

Abstract
Background
Silencing mutant huntingtin mRNA by RNA interference (RNAi) is a therapeutic strategy for Huntington’s disease. RNAi induces specific endonucleolytic cleavage of the target HTT mRNA, followed by exonucleolytic processing of the cleaved mRNA fragments.
Objectives
We investigated the clearance of huntingtin mRNA cleavage products following RNAi, to find if particular huntingtin mRNA sequences persist. We especially wanted to find out if the expanded CAG increased production of a toxic mRNA species by impeding degradation of human mutant huntingtin exon 1 mRNA.
Methods
Mice expressing the human mutant HTT transgene with 128 CAG repeats (YAC128 mice) were injected in the striatum with self-complementary AAV9 vectors carrying a miRNA targeting exon 48 of huntingtin mRNA (scAAV-U6-miRNA-HTT-GFP). Transgenic huntingtin mRNA levels were measured in striatal lysates after two weeks. For qPCR, we used species specific primer-probe combinations that together spanned 6 positions along the open reading frame and untranslated regions of the human huntingtin mRNA. Knockdown was also measured in the liver following tail vein injection.
Results
Two weeks after intrastriatal administration of scAAV9-U6-miRNA-HTT-GFP, we measured transgenic mutant huntingtin in striatum using probes targeting six different sites along the huntingtin mRNA. Real time PCR showed a reduction of 29% to 36% in human HTT. There was no significant difference in knockdown measured at any of the six sites, including exon 1. In liver, we observed a more pronounced HTT mRNA knockdown of 70% to 76% relative to the untreated mice, and there were also no significant differences among sites.
Conclusions
Our results demonstrate that degradation is equally distributed across the human mutant huntingtin mRNA following RNAi-induced cleavage.
Keywords: Huntington’s disease, RNAi, mRNA degradation, CAG repeats
Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder caused by expansion of trinucleotide repeats (CAG) in the first exon of the huntingtin (HTT) gene [1]. In general, the longer the CAG expansion, the earlier the disorder is manifest[2, 3]. RNAi and anti-sense oligonucleotides are promising treatments for Huntington’s disease. Theoretical concerns of incomplete clearance attend exonucleolytic processing of mutant huntingtin mRNA. It is generally accepted that N-terminal huntingtin protein fragments are more toxic than full-length protein [4–6]. Truncated mutant huntingtin protein, particularly encoded by exon 1 with expanded CAG repeats, is sufficient to cause neuronal degeneration [7]. Alternate strand translation in exon 1 of mutant huntingtin mRNA can produce toxic protein fragments[8]. Expanded CAG repeats in the huntingtin mRNA, through aberrant interactions, might contribute to the pathogenesis of HD[8]. Expanded CAG repeats could produce small CAG-repeated RNAs that are neurotoxic[9]. Therefore, complete exonucleolytic processing is a goal of RNAi.
Previously, we have demonstrated that 5 siRNAs targeting heterozygous single nucleotide polymorphisms of mutant huntingtin mRNA could provide allele-specific silencing for at least 75% of HD patients [10]. The candidate target sites with significant SNP heterozygosity (H>10%) are located downstream and at a distance (3 to 11 kilo-bases) from CAG repeats[11]. Following RNAi-based endonucleolytic cleavage by guide strand containing Argonaute2 complex, mRNAs are degraded through the exonucleolytic process in both the 5′ to 3′ and the 3′ to 5′ directions[12]. We were curious whether the expanded CAG repeats alter the exonucleolytic process. Using the YAC128 HD mouse model, we measured residual mutant HTT mRNA after AAV-miRNA treatment to address this question.
Materials and methods
HD model mouse and viral vector expressing miRNA against HTT
We used yeast artificial chromosome (YAC) transgenic mice expressing full-length HTT mRNA with 128 CAG repeats (YAC128)[13]. YAC128 mice were produced by crossing heterozygote females with wild-type FVB males purchased from Jackson Laboratory (Bar Harbor, ME). Genotyping was performed by PCR following the Jackson Laboratory guidelines on total genomic DNA isolated from tail snips. The mice were housed up to four per cage with food and water available. Animal care and handling were in accordance with institutional regulations for animal care and were approved by the Animal Investigation Committee of the University of Massachusetts Medical School.
