

Abstract
Background and Aims
Single-gene mutations cause syndromes of intrahepatic cholestasis, but previous multi-gene mutation screening in children with idiopathic cholestasis failed to fulfill diagnostic criteria in about two-thirds of children. In adults with fibrosing cholestatic disease, heterozygous ABCB4 mutations were present in 34% of patients. Here, we hypothesized that children with idiopathic cholestasis have a higher frequency of heterozygous non-synonymous gene sequence variants.
Methods
We analyzed the frequency and types of variants in 717 children in whom high-throughput sequencing of the genes SERPINA1, JAG1, ATP8B1, ABCB11, and ABCB4 was performed as part of an evaluation for intrahepatic idiopathic cholestasis. The frequency of non-synonymous variants (NSVs) was compared to those of 1092 control subjects enrolled in the 1000-Genome-Project.
Results
The frequency of NSVs in single genes was similar between disease (25%) and controls (26%, P=0.518). In contrast, double or triple NSVs in 2 or more genes were more frequent in disease (N= 7%) than controls (N=4.7%, P=0.028). Detailed review of clinical and laboratory information in a subgroup of double or triple heterozygous patients revealed variable GGT levels and severity of pruritus, with liver biopsies showing stage 2–3 fibrosis.
Conclusion
Children with intrahepatic idiopathic cholestasis have a higher frequency of double or triple NSVs in SERPINA1, JAG1, ATPB1, ABCB11, or ABCB4. These findings raise the potential role for gene-gene relationships in determining the phenotype of cholestatic liver disease in children.
Keywords: Liver, children, cirrhosis, jaundice, mutations
INTRODUCTION
The identification of genetic mutations in children with inherited syndromes of intrahepatic cholestasis has the potential to establish specific diagnoses and allow for the development of treatment protocols that take into account the genetic makeup of individual patients.1–6 In this context, the availability of clinical laboratories (listed on www.ncbi.nlm.nih.gov/sites/GeneTests) that sequence genes in the search for mutations makes genetic testing a reality in clinical practice. To enable a thorough sequence analysis of gene groups simultaneously, we developed a hybridization-based high density microarray that resequences the nucleotide signature for the five genes known to cause the most common forms of inherited cholestasis: SERPINA1 (for alpha-1-antitrypsin [A1AT] deficiency), JAG1 (for Alagille syndrome), ATP8B1 (for progressive familial intrahepatic cholestasis type 1, PFIC1), ABCB11 (for PFIC2), and ABCB4 (for PFIC3).7 Using this sequencing tool, we reported previously that gene sequence analysis failed to identify biallelic disease-causing mutations in SERPINA1, ATP8B1, ABCB4, or ABCB11, or monoallelic for JAG1 in at about two-thirds of children with intrahepatic cholestasis of unknown etiology.8
The lack of a genetic diagnosis in the majority of patients with cholestatic syndromes reflects, at least in part, the incomplete knowledge of how genes regulate the phenotype of intrahepatic cholestasis. For example, patients with clinical and biochemical features of ABCB11/BSEP deficiency may carry only one mutant allele, instead of the two-allele involvement that would be expected to occur in affected patients due to the recessive inheritance pattern of the disease.9 Further, single heterozygous mutations in ABCB4/MDR3 have been reported in 34% of in adults with idiopathic fibrosing cholestasis.10 Based on these data and on the functional relatedness of these genes11, we hypothesized that children with idiopathic cholestasis have a high frequency of non-synonymous heterozygous nucleotide sequence variants. Testing this hypothesis, we found double- and triple heterozygosity in a large cohort of cholestatic children, as well as biallelic disease-causing mutations in two genes simultaneously in a subgroup of children.
PATIENTS AND METHODS
Patients
The frequency and types of sequence variants were analyzed in 717 children in whom sequencing of the genes SERPINA1, JAG1, ATP8B1, ABCB11, or ABCB4 was performed as part of a clinical evaluation for idiopathic cholestasis. Of the 717 patients, 20 were cared for at the Pediatric Liver Care Center of Cincinnati Children’s Hospital Medical Center (CCHMC), and the remaining 697 subjects were from practitioners at other U.S. institutions or clinical practice groups. Information available for review included age, diagnosis, clinical features, presumptive diagnosis (if applicable), history of cholestasis, and high or low γ-glutamyl transpeptidase (GGT) for all subjects. Subjects receiving care at CCHMC had additional detailed clinical information, biochemical markers, radiological evaluation, histopathology and electron microscopy. When available, liver biopsy specimens were analyzed for grade of inflammation, stage of fibrosis, presence or absence of bile duct proliferation and giant cell transformation. This study was approved by the Institutional Review Board of Cincinnati Children’s Hospital Medical Center.
