

Abstract
The structure and functions of the M2 protein from Influenza A are sensitive to pH, cholesterol, and the antiinfluenza drug Amantadine. This is a tetrameric membrane protein of 97 amino-acid residues that has multiple functions, among them as a proton-selective channel and facilitator of viral budding, replacing the need for the ESCRT proteins that other viruses utilize. Here, various amino-acid-specific-labeled samples of the full-length protein were prepared and mixed, so that only interresidue 13C-13C cross peaks between two differently labeled proteins representing interhelical interactions are observed. This channel is activated at slightly acidic pH values in the endosome when the His37 residues in the middle of the transmembrane domain take on a +2 or +3 charged state. Changes observed here in interhelical distances in the N-terminus can be accounted for by modest structural changes, and no significant changes in structure were detected in the C-terminal portion of the channel upon activation of the channel. Amantadine, which blocks proton conductance by binding in the aqueous pore near the N-terminus, however, significantly modifies the tetrameric structure on the opposite side of the membrane. The interactions between the juxtamembrane amphipathic helix of one monomer and its neighboring monomer observed in the absence of drug are disrupted in its presence. However, the addition of cholesterol prevents this structural disruption. In fact, strong interactions are observed between cholesterol and residues in the amphipathic helix, accounting for cholesterol binding adjacent to a native palmitoylation site and near to an interhelix crevice that is typical of cholesterol binding sites. The resultant stabilization of the amphipathic helix deep in the bilayer interface facilitates the bilayer curvature that is essential for viral budding.
Introduction
Since the first membrane protein and peptide structures obtained in the presence of lipids (1, 2), the influence of the membrane environment on protein structure has been the subject of considerable interest (3, 4). Well before membrane protein structures were characterized, Anfinsen (5) made it clear that protein structure was the result of the totality of interactions including those within the protein and between the protein and its environment. Structures of M2 protein constructs from Influenza A that have been characterized in detergent crystal lattices, detergent micelles, proteoliposomes composed of liquid crystalline lipid bilayers, and raftlike lipid compositions have extended our understanding of the environmental influences on membrane protein structure (6, 7, 8, 9, 10, 11, 12, 13, 14). Here, we present structural restraints obtained from the full-length M2 (M2FL) proton channel in lipid bilayers describing the structural influence of cholesterol, protons, and drug binding on the helices of the M2 conductance domain (M2CD) using solid-state nuclear magnetic resonance (NMR) spectroscopy.
While details of the M2 structure are still debated, especially those associated with the conductance mechanism (15, 16, 17, 18), the oligomeric structure is well documented as a left-handed tetramer, with some dimer-of-dimer character from four monomers, each with a single transmembrane (TM) helix followed by an amphipathic helix in a juxtamembrane position in the viral interior. The structure of the N- and C-termini of the protein are largely unknown, except for strong evidence for β-sheet content in the first 20 residues (19) and recent evidence that a few residues, and maybe many more in the C-terminus, are disordered (20).
Solid-state NMR (ssNMR) is the primary technique for characterizing membrane protein structure in nativelike bilayer environments at atomic resolution. Recent ssNMR structures include the structure of phospholamban (21) and its phosphorylated pentameric form (22); a mercury transporter, MerF (23); a chemokine receptor, CXCR1 (24); the Mycobacterium cell division protein, CrgA (25); sensory rhodopsin (26); and the trimeric autotransporter, YadA (27). Many more publications provide detailed structural results on other membrane proteins. Such studies are still compromised by the simplified models of the very complex native membrane environment. In important efforts to address this challenge with the M2 protein, Hu et al. (28) and Cady et al. (29) have modeled the viral lipid environment using sphingomyelin, DPPC, DPPE, and cholesterol in a molar ratio of 28:21:21:30 (known as VM) or sphingomyelin, POPC, POPE, and cholesterol in a molar ratio 25.6:25.6:25.6:23 (known as VM+). However, M2CD does not bind amantadine (AMT, an antiinfluenza drug) in either of these environments, suggesting that these environments do not represent nativelike M2 environments (29). From functional assays conducted by Schroeder et al. (30), AMT inhibits proton flux through M2FL in liquid crystalline lipid bilayers containing 30% cholesterol, so it does not appear to be the high concentration of cholesterol that is responsible for the lack of AMT binding in Hong’s studies (30). Recently, it has become clear that M2 prefers an environment on the edge of the raftlike domains where the pyramidal-shaped M2CD conformation induced by its amphipathic helix interacting with the lipid bilayer facilitates viral budding (30, 31, 32). Indeed, a major strength of the solid-state NMR technology for membrane protein structure is the flexibility for preparing samples in a wide variety of pH and ionic strength conditions and lipid compositions.
