may be accurate in vivo markers of SCH772984 sensitivity.
We next expanded our analysis to include an orthotopic xenograft mouse model of PDAC, as stromal cells are known barriers to drug delivery/efficacy in the pancreas. We observed that SCH772984 decreased pERK, pRSK, and MYC levels and reduced tumor size (Figures 7G, 7H, 7I, and S6H). Since we showed previously that Ral inhibition suppressed pancreatic tumor growth (Lim et al., 2006), we also tested the combination of SCH772984 with dinaciclib, a CDK inhibitor that blocks pancreatic tumor growth by suppressing KRAS-Ral signaling (Feldmann et al., 2011; Feldmann et al., 2010) This combination resulted in a more significant reduction in tumor growth than either treatment alone.
Finally, because we had observed that the clinical candidate ERK inhibitor BVD-523 also synergized with the PI3K inhibitor AZD8186 in vitro, we sought to determine its efficacy in vivo. We observed markedly reduced tumor growth with this combination (Figure S6I). Importantly, western blot analysis and tumor staining revealed ERK target inhibition, as measured by decreases in pERK, pRSK, pS6, and p4E-BP1 levels (Figure S6J and S6K). Together, our data suggest that ERK inhibition alone or in combination with other targeted agents may be beneficial for treatment of PDAC patients.
Identification of Resistance Mechanisms to Short-term SCH772984 Treatment
To assess the mechanistic basis of de novo resistance (Figure 1A), we used two strategies. First, we applied reverse-phase protein array (RPPA)-based pathway activation mapping analysis (Liotta et al., 2003) to a panel of sensitive and de novo resistant cell lines to measure the basal activation/phosphorylation state of 135 key signaling proteins involved in cell proliferation, autophagy, apoptosis, survival, migration and adhesion. We identified a distinct signature that distinguished sensitive versus resistant cell lines (Figure S7A). Within this signature, there was a significant (p<0.05) activation of the PI3K-AKT-mTOR signaling network, with increased phosphorylation/activation of AKT and of multiple AKT substrates (e.g., p27, NF-κB, as well as mTOR substrates such as 4E-BP1, EIF4G, S6 ribosomal protein) (Figure S7A). In agreement with our observation that inhibitors of PI3K signaling synergistically enhanced SCH772984 inhibitory activity in sensitive cell lines, RPPA pathway mapping analyses identified PI3K-AKT-mTOR signaling as a key driver of ERK inhibitor resistance.
Second, we applied a kinome siRNA library screen to identify genes displaying synthetic lethality with SCH772984. Using two SCH772984-resistant PDAC cell lines (CFPAC-1 and SW1990), we performed two independent kinome-wide siRNA screens (711 genes) in the absence or presence of SCH772984. Subsequent validation analyses in three PDAC cell lines (CFPAC-1, SW1990 and DAN-G) verified 24 genes whose targeting by at least two different siRNAs enabled SCH772984 to suppress anchorage-dependent growth in otherwise resistant cell lines (Figure S7B and S7C). Further validation showed that siRNA suppression along with SCH772984 treatment decreased cell growth by greater than 70% (Figure S7D–G). We identified new genes as well as known modulators of MAPK-inhibitor resistance (Johannessen et al., 2010). These analyses reveal the striking heterogeneity in the signaling mechanisms that can drive de novo resistance to ERK inhibition.
Since the majority of kinases identified in our screen have not been associated previously with resistance to RAF or MEK inhibitors, we performed Ingenuity Pathway Analysis on our hits to determine their possible relevance to ERK signaling. This analysis identified recognized associations with ERK signaling in 13 of the 24 kinases (Figure 8). These results indicate that multiple and diverse mechanisms will likely drive de novo and/or acquired resistance to ERK inhibition.
Figure 8. Identification of Protein Kinases That Regulate Resistance of PDAC Cell Lines to SCH772984.

Pathway analysis based on Ingenuity Pathway Knowledge Base. The highest scoring networks (Post-translational Modification, Cell Morphology, Cellular Assembly and Organization; score 29, p-values < 0.05) were obtained from the proteins identified in our kinome-wide siRNA SCH772984 resistance screen. A black solid line represents a direct relationship between two nodes. A dotted line represents an indirect interaction between two nodes. Shaded nodes are the genes identified in our screen.
See also Figure S7.