Chip hybridization and analysis
DNA was isolated from peripheral blood using the Gentra Purification Kit (Qiagen, Venlo, The Netherlands), according to the manufacturer’s protocol. Then, DNA samples served as templates in long-and short-range high-fidelity PCRs to amplify selected domains of target genes, followed by hybridization with the JaundiceChip, detection of biotin-labeled signals by the GeneChip 3000 Scanner, capture with the Affymetrix GeneChip® Operating Software, and analysis with the Affymetrix GeneChip® Sequence Analysis Software (GSEQ) as described by us previously.7
Gene Mutation Survey
The nucleotide sequence readout was reviewed and sequence variants were included in the analysis if they altered the amino acid sequence (non-synonymous variants). Among these, variants were considered clinically significant and called “mutation” if they were reported in the literature as non-synonymous variants linked to the disease phenotype. Variants were also called “mutation” if they were highly conserved among species, occurred in <2% of the normal population, and predicted to impair the function of the encoded protein based on the computational methods Grantham Score12, SIFT (Sorting Intolerant From Tolerant)13, and PolyPhen11 classified as “damaging.” If this analysis showed inconsistent results, they were called variants of unknown significance (VUS), as described previously.8,14 Nucleotide changes within 10 base pairs of the intron/exon boundaries (splice sites) were checked using NetGene(http://www.cbs.dtu.dk/biolinks/pserve2.php), a gene finder and intron splice site prediction algorithm hosted by the Center for Biological Sequence Analysis in Denmark.8 The frequency of variants in control population was retrieved from 1000_Genome-Project, an extensive data base containing detailed gene sequence variants in healthy subjects available publically available at www.1000genomes.org. At the time of data analysis, the 1092 healthy subjects consisted of 48.1% males (N=525) and 51.9% females (N=567); race was reported as East Asian (26%, N=286), African (23%, N=247), European (35%, N=378), and American ancestry (16%, N=181).
Statistical Analysis
The frequency of nucleotide variants as single, double and triple heterozygous was examined and compared for statistical significance between the patient and control groups using Chi-Square Test, with significance set at P<0.05.
RESULTS
Increased frequency of double or triple heterozygous variants
Patient age at time of gene sequencing ranged from 6 weeks to 17 years old. Single-gene NSVs and those involving at least two genes occurred in the study cohort and control population. The frequency of any NSVs, regardless of the predicted impact of the variant to the biology of the encoded protein, was similar in the patient and control groups (disease: 32% vs control 31%, P=0.628) (Figure 1). Focusing only on NSVs that fulfilled the significance criteria (i.e., mutation or VUS), the frequency of patients with NSVs in only 1 gene was similar between the two groups (disease: 25% vs control 26%, P=0.518; Table 1). In contrast, NSVs in ≥2 genes were notably higher in the study cohort compared to the control subjects (disease 7% or 51 of 717 vs control 4.7% or 51 of 1092, P=0.028; Table 1 and Supplementary Table 1).
Figure 1.
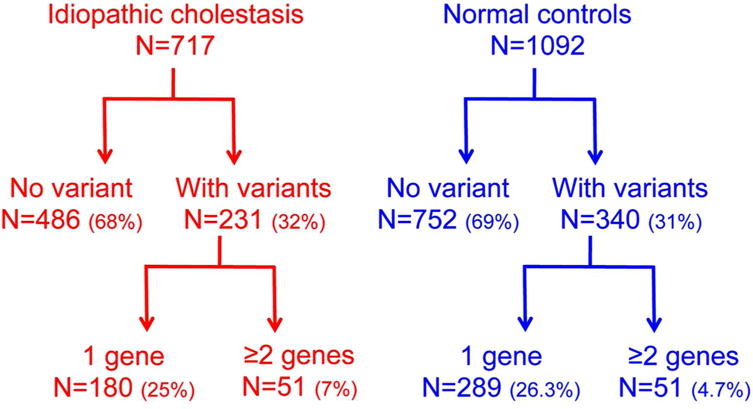
Flow diagram showing the frequency of nucleotide variants in children with idiopathic cholestasis and normal controls.