Potentially facilitating the location of the M2 protein on the periphery of raftlike domains are the relatively short and significantly tilted TM helices that will be better accommodated in the lipid bilayers surrounding the raftlike domains (30). While it has been suggested that M2 binds cholesterol, many of these claims are based on cholesterol-recognition amino acid-consensus sequence (CRAC) motifs occurring in the amphipathic helix of numerous influenza strains (33). However, it is unlikely that these sequences are predictive of cholesterol binding, because these motifs were originally characterized for TM helices. Furthermore, in the influenza A/Udorn 307/72 H3N2 strain (the M2 sequence studied here), the amino-acid sequence does not meet all of the requirements for a CRAC domain. However, the M2 amphipathic helix has a Cys50 residue (mutated in our constructs to Ser) that is natively palmitoylated, a feature that is often associated with cholesterol binding (34).
In this article, we demonstrate the application of mixed/asymmetrically isotopic labeled samples (18, 35), in which two different 13C labeled preparations of M2FL are mixed in a 1:1 molar ratio to selectively observe cross peaks between monomers in this tetrameric structure. For this, 13C-13C dipolar-assisted rotational resonance (DARR) experiments (36) are performed to observe local interactions between helices. Consequently, no such cross peaks will arise from within a monomer, and the cross peaks will only arise when the different labeled monomers are adjacent to each other in the tetrameric structure. The 1:30 molar ratio of protein monomer/lipid represents a 1:120 tetramer/lipid, which is more than adequate to suppress any tetramer-to-tetramer cross peaks, because there is no propensity for the tetramers to form a higher-order oligomeric structure. However, the dilute protein and dilute pairs of labels leads to limited sensitivity for these experiments.
Materials and Methods
Isotopically labeled M2FL protein was expressed with N-terminal histidine-6 tag from the BL21 RP codon plus Escherichia coli. Initial cultures were grown in rich media. Two liters of cells were pelleted and washed with 50 mL of M9 minimal media before growing 1 L of M9 culture supplemented with a 100 mg of an isotopically labeled amino acid (Cambridge Isotope Labs, Tewksbury, MA; Sigma-Aldrich, Miamisburg, OH). Purification was carried out using Empigen-BB solubilized M2FL after cell disruption with a French press. After high-g centrifugation the soluble fraction was loaded onto a nickel affinity column and purified M2FL was eluted from the column at pH 8.0 with 0.5% wt/vol octylglucoside detergent in Tris buffer. This purified protein was then treated with 5 mM dithiothreitol for obtaining the protein in monomeric form. Additional details are provided in the Supporting Material.
A 4:1 molar ratio of DMPC and DMPG lipids was used for reconstitution of the protein in lipid bilayers, and when cholesterol was added a molar ratio of 1:4 (cholesterol/other lipids or ∼20% cholesterol lipid fraction) was used. For reconstitution of M2FL into these lipid environments, the lipid mixture was made 3% (wt/vol) with the octylglucoside followed by the addition of a 1:1 molar ratio of two different amino acid-labeled M2FL preparations to yield a protein/lipid of 1:30. The detergent was removed by dialysis and confirmed by HPLC using evaporative light scattering detection (37, 38). The final buffer was either 20 mM HEPES buffer for pH 7.5 or 20 mM citrate buffer for pH 5.0. For AMT-binding experiments, the drug was added after the removal of detergents to the resulting proteoliposome suspension at a 1:2 molar ratio of channel/drug. Labeled cholesterol was obtained from Sigma-Aldrich (Miamisburg, OH).
Proteoliposomes containing M2 were packed into 3.2 mm-thin walled magic-angle spinning rotors. Spectra were acquired on a Bruker Avance 600.1 MHz (14.1 T) NMR spectrometer (Bruker, Billerica, MA) at 262 K using an NHMFL Low-E triple resonance probe for minimal sample heating and optimized sensitivity (39, 40). The observation of intermonomer structural restraints were obtained with DARR experiments (36) using mixing times of 300 or 500 ms. Different spinning rates (9–12.2 kHz) were used to avoid spinning side-band overlap with the potential cross peaks. All 13C chemical shifts were referenced externally, using 13C′ labeled glycine sample, by setting the carbonyl carbon resonance to 178.4 ppm relative to TMS.