YAC128 mice at the age of about three months were used for injection. Self-complementary adeno-associated virus serotype 9 (scAAV 9) with an insert of GFP was used to deliver U6-driven miRNA against huntingtin mRNA (scAAV9-U6-miRNA-HTT-GFP). The viral vector was validated for its efficiency and specificity. The distribution of the viral vector is confirmed by immunohistochemical staining using antibody against GFP (Figure 1d). The target site of the huntingtin mRNA, complementary to vector-produced miRNAs, is located in the middle of the long transcript at exon 48. The right striatum of YAC128 mice (n=4) were injected using a micro-pump syringe with 3μl of scAAV9-U6-miRNA-HTT-GFP at a titer of 5×1012 vg/μl for a total dose of 1.5×1013 vg/mouse. Following injection, males were housed singly and females were housed with littermates. The injected mice were sacrificed at two weeks and the striatal tissues of both sides were removed and stored at −80 °C. To examine transgenic huntingtin mRNA degradation in liver, we also injected the scAAV9-U6-miRNA-HTT-GFP through tail vein (3×1011 vg per mouse). Liver tissue samples were also collected for mice (n=4) with tail-vein injection at 2 weeks and stored at −80 °C for further analysis. Liver samples of non-injected YAC128 mice with same age were used as control.
Figure 1.
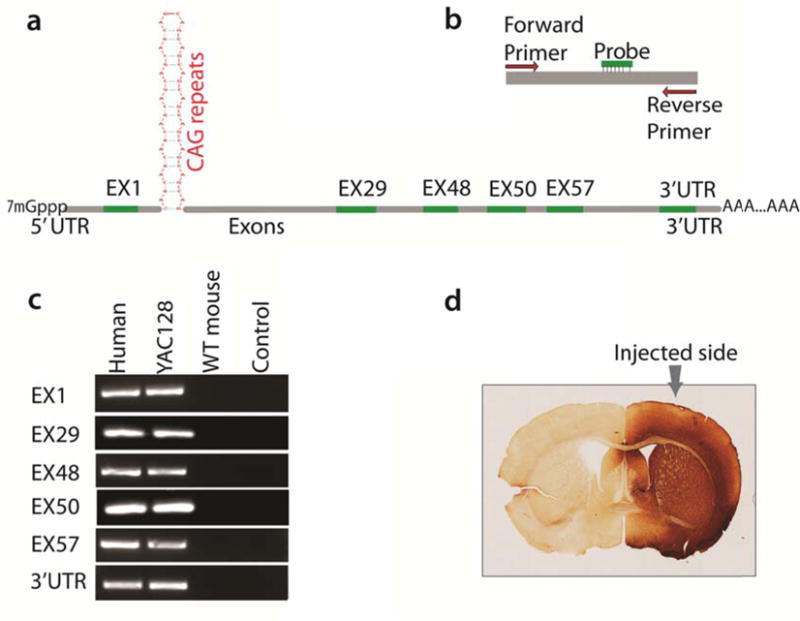
Selected Sites along the mutant HTT mRNA for quantitative PCR to estimate the regional abundance of the mRNA. a. Schematic drawing showing the positions of the qPCR amplicon on HTT mRNA; b. Primers and probe combination of species-specific qPCR, note both primers, as well as the probe, are perfectly match to human HTT but with mismatches, or deletions to mouse homologous Htt to enhance the species-specificity; c. Gel images showing that real-time PCR for each part is species-specific to transgenic human HTT without interference by mouse endogenous Htt. Note that the primer combinations work efficiently and specifically with human HTT input (human samples and YAC128 mouse samples); d. Immunohistochemical staining with antibody against GFP showing the vector was expressed unilaterally, with no noticeable spreading to the other hemisphere that is used as an internal control.