Table 1.
Frequency of variants in children with idiopathic intrahepatic cholestasis (disease) and unaffected controls
| Disease | Control | P value** | |
|---|---|---|---|
| Any variant* | 32% (230 of 717) | 31% (340 of 1092) | 0.628 |
| Mutations and VUS in 1 gene | 25% (180 of 717) | 26% (289 of 1092) | 0.518 |
| Mutations and VUS in ≥2 genes | 7% (50 of 717) | 4.7% (51 of 1092) | 0.028 |
Any variant = Mutations + variants of unknown significance (VUS)
Based on Chi-Square test
Among the 51 patients with NSVs in ≥2 gene, 6 patients had variants in 3 genes, which was not present in controls (triple heterozygous subjects; disease: 6 of 717 or 0.8% vs control: 0%, P=0.0025). These data pointed to an overall increase in gene sequence variants in children with idiopathic cholestasis, with a specific increase in variants in two or more genes, which raised the possibility that functional heterozygosity may be related to disease phenotypes.
Coexistence of genetic diagnosis and heterozygous variants in additional genes
To examine how the coexistence of variants in ≥2 genes may relate to disease phenotypes, we assigned a specific molecular diagnosis based on the presence of a single-allele mutation in JAG1 or biallelic mutations in ATP8B1, ABCB11, ABCB4, or SERPINA1. We were able to assign a molecular diagnosis in 54 subjects (54 of 717 or 7.5%); as expected this diagnostic criteria was not found in the healthy control population of the 1000_Genome-Project (0 of 1092). Of the subjects with a molecular diagnosis, 11 had variants in one or more additional genes (Figure 2 and Supplementary Table 1). Interestingly, one of the subjects carried a biallelic mutation in both ATP8B1 (c.2546 G>A [R849Q] in both alleles) and ABCB4 (c.1436 C>T [P479L] in both alleles). These data suggest that the presence of a molecular diagnosis does not rule out the coexistence of heterozygosity of other gene(s), or of genetic variants that potentially fulfill criteria for mutation in another gene.
Figure 2. Co-existence of disease and heterozygous gene variants.
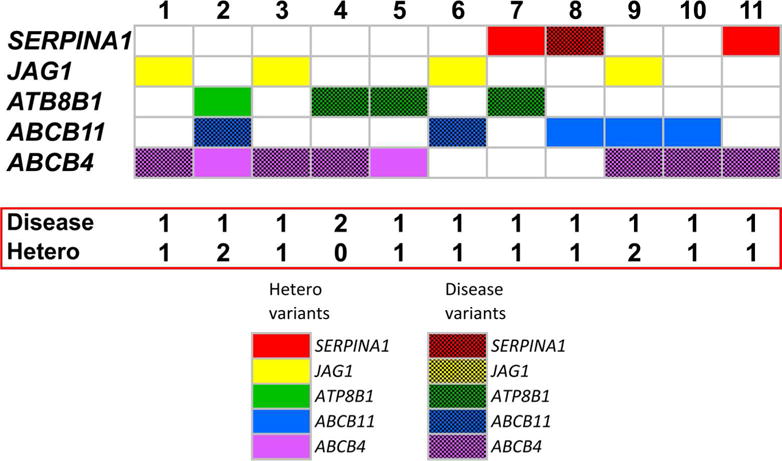
Identification of affected genes (defined as carrying disease-causing variants in two alleles for SERPINA1, ATP8B1, ABCB11 or ABCB11, or single allele for JAG1) that coexist with heterozygous non-synonymous variants in 1–2 other genes in children with idiopathic cholestasis. Seven patients have an affected gene plus a heterozygous allele in one or two other genes. Patient #4 has two affected genes. The solid color-rectangles represent heterozygous variants; check-board rectangles represent a single-allele variant in JAG1 or two-allele disease variants in the other genes. Mutations of patients 1–11 are described in Supplementary Table 1.