Results and Discussion
The labeling strategy was based on the M2CD structure (PDB: 2L0J (10)) and the amino-acid sequence from the influenza A/Udorn 307/72 H3N2 strain of M2 (Fig. 1 A). Five different amino acid-specific 13C-labeled protein preparations were expressed and purified using uniformly 13C labeled Ala, Ile, Leu, Phe, and Val. The distributions of the labels are shown in Figs. 1 A and in S1. The two-dimensional magic-angle spinning ssNMR spectra are shown for each of these single amino acid-labeled samples illustrating that there is no observable scrambling of the labels. Four different mixed labeled samples were prepared by combining pairs of amino acid-labeled samples in a 50:50 mixture: Ala-Ile, Ile-Leu, Val-Leu, and Phe-Leu. The first three samples were prepared to search for unambiguous interhelical distance restraints between amino acids of different types in the TM helices and the last sample was prepared for a similar search between the amphipathic helices and the adjacent monomer.
Figure 1.

(A) The amino-acid sequence of M2 protein highlighting the 13C amino-acid labels of the various samples used here. Note that only one amino-acid type is labeled in each preparation. The N-terminus (residues 1–22) is extramembranous and has significant β-sheet structure (19). The TM helix is residues 23–46. The juxtamembrane amphipathic helix is residues 47–62 and the C-terminus, for which there is little information about the structure, is residues 63–97. (B) The PDB: 2L0J structure of the M2CD (residues 22–62) in the lipid environment in which it was characterized (10). One of four monomers is colored yellow. For the nonprotein atoms, the carbons are green and oxygens are red.
In considering the cross-peak possibilities, we and others have found that the TM domain resonances are stronger than resonances from the terminal regions of the protein. Consequently, in general it can be anticipated that our observed cross peaks would be dominated by magnetization transfer within the conductance domain. In addition, the only ambiguities for Ala29 and Ala30 residues in the TM helix are two Ala residues in the C-terminus where there is a single Ile residue (Ile94). This Ile residue is directly involved in the virus’s mechanism to evade autophagy by binding the essential autophagy protein, LC3, to the F91xxI94 sequence—an interaction recognized as monomer-monomer protein interaction (41). Consequently, it is very unlikely that this site would be in close proximity to an adjacent monomer for an Ala-Ile interaction, and therefore any observed interactions between Ala and Ile residues are likely to be associated with packing of the TM helices. The Ile-Leu residues in the N-terminus are far apart in the monomer, and therefore they are likely to be far apart in the tetramer, where there is suspected β-structure (19). There is a pair of these residues in the C-terminus (Ile94; Leu96), but again it is likely that this sequence acts as a monomer to bind LC3 and therefore it is unlikely to form an Ile-Leu cross peak in the 13C correlation spectra. The Val-Leu pair presents a possible interaction in the N-terminus if intermonomer β-strand hydrogen bonding occurs, but even here the distance between Cα sites would be ≫10 Å for a parallel sheet structure. In the C-terminus, the Val residue (Val92) is also in the LC3 interacting region and unlikely to have intermonomer interactions with Leu96 as described above. The fourth sample was Phe-Leu labeled, and the only possible intermonomer interactions between these residues are those involving the Phe residues of the amphipathic helix, because the only terminal region Phe residue is Phe91 in the LC3 binding site. Below we will only discuss the ambiguities that might arise within the conductance domain (residues 23–62) of the full-length protein.
Proton binding
In studies of the M2FL protein in lipid bilayer preparations the structural influences of proton, AMT, and cholesterol binding are assessed through the use 13C-13C distance restraints between monomers. At high pH (≥7.5) the four histidines (His37) responsible for proton transport are in their neutral state, and at low pH (5.0) the proton channel is activated by having the histidine tetrad charged with a mix of +2 and +3 states (42, 43, 44). The structural influence of pH activation has been assessed by observing each of these mixed labeled samples at pH 7.5 and 5.0 (Figs. 2 and S2) with a spectral slice through a well-resolved resonance of one of the two amino acid-labeled monomers. Based on the structure of the M2CD in lipid bilayers (PDB: 2L0J) at pH 7.5, the minimal Cα-Cα distance between Val and Leu residues is the Val28 and Leu26 distance of 8.1 Å (Fig. 3 A; Table 1). In the DARR spectrum at a 500 ms mixing time, a cross peak is not observed (Fig. 2 A, pH 7.5), while at pH 5.0 this cross peak is observed. For Ala Cα and Ile Cγ1, the minimal distance in PDB: 2L0J is 3.6 Å between residues Ala30 and Ile35, respectively. This cross peak is observed at pH 7.5, but not at pH 5.0 (Fig. 2 B). Based on the PDB: 2L0J structure, two Ile and Leu Cα interactions could contribute to observed cross peaks: an 8.1 Å distance between residues Ile32 and Leu26, and a 7.7 Å distance between residues Ile33 and Leu38. An Ile-Leu cross peak is observed from both pH 7.5 and 5.0 samples.