RNA extraction and Reverse transcription
Total RNA was isolated from 50–100 mg tissue using MirVana miRNA Isolation Kit (Life Technologies, NY) following the manufacturer’s instructions and at final step eluted in water. RNA concentration was measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific Inc. MA) and RNA was stored at −80°C. To eliminate unwanted genomic DNA, total RNA was incubated with RNase-free DNase I (Invitrogen, Carlsbad, CA) for 30 min at 37°C, EDTA was added (5 mM final concentration), and then DNase was inactivated at 65°C for 10 min. Total RNA (1 μg) was then used to synthesize first strand cDNA by using Transcriptor transcriptase (Transcriptor RT, Roche Applied Science, Indianapolis, IN) and oligo (dT) primers (Invitrogen, Carlsbad, CA). The resulting full-length cDNA was stored at −20°C. To confirm the full-length synthesis of the transgenic huntingtin cDNA quality, we performed a PCR amplification of CAG repeat-containing region of huntingtin exon 1 using LA-Taq with GC-rich buffer (Takara, Japan) [14]. Reaction products were separated by electrophoresis on a 1.5% agarose gel with ethidium bromide (0.5 μg/ml) in Tris-acetic acid-EDTA buffer (TAE, pH 8.3), examined and photographed under UV light.
Species-specific Quantitative PCR and TaqMan probes
To examine the transgenic human huntingtin, we designed a series of species-specific quantitative PCR assay that detects transgenic human HTT mRNA without the interference from the endogenous mouse huntingtin mRNA. We aligned the human HTT cDNA and the mouse homologue together and designed our assays to target areas that differ between human and mouse huntingtin (Figure 1b, Table 1). Assays were designed to target sites in coding region: exon1, exon 29, exon48, exon 50, exon57 and in the 3′ UTR. The 3′ UTR of huntingtin mRNA is quite long and displays different patterns of polyadenylation among tissue types [15]. We chose to examine the region that is common to both 3′-UTR isoforms found in brain and in liver. Primer-probe combinations are highly specific to the human huntingtin (Figure 1c). We performed real-time PCR using Mastercycler Ep Realplex (Eppendorf, Hauppauge, NY). The 20-μl-reaction mixture contained 10 μl 2× Type-it Fast SNP Probe PCR Master Mix (Qiagen, Valencia, CA), forward and reverse primers (2 μM each), and probes (150 nM final concentration), 1 μl cDNA, and H2O up to 20 μl. Reactions were run in triplicate in 96-well plates (Twin-tech, Eppendorf). The thermal cycle program was 5 min at 95°C followed by 40 amplification cycles (95°C for 15 s, 62°C for 15 s and 72°C for 30 s), then 30°C forever. Mouse hypoxanthine phosphoribosyltransferase gene (HPRT, Applied Biosystems, CA) was used as a housekeeping control in all the experiments. All experiments were repeated twice. Relative knock-down of HTT mRNA was calculated based on delta-delta Ct obtained for each sample. Statistical significance (P values < 0.05) of the transgenic HTT mRNA among samples was assessed by t-test, two-way ANOVA analysis using Prism 6 for Mac (Ver. 6c).
Table 1.
Primers and probes used in real-time PCR
| Name | Sequence (5′—3′) | Length |
|---|---|---|
| E1F1 | GTGCTGAGCGGCGCCGCGAGTC | 22 |
| E1R2 | GGACTTGAGGGACTCGAAGGC | 21 |
| PROBE-E1 | CCTCCGGGGACTGCCGTGC | 19 |
| E29F1 | ACGCTAACTACAAGGTCACGCTG | 22 |
| E29R2 | TCCCAATGTCCTGCAGTGTGGC | 21 |
| PROBE-E29 | AGGGTTTCTtCGCTCAGCC | 19 |
| E48F1 | CCTAAGCCTGCTAGCTCCATGC | 22 |
| E48R1 | GAGCTGCTGCACGGTGCCGC | 20 |
| PROBE-E48 | GATGAGTGAAATTTCTGGT | 19 |
| E50F1 | CGTGGTCTCCTCCACAGAGTTT | 22 |
| E50-R1 | TGCAGTGATATACTTAGGAT | 20 |
| PROBE-E50 | AGTCCAGAAAGAAGGACA | 19 |
| E57F1 | CGCCATGGTGGGAGAGACTGTG | 22 |
| E57R1 | CCACATGGCAGAGACACGCACG | 22 |
| PROBE-E57 | CTCCGGCTACTACAGGTGC | 19 |
| 3′UTR F1 | CGCCATGGTGGGAGAGACTGTG | 22 |
| 3′UTR R1 | CCACATGGCAGAGACACGCACG | 22 |
| PROBE-3′UTR | TGGAAGTCTGCGCCCTTGT | 19 |
E: exon; F: forward; R: reverse
Results
At two weeks, AAV would not be expected to reach full expression of miRNA. Our intention was to avoid oversaturation of the vector-expressed miRNAs that occurs after two weeks[16], as well as long-term equilibration of RNA interference.