Variable phenotypes in double or triple heterozygous variants
To obtain insight into the relationship between double or triple heterozygous variants and disease phenotypes, we reviewed the clinical, laboratory, and histological features in the 10 patients followed in the Pediatric Liver Care Center at Cincinnati Children’s Hospital Medical Center (Figure 3); detailed analysis of other patients with a similar pattern of NSVs was not possible because there was limited information on their clinical features. Most NSVs were identified in the genes ABCB4 (6 of 10 patients) and SERPINA1 (7 of 10 patients). Patient age ranged from 1.5 month to 16 years and history of cholestasis for 1 month to 2 years. The persistent increase of aminotransferases and/or elevated direct bilirubin, GGT or bile acid levels formed the clinical rationale for obtaining a liver biopsy and gene mutation screen (Figure 3 and Supplementary Table 2); there was no evidence of obesity, no history of drug exposure, and negative evaluation for infections (examples: hepatitis A, B, C), metabolic (examples: alpha-1- antitrypsin deficiency, cystic fibrosis), autoimmune (negative auto-antibody panel), vascular, or biliary malformations by ultrasound of the liver and abdomen with Doppler. Liver biopsies, available for review in 8 of the patients, were evaluated for staging of fibrosis, grading of inflammation, and presence of bile duct proliferation and giant cell transformation. The most common findings were stage 2-3 (out of 4) fibrosis (6 of 8 patients) and grade 1 (out of 3) inflammation (7 of 8 patients), with only 3 subjects with bile duct proliferation and 3 with giant cell transformation (Figure 3 and Supplementary Table 2).
Figure 3. Clinical features of patients with disease and/or heterozygous variants in ≥2 genes.
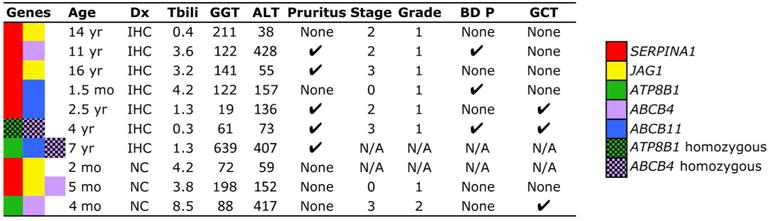
Age, clinical diagnosis, total bilirubin (Tbili, mg/dL), serum glutamyl transpeptidase (GGT, IU/L) and alanine aminotransferase (ALT, IU/L), presence or absence of pruritus, histological stage (1–4) or grade (1–3), presence of bile duct proliferation (BD-P) and giant cell transformation (GCT) in 10 patients with intrahepatic cholestasis. The solid color-rectangles represent heterozygous variants; check-board rectangles represent a single-allele variant in JAG1 or two-allele disease variants in the other genes. IHC = intrahepatic cholestasis, NC = neonatal cholestasis, Dx = diagnosis, M = male, F = female, mo = month, yr = year, NA = not available.
Interestingly, one of the 10 patients carried biallelic mutations in ABCB4 (c.1463C>T [P479L]) and biallelic VUS in ATP8B1 (c.2546G>A [R849Q]) simultaneously (6th patient in Figure 3 and in Supplementary Table 2). This patient, a 4-year-old male with ongoing severe pruritus and cholelithiasis had persistent elevation of serum bile acid (37–97 μmol/L), GGT of 61 IU/L and ALT 73. Despite the mutations in ABCB4, his highest serum GGT was 61 IU/L (more consistent with ATP8B1/FIC1 deficiency). At the time of genetic testing, his liver had stage 3 fibrosis, grade 1 inflammation, and bile duct proliferation (Figure 4), features more typical of ABCB4/MDR3 deficiency.
Figure 4. Histological features of the liver in a child with biallelic mutations in both ATP8B1 and ABCB4.
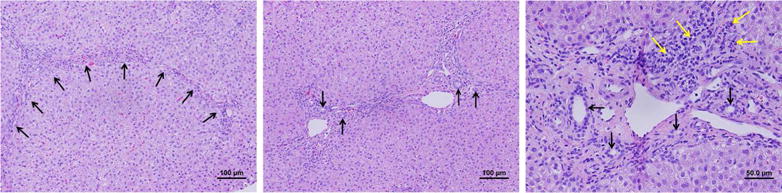
Hematoxylin/eosin staining of a liver biopsy obtained at the time of diagnosis and genetic testing of a child with intrahepatic cholestasis carrying biallelic mutations in ATP8B1 portal-to-portal fibrosis (arrows); the middle panel and right panels show bile duct proliferation (arrows) and mild portal inflammation (yellow arrows).