To investigate mechanisms of acquired resistance to ERK inhibition, we applied an innovative gain-of-function screen (aka Cancer Toolkit, CTK (Martz et al., 2014)) to identify activated signaling components that can cause acquired resistance. The CTK is a library of barcoded expression constructs consisting of key genes in 20 of the oncogenic pathways most commonly activated in cancer. We examined potential mechanisms of acquired resistance to both SCH772984 and the clinical candidate BVD-523. Interestingly, we found that CTK-induced acquired resistance to SCH772984 and BVD-523 yielded many overlapping hits, which included PI3K, Notch, and p38 signaling cascades (Figure S7H–J). With inhibitors of Notch and p38 under clinical evaluation, their application together with ERK inhibitors may be tested rapidly to determine if these combinations delay onset of acquired resistance.
DISCUSSION
Clinical experiences with RAF and MEK inhibitors in BRAF-mutant melanomas emphasize that the RAF-MEK-ERK cascade is not a simple linear signaling pathway (Samatar and Poulikakos, 2014). Clearly, each level of the cascade is subject to distinct regulatory mechanisms, with inhibition at each level leading to distinct compensatory mechanisms and drivers of inhibitor resistance. Furthermore, Raf and MEK inhibitors that induce loss of ERK-mediated negative feedback mechanisms, leading to increased pathway flux and ERK reactivation (Nissan et al., 2013) have been largely ineffective in RAS-mutant cancers. Our recent finding that ~50% of RAS-mutant human cancer cells are responsive to the ERK-selective inhibitors SCH772984 and BVD-523 (clinical candidate), despite their resistance to MEK inhibitors (Morris et al., 2013), suggests that inhibition of the pathway at the level of ERK may be advantageous (Hatzivassiliou et al., 2012).
An unexpected finding from our studies was the different mechanisms of growth suppression seen upon short-term (24–72 hr) versus long-term (1–2 weeks) ERK inhibitor treatment. Conventionally, mechanisms of growth suppression by signaling inhibitors are characterized by short-term analyses, yet clinical application of such inhibitors involves persistent long-term treatment. As with MEK inhibitors (Alagesan et al., 2015), short-term treatment (24–72 hr) with this ERK inhibitor induced growth suppression that was associated with perturbation of progression through G1 and induction of apoptosis. In contrast, we found that long-term treatment (weeks) instead caused a senescence-like growth suppressive phenotype, which was dependent on the proteasomal degradation of MYC. Since clinical use of ERK inhibitors will certainly require long-term treatment, the induction of this phenotype may be the more clinically relevant mechanism of action. There is emerging appreciation that drug-induced senescence may be part of an important and effective approach for cancer treatment (Nardella et al., 2011) by rendering cancer cells sensitive to cytotoxic drugs.
Our application of unbiased chemical library screening and phosphoprotein profiling independently identified the PI3K-AKT-mTOR pathway as a key mediator of ERK inhibitor sensitivity. However, while concurrent treatment with MEK and PI3K inhibitors has recently been explored in mouse models of pancreatic cancer, only modest anti-tumor activity was observed (Alagesan et al., 2015; Junttila et al., 2015). Furthermore, clinical trial evaluations of MEK and PI3K inhibitor combinations also suggest that normal tissue toxicity may limit this combination (Shimizu et al., 2012; Tolcher et al., 2015). Clinical evaluation of PI3K and ERK inhibitor combinations will be needed to determine whether targeting ERK rather than MEK will overcome these limitations.
We found that a significant subset of KRAS-dependent pancreatic cancer cell lines exhibited de novo resistance to this ERK inhibitor. Our RPPA phosphoprotein profiling analyses identified a PI3K-AKT-mTOR activation signature that distinguished sensitive versus resistant cell lines. Mutations activating the PI3K-PTEN-AKT signaling cascade have been associated with de novo resistance to MEK inhibition in RAS-mutant cancers (Wee et al., 2009). However, most of our ERK inhibitor-resistant cell lines did not contain such mutations. Furthermore, concurrent PI3K inhibition, which conferred increased growth inhibition in ERK inhibitor-sensitive cells, could not overcome ERK inhibitor resistance in the de novo resistant cells. Thus, increased PI3K signaling together with additional mechanisms must drive de novo resistance.