Striatum
Tissue samples were collected at two weeks after scAAV-miRNA injection. Using the species-specific qPCR, we examined the transgenic human huntingtin mRNA levels in the striatum of YAC128 mice. To control for mouse-to-mouse variation, the non-injected striatum was used as control. Real-time PCR revealed that mRNA levels at the artificial miRNA target site in exon 48 decreased by 32% when compared to the non-injected striatum. We also measured huntingtin mRNA at two sites upstream of the RNAi cut site. Using a qPCR assay directed at exon 1, we saw a 31% decrease in huntingtin mRNA and in exon 29 we saw a reduction of 38% relative to the untreated side. We designed three additional assays downstream of the miRNA cut site: two in the coding region (exon 50 and 57) and one in the 3′ UTR. Quantitative PCR revealed that mRNA levels decreased by 31% in the region of exon 50, and by 30% in the region of exon 57. Overall, we conclude that transgenic mutant HTT mRNA level decreased by 30% in striatum with scAAV9-U6-miRNA-HTT-GFP treatment in comparison to the non-injected side (Figure 2). Statistical analysis reveals that there is no significant difference among any of the qPCR-flanked sites.
Figure 2.
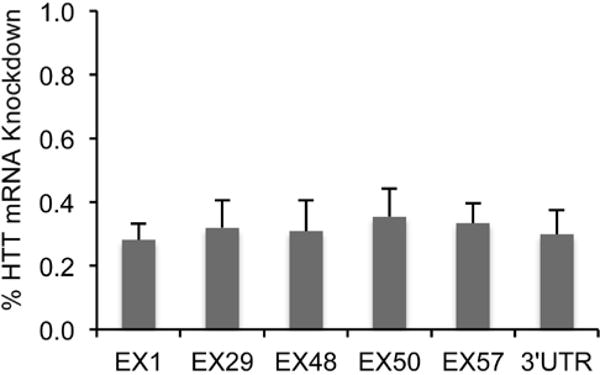
Real-time PCR results showing that scAAV9 delivered miRNA against HTT knocks down the HTT mRNA in treated striatum estimated at different parts along the HTT mRNA.
Liver
We wanted to know if the degradation of mutant huntingtin mRNA in liver where the miRNA induces robust knockdown is comparable to degradation in striatum. The same qPCR experiments were performed to measure mRNA levels in liver samples. Two weeks after tail vein injection of scAAV9-U6-miRNA-HTT, mutant HTT mRNA level reduced 73% at the RNAi cut site (exon 48). In two upstream sites we examined, mRNA level decreased by 74% on exon 1, and by 72% for exon 29. Similarly, three sites in the downstream fragments show a significant decrease by 76% at exon 50, 72% at exon 57, and 70% at 3′-UTR (Figure 3). As with striatal samples, there were no significant differences among target areas examined.
Figure 3.
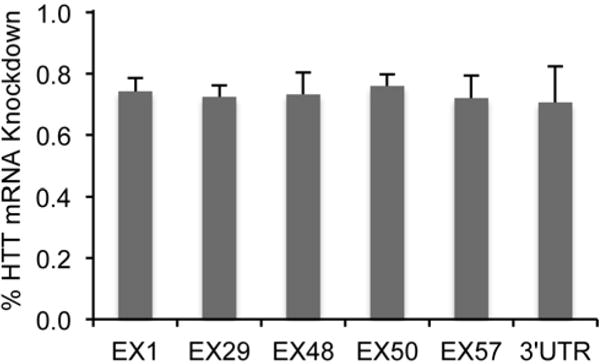
Real-time PCR results showing relative knock-down of HTT mRNA in liver by tail-vein injection of scAAV9 delivered miRNA against HTT in YAC128 mice (non-injected YAC128 mice with same age were used as controls).