Figure 2.

Structural influences of pH on M2FL. Spectral slices obtained at 262 K from 13C-13C DARR spectra of 13C amino acid-labeled mixed M2 samples at pH 7.5 and 5.0. (A) Spectral slices through Val-Cα frequency (66.8 ppm) using a mixing time of 500 ms and a 10.8 kHz spinning rate. (Dashed line) Leu-Cα frequency (∼58 ppm). (B) Spectral slices through Ala-Cα frequency (55.8 ppm) using a mixing time of 300 ms and a 12.2 kHz spinning rate. (Dashed line) Ile-Cγ1 frequency (∼27 ppm). (C) Spectral slices through Leu-Cα frequency (58.0 ppm) using a mixing time of 500 ms and a 12.2 kHz spinning rate. (Dashed line) Ile Cα frequency (65 ppm). (D and E) Spectral slices through Leu-Cγ frequency (26.6 ppm) using a mixing time of 500 ms and a 9.0 kHz spinning rate. (Dashed lines) Phe-Cγ (∼138 ppm) and Phe-Cδε (∼130 ppm) frequencies.
Figure 3.

Interhelical distances measured in PDB: 2L0J of the M2 conductance domain structure characterized by solid-state NMR in a lipid bilayer environment. (A) Distances between the TM helices. (B) Distances between the juxtamembrane helices or between the juxtamembrane helix and the TM helix. Note in the experimental samples that the labeled Phe and Leu are not on the same protein monomer.
Table 1.
pH Dependence of Cross Peaks and Correlation with Conductance Domain Structure PDB: 2L0J
| Residues Involved | Distance Range in PDB: 2L0J (Å) | pH 7.5 Cross Peaks Observed | pH 7.5 Agreement with PDB: 2L0J Distances | pH 7.5 Cross Peaks Observed |
|---|---|---|---|---|
| Leu26Cα-Val28Cα | 8.1–8.2 | — | + | + |
| Ala30Cα-Ile35Cγ2 | 3.6–3.9 | + | + | — |
| Leu26Cα-Ile32Cα | 7.5–8.1 | +b | +b | +b |
| Leu38Cα-Ile33Cα | 7.7–8.2 | +b | +b | +b |
| Leu46Cγ-Phe54Cδε | 4.6–7.0a | +c | +c | +c |
| Leu59Cγ-Phe48Cδε | 5.2–6.7a | +c | +c | +c |
| Leu46Cγ-Phe54Cγ | 6.4–7.4a | +d | +c | +c |
| Leu59Cγ-Phe48Cγ | 5.0–5.7a | +c | +c | +c |
| L59V Leu46Cγ-Phe54Cδε | 4.6–7.0a | + | + | — |
| L59V Leu46Cγ-Phe54Cγ | 6.4–7.4a | — | — | — |
Measured range for three out of four residue pairs in the PDB: 2L0J WT protein.
The observed interaction could be the result of a short distance for either of these two Leu/Ile pairs.
The observed interaction could be the result of a short distance for either of these two Leu/PheCde pairs.
The observed interaction could be the result of a short distance for either of these two Leu/PheCg pairs.
The specific labels and relatively long mixing times permit spin diffusion over relatively long distances (45) and therefore, with the possible exception of the ambiguous Ile-Leu restraints (Fig. 2 C), the pH 7.5 data is consistent with the PDB: 2L0J structure of M2CD obtained at the same pH. Despite the long distances in the PDB: 2L0J structure for the Ile32-Leu26 and Ile33-Leu38 Cα-Cα pairs (Fig. 3 A), the combination of a modest change in the full-length protein structure at the helix-helix interface and/or a slightly different tilt or rotation angle for the helix at one or both of these sites could better account for the observed cross peaks. Large aliphatic residues (L26, I33, I35, and L38) are in the helical interface, and consequently, a change in side-chain rotameric states for any of these residues could significantly change the helix packing and alter the distances by a couple of Ångstroms, accounting for the pH 7.5 results in Fig. 2 tabulated in Table 1. Recently, conformational heterogeneity in the neutral His37 tetrad has been observed, and it was suggested that it could have been induced by heterogeneity in helix packing (44). In other words, it is not expected that a single set of rotameric states represents the native structure at even the low temperatures used here.