In support of such additional mechanisms, our kinome siRNA library screen identified a striking diversity of protein kinases that, when inactivated, overcame ERK inhibitor resistance. Although there was but one overlap in the hits from two full kinome siRNA library screens, validation of the combined hits showed that the majority drove resistance in a third cell line. This striking heterogeneity is not unexpected in light of the fact that there are numerous alterations in non-KRAS genes that are not widely shared among PDAC tumors (Waddell et al., 2015). This may be why both ERK-dependent and -independent pathways can overcome KRAS addiction. Recent studies identified the transcription factor YAP1 as one mechanism that can overcome loss of mutant K-Ras in KRAS-mutant PDAC (Kapoor et al., 2014; Shao et al., 2014). While these studies provided compelling evidence in support of YAP1, additional modulators are likely to be revealed. Future studies will be needed to expand on these observations.
Importantly, we employed a genetic gain-of-function screen to identify mechanisms of acquired resistance to ERK inhibition. SCH772984 and BVD-523 shared several hits from the screen, namely, PI3K, Notch and p38. None of these has previously been identified as a modulator of ERK resistance (Goetz et al., 2014; Vogel et al., 2015). Together, our results stress that many distinct ERK inhibitor-based combinations will be needed for efficacy across different KRAS-mutant PDAC populations. Elucidation of the molecular determinants of ERK inhibitor response will be critically needed to accomplish our goal of achieving effective personalized medicine for KRAS-mutant PDAC patients.
EXPERIMENTAL PROCEDURES
Mouse Xenograft Studies
The mouse xenograft assays are described in Supplemental Experimental Procedures. All animal experiments conformed to the guidelines of the Animal Care and Use Committee of Johns Hopkins University and animals were maintained in accordance to guidelines of the American Association of Laboratory Animal Care. For PDAC cell line xenografts, animal procedures were performed in accordance with the rules set forth in the NIH Guide For The Care And Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Merck.
Cell Lines and Reagents
Information on cell lines and culture conditions is presented in Supplemental Experimental Procedures.
Expression Constructs and Antibodies
The sources of expression plasmids, siRNA and antibodies are described in Supplemental Experimental Procedures.
Small Molecule Inhibitors
The sources of SCH772984 and AZD6244/selumetinib, and GI50 determinations are provided in Supplemental Experimental Procedures.
Growth Assays
Growth, senescence-associated β-galactosidase staining and cell cycle analyses are described in Supplemental Experimental Procedures.
Drug Sensitivity and Resistance Testing (DSRT)
Protocols for the chemical library screen are described in Supplemental Experimental Procedures.
Reverse-Phase Protein Array (RPPA) Analysis
Protocols for RPPA analyses are described in Supplemental Experimental Procedures.
Kinome-wide siRNA Library Screen
The library and protocols for the kinome siRNA screen are described in Supplemental Experimental Procedures.
Supplementary Material
Significance.
The RAF-MEK-ERK protein kinase cascade is a well-validated driver of KRAS-dependent cancer growth. Despite this key role, pharmacologic inhibitors of RAF and MEK have been ineffective in inhibiting the growth of KRAS-mutant cancers, in large part due to dynamic signal reprogramming leading to ERK reactivation. We addressed the possibility that direct ERK inhibition may be a more effective therapeutic approach for KRAS-mutant pancreatic cancer. We identified a growth suppression mechanism due to ERK inhibition that involves degradation of the MYC oncoprotein and that is associated with induction of a senescence-like phenotype. We also identified PI3K-AKT-mTOR signaling as a critical modulator of ERK-inhibitor sensitivity. Our findings support a distinct and advantageous therapeutic value in targeting ERK.
Highlights.
Bimodal action: differential consequences of short- versus long-term ERK inhibition
ERK inhibitor sensitivity is associated with MYC degradation in vitro and in vivo
PI3K-AKT-mTOR signaling drives basal resistance to ERK inhibitor treatment
Resistance mechanisms are complex and display significant inter-tumor heterogeneity
Acknowledgments
Support was provided by grants from the National Institutes of Health (CA42978 and CA179193), the Lustgarten Pancreatic Cancer Foundation, and the Pancreatic Cancer Action Network (C.J.D.), and a National Cancer Institute fellowship (T.K.H.). We thank Kris Wood (Duke) for the Cancer Toolkit, Juan Belmonte for plasmid pMSCVpuro-Fag-cMyc T58A, and BioMed Valley Discoveries (BVD) for providing BVD-523.