Discussion
Messenger RNAs are degraded by the following exonucleolytic pathways: (1) poly-A tail shortening followed by 3′ to 5′ decay mediated by exosome complex, (2) decapping and decay by XRN1-like enzymes (5′ to 3′ exonucleolytic process), and (3) endonucleolytic cleavage (e.g. RISC mediated RNAi cleavage) followed by exonucleolytic reactions mediated by the same components also involved in pathways (1) and (2) [12]. RNAi introduces an intended, well-designed endonucleolytic cleavage. RNAi as a mechanism of microRNA exists in eukaryotic organisms as part of a genetic regulatory system [12, 17, 18]. Using RNAi to induce degradation of mutant huntingtin mRNA is a promising therapeutic to treat Huntington’s disease. Because expanded CAG repeats might alter the folding of mutant huntingtin mRNA, we are concerned that exon 1 of mutant huntingtin might resist exonucleolytic cleavage initiated by RNAi; if so, 5′ RNA fragments might accumulate. The elongated CAG repeats would alter the structure of the upstream fragment and hinder the 3′ to 5′ exonucleolytic processing. Our results show that there is no significant difference among the 6 regions along the huntingtin mRNA, including exon 1. The exonucleolytic process of mutant huntingtin mRNA continues unabated.
Mis-splicing of the transgenic mutant huntingtin can produce a short form of mutant huntingtin transcripts, facilitated by the presence of exon 1 with poly (A) [19]. The scAAV-U6-miRNA-HTT-GFP we have used in this study induces cleavage in exon 48, so that exonucleolytic activity would not affect this aberrant mutant huntingtin mRNA fragment. Our exon 1 primer-probe combinations would not distinguish between exon 1 mRNA fragments from long mutant huntingtin mRNA and the aberrant fragment.
At two weeks, the difference between brain and liver RNAi is noteworthy, with silencing of liver mutant huntingtin more than in brain. We speculate that that uptake of AAV9-miRNA in hepatocytes is more consistent than occurs in neurons, even with direct brain injection of AAV, thereby accounting for higher knockdown of mutant huntingtin in liver. An alternative, though less likely, possibility is that RNAi in hepatocytes is more efficient than in neurons.
Neurodegenerative disorders caused by trinucleotide repeat expansions might involve RNA-mediated mechanisms [20–22]. Expanded CAG repeats could form an inverse-repeat-like folding, which not only alter the confirmation of RNA, but also could be substrates for production of CAG repeat small RNAs. Those RNAs were found to be neurotoxic[22]. Our finding indicates that RNAi-mediated mutant huntingtin cleavage results in complete degradation of the huntingtin mRNA. There is no noticeable accumulation of exon 1 with expanded CAG repeats after miRNA administration.
Acknowledgments
This study was supported by NIH NS 38194 and CHDI. WL, MD and NA designed the experiments. WL, EP, LK and KC performed the experiments. CM designed the viral vector. WL, EP, MD and NA wrote the manuscript.
Footnotes
Conflict of Interest: The authors have no conflict of interest to report.