DISCUSSION
Analyzing the frequency of NSVs in individual genes SERPINA1, JAG1, ATP8B1, ABCB11, or ABCB4, which were sequenced simultaneously as part of diagnostic evaluation in a large cohort of children with idiopathic cholestasis, we found no differences between patients and the healthy subjects sequenced as part of the 1000 Genome Project. However, considering only NSVs that fulfilled the significance criteria for potential clinical relevance, children with idiopathic cholestasis had higher frequency of variants in ≥2 genes than controls. In this group, most variants were double heterozygous, but six subjects were triple heterozygous, a finding not present in the control subjects. Interestingly, analysis of patients whose mutation screen established a genetic diagnosis also found NSVs in one or more additional genes, and one of the patients had biallelic mutation or VUS in both ATP8B1 and ABCB4.
The contribution of genetic heterozygosity to the development of cholestasis was initially reported for ABCB4 in mothers with intrahepatic cholestasis of pregnancy,15 and later in other canalicular transporters.16 Heterozygous ABCB4 mutations have also been associated with fibrosing cholestatic disease in adults with variable biochemical and clinical abnormalities, but shared features of fibrosis.10 In a gene sequencing analysis of 51 children with intrahepatic cholestasis of unknown etiology, we reported a 20% frequency of heterozygous non-synonymous variants in single genes.8 In the current larger cohort of 717 children, the overall frequency of single variants did not differ from controls, pointing to the importance of the inclusion of an adequate size of a control population. Although double heterozygosity is also found in healthy controls, its frequency is much greater in children with intrahepatic cholestasis of unknown etiology. In fact, the 7% frequency is probably an underestimation because the hybridization-based sequencing technology of the JaundiceChip has a lower call rate for heterozygous variants, and may miss heterozygous deletions and insertions.7
A limitation of our study is the lack of detailed clinical history in most of the patients screened for genetic variants; the test requisition form completed by practicing pediatric gastroenterologists stated intrahepatic cholestasis, implying a long duration that led to the request for the gene mutation screen. As such, the available data do not allow for a careful analysis of the potential relationships between genotype and clinical phenotype of all patients, or genotype and histological features in the entire cohort, but our study design enabled the exploration of this relationships in a subgroup of 11 patients followed at our Center, in whom comprehensive information was available for review. The most consistent finding was stage 2–3 fibrosis in 6 of 8 patients, and there was substantial variability in the form of clinical presentation (neonatal cholestasis, persistent elevation of liver enzymes, and idiopathic liver disease in older children), degree of pruritus, and levels of GGT. Notably, one patient had biallelic mutation or VUS in both ATP8B1 and ABCB4. Consistent with the expected lower levels of serum GGT in patients carrying mutations or VUS in ATP8B1, the highest serum GGT was 61 IU/L. Histologically, however, the liver displayed expanded profiles of interlobular bile ducts, a finding most typically associated with mutations of ABCB4.17 The additional finding that patients carrying single-allele mutations in JAG1 or biallelic mutations in the other 4 genes may also carry non-synonymous variants in at least one other gene raises the possibility that these variants may serve as modifiers of clinical phenotype or severity of liver disease. While this possibility is suggested by the low frequency of this finding in the large control cohort, such a role as modifiers of liver disease can only be demonstrated by a detailed analysis of the relationship between nucleotide variants and clinical, laboratory and histological data in a much larger cohort.
In summary, a mutation screen in children with intrahepatic cholestasis of unknown etiology found a high frequency of double and heterozygous variants in SERPINA1, JAG1, ATP8B1, ABCB11, or ABCB4. Although the clinical relevance of these findings is not fully known, they point to a potential influence on the clinical phenotype. Further, the identification of a patient with bi-allelic mutations in two genes simultaneously and the presence of variants of additional gene(s) in patients carrying a single-gene diagnosis suggest that the coexistence of variants may contribute to the variability in disease presentation and/or progression.