Footnotes
Supplemental information includes Supplemental Experimental Data (seven figures, and two tables), Supplemental Experimental Procedures and Supplementary References.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alagesan B, Contino G, Guimaraes AR, Corcoran RB, Deshpande V, Wojtkiewicz GR, Hezel AF, Wong KK, Loda M, Weissleder R, et al. Combined MEK and PI3K Inhibition in a Mouse Model of Pancreatic Cancer. Clin Cancer Res. 2015;21:396–404. doi: 10.1158/1078-0432.CCR-14-1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonet C, Giuliano S, Ohanna M, Bille K, Allegra M, Lacour JP, Bahadoran P, Rocchi S, Ballotti R, Bertolotto C. Aurora B is regulated by the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling pathway and is a valuable potential target in melanoma cells. J Biol Chem. 2012;287:29887–29898. doi: 10.1074/jbc.M112.371682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell ME, DeNicola GM, Martins CP, Jacobetz MA, Maitra A, Hruban RH, Tuveson DA. Cellular features of senescence during the evolution of human and murine ductal pancreatic cancer. Oncogene. 2012;31:1599–1608. doi: 10.1038/onc.2011.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano E, Sheridan C, Thin MZ, Nye E, Spencer-Dene B, Diefenbacher ME, Moore C, Kumar MS, Murillo MM, Gronroos E, et al. Requirement for interaction of PI3-kinase p110alpha with RAS in lung tumor maintenance. Cancer Cell. 2013;24:617–630. doi: 10.1016/j.ccr.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschenes-Simard X, Gaumont-Leclerc MF, Bourdeau V, Lessard F, Moiseeva O, Forest V, Igelmann S, Mallette FA, Saba-El-Leil MK, Meloche S, et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013;27:900–915. doi: 10.1101/gad.203984.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–224. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, Hieber M, Arbeiter A, Klein S, Kong B, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23:406–420. doi: 10.1016/j.ccr.2013.01.023. [DOI] [PubMed] [Google Scholar]
- Farrell AS, Sears RC. MYC Degradation. Cold Spring Harb Perspect Med. 2014;4 doi: 10.1101/cshperspect.a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann G, Mishra A, Bisht S, Karikari C, Garrido-Laguna I, Rasheed Z, Ottenhof NA, Dadon T, Alvarez H, Fendrich V, et al. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol Ther. 2011;12:598–609. doi: 10.4161/cbt.12.7.16475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann G, Mishra A, Hong SM, Bisht S, Strock CJ, Ball DW, Goggins M, Maitra A, Nelkin BD. Inhibiting the cyclin-dependent kinase CDK5 blocks pancreatic cancer formation and progression through the suppression of Ras-Ral signaling. Cancer Res. 2010;70:4460–4469. doi: 10.1158/0008-5472.CAN-09-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germann U, Furey B, Roix J, Marklan W, Hoover R, Aronov A, Hale M, Chen G, Martinez-Botella G, Alargova R, Fan B, Sorrell D, Meshaw K, Shapiro P, Wick M, Benes C, Garnett M, DeCrescemzp G, Namchuk M, Saha S, Welsch D. The selective ERK inhibitor BVD-523 is active models of MAPK pathway-dependent cancers, including those with intrinsic and acquired drug resistance. Proceedings of the 106th Annual Meeting of the American Association for Cancer Research.2015. [Google Scholar]
- Goetz EM, Ghandi M, Treacy DJ, Wagle N, Garraway LA. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 2014;74:7079–7089. doi: 10.1158/0008-5472.CAN-14-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–968. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- Hancox U, Cosulich S, Hanson L, Trigwell C, Lenaghan C, Ellston R, Dry H, Crafter C, Barlaam B, Fitzek M, et al. Inhibition of PI3Kbeta Signaling with AZD8186 Inhibits Growth of PTEN-Deficient Breast and Prostate Tumors Alone and in Combination with Docetaxel. Mol Cancer Ther. 2015;14:48–58. doi: 10.1158/1535-7163.MCT-14-0406. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, Hewitt JF, Zak M, Peck A, Orr C, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501:232–236. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Liu B, O’Brien C, Spoerke JM, Hoeflich KP, Haverty PM, Soriano R, Forrest WF, Heldens S, Chen H, et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther. 2012;11:1143–1154. doi: 10.1158/1535-7163.MCT-11-1010. [DOI] [PubMed] [Google Scholar]
- Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Maelandsmo GM, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila MR, Devasthali V, Cheng JH, Castillo J, Metcalfe C, Clermont AC, Otter DD, Chan E, Bou-Reslan H, Cao T, et al. Modeling targeted inhibition of MEK and PI3 kinase in human pancreatic cancer. Mol Cancer Ther. 2015;14:40–47. doi: 10.1158/1535-7163.MCT-14-0030. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. 2014;158:185–197. doi: 10.1016/j.cell.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KH, Counter CM. Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance. Cancer Cell. 2005;8:381–392. doi: 10.1016/j.ccr.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Lim KH, O’Hayer K, Adam SJ, Kendall SD, Campbell PM, Der CJ, Counter CM. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol. 2006;16:2385–2394. doi: 10.1016/j.cub.2006.10.023. [DOI] [PubMed] [Google Scholar]
- Liotta LA, Espina V, Mehta AI, Calvert V, Rosenblatt K, Geho D, Munson PJ, Young L, Wulfkuhle J, Petricoin EF., 3rd Protein microarrays: meeting analytical challenges for clinical applications. Cancer Cell. 2003;3:317–325. doi: 10.1016/s1535-6108(03)00086-2. [DOI] [PubMed] [Google Scholar]
- Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–1409. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- Martz CA, Ottina KA, Singleton KR, Jasper JS, Wardell SE, Peraza-Penton A, Anderson GR, Winter PS, Wang T, Alley HM, et al. Systematic identification of signaling pathways with potential to confer anticancer drug resistance. Sci Signal. 2014;7:ra121. doi: 10.1126/scisignal.aaa1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742–750. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- Murillo MM, Zelenay S, Nye E, Castellano E, Lassailly F, Stamp G, Downward J. RAS interaction with PI3K p110alpha is required for tumor-induced angiogenesis. J Clin Invest. 2014;124:3601–3611. doi: 10.1172/JCI74134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nature Rev Cancer. 2011;11:503–511. doi: 10.1038/nrc3057. [DOI] [PubMed] [Google Scholar]
- Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318–330. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissan MH, Rosen N, Solit DB. ERK pathway inhibitors: How low should we go? Cancer Discov. 2013;3:719–721. doi: 10.1158/2159-8290.CD-13-0245. [DOI] [PubMed] [Google Scholar]
- Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, Ohashi Y, Sharrocks AD, Peters G, Hara E. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature. 2001;409:1067–1070. doi: 10.1038/35059131. [DOI] [PubMed] [Google Scholar]
- Pemovska T, Kontro M, Yadav B, Edgren H, Eldfors S, Szwajda A, Almusa H, Bespalov MM, Ellonen P, Elonen E, et al. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 2013;3:1416–1429. doi: 10.1158/2159-8290.CD-13-0350. [DOI] [PubMed] [Google Scholar]
- Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014;158:171–184. doi: 10.1016/j.cell.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, Gunn S, Smetzer L, Mays TA, Kaiser B, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18:2316–2325. doi: 10.1158/1078-0432.CCR-11-2381. [DOI] [PubMed] [Google Scholar]
- Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, Settleman J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging Ras Back in the Ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- Tolcher AW, Khan K, Ong M, Banerji U, Papadimitrakopoulou V, Gandara DR, Patnaik A, Baird RD, Olmos D, Garrett CR, et al. Antitumor Activity in RAS-Driven Tumors by Blocking AKT and MEK. Clin Cancer Res. 2015;21:739–748. doi: 10.1158/1078-0432.CCR-14-1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CJ, Smit MA, Maddalo G, Possik PA, Sparidans RW, van der Burg SH, Verdegaal EM, Heck AJ, Samatar AA, Beijnen JH, et al. Cooperative induction of apoptosis in NRAS mutant melanoma by inhibition of MEK and ROCK. Pigment Cell Melanoma Res. 2015;28:307–317. doi: 10.1111/pcmr.12364. [DOI] [PubMed] [Google Scholar]
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, Sellers WR, Lengauer C, Stegmeier F. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286–4293. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci USA. 2007;104:13028–13033. doi: 10.1073/pnas.0701953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.