Although no significant pH dependence in the N-terminal helical structure has been observed previously from truncated constructs of the M2 protein (46), here it is clear that a structural change does occur in the full-length protein based on specific interhelical distance differences upon channel activation. At pH 5.0, Val28-Leu26 are closer together, while Ala30-Ile35 are further apart and the ambiguous Leu-Ile results remain constant; however, compensating differences between these ambiguous pairs could occur. This structural perturbation could be the result of a small decrease in the helical tilt or small change in the rotational orientation of the helices.
For the amphipathic helix, cross peaks are observed at pH 7.5 between the Phe ring carbons and Leu Cγ sites (Fig. 2 D). The M2CD PDB: 2L0J structure is considered an excellent model for this portion of the protein, because the amphipathic helix has the tilt and rotation angle as previously characterized for the full-length protein at pH 8.0 (47). From the PDB: 2L0J structure, two possible interactions (Phe48-Leu59 and Leu46-Phe54) could contribute to these cross peaks (Fig. 3 B). Both of these are potential sources for the observed cross-peak intensity that represents interactions between the C-terminus of the amphipathic helix (Leu59 and Phe54) and the start of the amphipathic helix (Phe48) or the C-terminus of the TM helix (Leu46). In the PDB: 2L0J structure, Leu46 Cγ-Phe54 Cδε distances range from 4.6 to 7.0 Å for three of the four residue pairs in this tetrameric structure; and for the Leu46 Cγ-Phe54, Cγ distances range from 6.4 to 7.4 Å (Table 1). In part, these increased distances account for the weaker Phe Cγ cross-peak intensity in Fig. 2, D and E. For three of the Phe48 Cδε-Leu59 Cγ pairs, distances range from 5.2 to 6.7 Å and for the Phe48 Cγ-Leu59 Cγ distances range from 5.0 to 5.7 Å. In Fig. 2 D, cross peaks from Leu Cγ to Phe Cδε and Phe Cγ are observed at both pH 7.5 and 5.0. When the Leu-labeled L59V M2 mutant was mixed with Phe-labeled M2WT, a cross peak was still observed to Phe Cδε (Phe54) from Leu46 Cγ (Fig. 2 E), but at substantially reduced intensity, suggesting that both pairs (Phe48-Leu59 and Leu46-Phe54) contribute to the strong Phe-Leu wild-type (WT) cross peaks at pH 7.5 in Fig. 2 D. However, the Leu46 Cγ-Phe54 Cγ cross peak is not observed, in part due to the weaker transfer to the nonprotonated Phe Cγ and the longer Leu46 Cγ-Phe54 Cγ distance (Table 1). Hence, even this distance subtlety seems to be consistent with the PDB: 2L0J structure. Interestingly, at pH 5.0, intensity values are observed for the WT sequences that are similar to those seen at pH 7.5. This appears to be at odds with electron spin resonance (ESR) results that noted different water accessibility at low pH for spin labels on the amphipathic helix and hence a different structural arrangement at low pH (48).
AMT binding
The binding of AMT results in no significant differences in the cross-peak intensities compared to protein samples at the same pH without AMT in the N-terminus of the TM helix (Figs. 4, A–C, and S3). This is consistent with the solid-state NMR studies of the truncated samples of the M2 protein (7, 11). The ESR studies using spin labels tethered to the N-terminus of the TM domain showed changes upon AMT binding that were interpreted as pore diameter restrictions, but a change in structure of the N-terminal residue could account for the spectral changes (49). The C-terminus of the TM helix was not explored directly by our 13C-13C strategy, because the helices are more splayed apart in this portion of the structure, resulting in longer distances between the TM backbones. However, changes in the structure induced by AMT binding in this region can be deduced from observations of cross peaks between the TM helix and the amphipathic helix using mixed 13C Phe and 13C Leu labeling (Fig. 4, D and E). Although at high pH, strong Phe-Leu cross peaks were observed in the absence of AMT, these cross peaks were not observed in the presence of the drug (Fig. 4 D). At low pH, strong cross peaks are observed with these labels both with and without AMT (Fig. 4 E; Table S1). Because this distance tethers the end of the amphipathic helix to the structural kink between the TM and amphipathic helix, it means that there is no change in the tilt of the C-terminal portion of the TM helix at low pH in the presence of AMT; however, at high pH, a significant change in tilt is possible upon AMT binding, as has been suggested by both solid-state NMR and ESR of the M2 TMD and M2CD, respectively (7, 49, 50).