Supplementary Material
Acknowledgments
This work was supported by the NIH grant T32 -DK007727 (to M.L.G.), DK093214 (to J.A.B.) and DK078392 (Gene and Protein Expression Core, the Bioinformatics Core, the Integrative Morphology Core, and the Clinical Component of the Digestive Disease Research Center in Cincinnati; to J.A.B.).
List of abbreviations
- NSV
non-synonymous variant
- VUS
variant of unknown significance
- BSEP
bile salt export pump
- MDR
multidrug resistance protein
- PFIC
progressive familial intrahepatic cholestasis
- A1AT
alpha-1-antitrypsin
- MDR
multidrug-resistance protein
- GGT
γ-glutamyl transpeptidase
Footnotes
Competing interests:
Jessica Connor and Kejian Zhang are employees of the Molecular Genetics Laboratory of Cincinnati Children’s Hospital Medical Center.
Author’s contribution:
MLG and RM curated and analyzed the data on gene variants and clinical phenotypes; JC and KZ analyzed the gene sequence database from the Molecular Genetics Laboratory and coexistence of gene variants; PD and RK performed statistical analyses and mined the control population in the 1000_Genome-Project; and KS conducted all murine experiments and data analysis; RS analyzed liver biopsy pathology; KZ, AM and JAB oversaw study design, data analysis, and validation experiments. All authors contributed to the draft of the manuscript.
References
- 1.Alissa FT, Jaffe R, Shneider BL. Update on progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 2008;46:241–52. doi: 10.1097/MPG.0b013e3181596060. [DOI] [PubMed] [Google Scholar]
- 2.Balistreri WF, Bezerra JA. Whatever happened to “neonatal hepatitis”? Clin Liver Dis. 2006;10:27–53. v. doi: 10.1016/j.cld.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Bezerra JA, Balistreri WF. Intrahepatic cholestasis: order out of chaos. [letter; comment] Gastroenterology. 1999;117:1496–8. doi: 10.1016/s0016-5085(99)70302-1. [DOI] [PubMed] [Google Scholar]
- 4.Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. The spectrum of liver diseases related to ABCB4 gene mutations: pathophysiology and clinical aspects. Semin Liver Dis. 2010;30:134–46. doi: 10.1055/s-0030-1253223. [DOI] [PubMed] [Google Scholar]
- 5.European Association for the Study of the L. EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol. 2009;51:237–67. doi: 10.1016/j.jhep.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Lang T, Haberl M, Jung D, et al. Genetic variability, haplotype structures, and ethnic diversity of hepatic transporters MDR3 (ABCB4) and bile salt export pump (ABCB11) Drug metabolism and disposition: the biological fate of chemicals. 2006;34:1582–99. doi: 10.1124/dmd.105.008854. [DOI] [PubMed] [Google Scholar]
- 7.Liu C, Aronow BJ, Jegga AG, et al. Novel resequencing chip customized to diagnose mutations in patients with inherited syndromes of intrahepatic cholestasis. Gastroenterology. 2007;132:119–26. doi: 10.1053/j.gastro.2006.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matte U, Mourya R, Miethke A, et al. Analysis of gene mutations in children with cholestasis of undefined etiology. Journal of pediatric gastroenterology and nutrition. 2010;51:488–93. doi: 10.1097/MPG.0b013e3181dffe8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strautnieks SS, Byrne JA, Pawlikowska L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134:1203–14. doi: 10.1053/j.gastro.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 10.Ziol M, Barbu V, Rosmorduc O, et al. ABCB4 heterozygous gene mutations associated with fibrosing cholestatic liver disease in adults. Gastroenterology. 2008;135:131–41. doi: 10.1053/j.gastro.2008.03.044. [DOI] [PubMed] [Google Scholar]
- 11.Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Human molecular genetics. 2001;10:591–7. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 12.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–4. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 13.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic acids research. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annual review of genomics and human genetics. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- 15.Dixon PH, Weerasekera N, Linton KJ, et al. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Human molecular genetics. 2000;9:1209–17. doi: 10.1093/hmg/9.8.1209. [DOI] [PubMed] [Google Scholar]
- 16.Kia L, Rinella ME. Interpretation and management of hepatic abnormalities in pregnancy. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2013;11:1392–8. doi: 10.1016/j.cgh.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 17.Jacquemin E, De Vree JM, Cresteil D, et al. The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology. 2001;120:1448–58. doi: 10.1053/gast.2001.23984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.