References
- 1.MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, MacFarlane H, Jenkins B, Anderson MA, Wexler NS, Gusella JF, Bates GP, Baxendale S, Hummerich H, Kirby S, North M, Youngman S, Mott R, Zehetner G, Sedlacek Z, Poustka A, Frischauf A-M, Lehrach H, Buckler AJ, Church D, Doucette-Stamm L, O’Donovan MC, Riba-Ramirez L, Shah M, Stanton VP, Strobel SA, Draths KM, Wales JL, Dervan P, Housman DE, Altherr M, Shiang R, Thompson L, Fielder T, Wasmuth JJ, Tagle D, Valdes J, Elmer L, Allard M, Castilla L, Swaroop M, Blanchard K, Collins FS, Snell R, Holloway T, Gillespie K, Datson N, Shaw D, Harper PS. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–83. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2.Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, Starr E, Squitieri F, Lin B, Kalchman MA, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet. 1993;4:398–403. doi: 10.1038/ng0893-398. [DOI] [PubMed] [Google Scholar]
- 3.Gusella JF, MacDonald ME. Huntington’s disease: CAG genetics expands neurobiology. Curr Opin Neurobiol. 1995;5:656–62. doi: 10.1016/0959-4388(95)80072-7. [DOI] [PubMed] [Google Scholar]
- 4.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 5.Juenemann K, Weisse C, Reichmann D, Kaether C, Calkhoven CF, Schilling G. Modulation of mutant huntingtin N-terminal cleavage and its effect on aggregation and cell death. Neurotox Res. 2011;20:120–33. doi: 10.1007/s12640-010-9227-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet. 2002;11:1911–26. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- 7.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 8.Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK, Smith DL, Faull RLM, Roos RAC, Howland D, Detloff PJ, Housman DE, Bates GP. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2366–70. doi: 10.1073/pnas.1221891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banez-Coronel M, Porta S, Kagerbauer B, Mateu-Huertas E, Pantano L, Ferrer I, Guzman M, Estivill X, Marti E. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS genetics. 2012;8:e1002481. doi: 10.1371/journal.pgen.1002481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfister EL, Kennington L, Straubhaar J, Wagh S, Liu W, DiFiglia M, Landwehrmeyer B, Vonsattel JP, Zamore PD, Aronin N. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr Biol. 2009;19:774–8. doi: 10.1016/j.cub.2009.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu W, Kennington LA, Rosas HD, Hersch S, Cha J-H, Zamore PD, Aronin N. Linking SNPs to CAG repeat length in Huntington’s disease patients. Nat Meth. 2008;5:951–3. doi: 10.1038/nmeth.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belostotsky D. mRNA turnover meets RNA interference. Molecular cell. 2004;16:498–500. doi: 10.1016/j.molcel.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 13.Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–67. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 14.Liu W, Kennington LA, Rosas HD, Hersch S, Cha JH, Zamore PD, Aronin N. Linking SNPs to CAG repeat length in Huntington’s disease patients. Nat Methods. 2008;5:951–3. doi: 10.1038/nmeth.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin B, Rommens JM, Graham RK, Kalchman M, MacDonald H, Nasir J, Delaney A, Goldberg YP, Hayden MR. Differential 3′ polyadenylation of the Huntington disease gene results in two mRNA species with variable tissue expression. Human molecular genetics. 1993;2:1541–5. doi: 10.1093/hmg/2.10.1541. [DOI] [PubMed] [Google Scholar]
- 16.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–41. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 17.Chen X. A silencing safeguard: links between RNA silencing and mRNA processing in Arabidopsis. Developmental cell. 2008;14:811–2. doi: 10.1016/j.devcel.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gazzani S, Lawrenson T, Woodward C, Headon D, Sablowski R. A link between mRNA turnover and RNA interference in Arabidopsis. Science (New York, N Y) 2004;306:1046–8. doi: 10.1126/science.1101092. [DOI] [PubMed] [Google Scholar]
- 19.Gipson TA, Neueder A, Wexler NS, Bates GP, Housman D. Aberrantly spliced HTT, a new player in Huntington’s disease pathogenesis. RNA Biology. 2013;10:1647–52. doi: 10.4161/rna.26706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li L-B, Bonini NM. Roles of trinucleotide-repeat RNA in neurological disease and degeneration. Trends in neurosciences. 2010;33:292–8. doi: 10.1016/j.tins.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ranum LPW, Day JW. Pathogenic RNA repeats: an expanding role in genetic disease. Trends in genetics: TIG. 2004;20:506–12. doi: 10.1016/j.tig.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 22.Banez-Coronel M, Porta S, Kagerbauer B, Mateu-Huertas E, Pantano L, Ferrer I, Guzman M, Estivill X, Marti E. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. 2012;8:e1002481. doi: 10.1371/journal.pgen.1002481. [DOI] [PMC free article] [PubMed] [Google Scholar]