Figure 4.

Structural influences on M2FL upon AMT binding. Spectral slices obtained at 262 K from 13C-13C DARR spectra of 13C amino acid-labeled, mixed M2 samples with and without AMT at two pH values. (A and B) Spectral slices through Val-Cα frequency (66.8 ppm) using a mixing time of 500 ms and a 10.8 kHz spinning rate. (Dashed line) Leu-Cα frequency, ∼58 ppm. (C) Spectral slices through Ile-Cα frequency (65.7 ppm) using a mixing time of 500 ms and a 12.2 kHz spinning rate. (Dashed line) Leu-Cα frequency ∼58 ppm. (D and E) Spectral slices through Leu-Cγ frequency (26.6 ppm) using a mixing time of 500 ms and a 9.0 kHz spinning rate. (Dashed lines) Phe-Cγ frequency ∼138 ppm and Phe-Cδε frequency ∼130 ppm.
Cholesterol binding
When cholesterol is added to the lipid composition of the membrane at pH 7.5, the binding of AMT has no influence on the cross peaks between Leu and Phe (Figs. 5 and S4). This result suggests that cholesterol is stabilizing the structure of the amphipathic helix in the lipid interface, as it is represented in the PDB structure, 2L0J. When cholesterol is labeled with 13C2,3,4 and mixed with 13C Phe-labeled M2FL, strong cross peaks are observed in slices through the cholesterol C2 resonance frequency to the ring carbons of Phe, as shown in Fig. 6, A and B. No cross peaks were observed in spectral slices observed through the C25 resonance of cholesterol labeled with 13C25,26,27 mixed with 13C Phe-labeled M2FL. These results suggest a standard orientation for cholesterol in the lipid bilayer, including for DMPC lipids used in these experiments (51), with the hydroxyl in the bilayer interface and the hydrocarbon tail buried in the interstices of the lipid bilayer.
Figure 5.
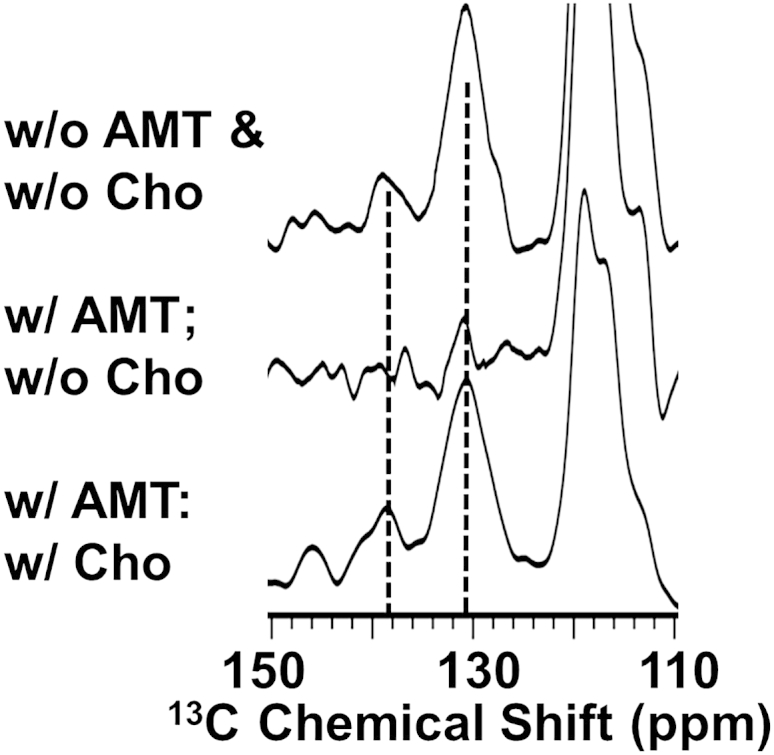
Influence of cholesterol on AMT-bound M2FL at pH 7.5. Slices from two-dimensional DARR spectra of 13C Phe:13C Leu mixed sample of M2FL in lipid bilayers at 262 K both with and without cholesterol and AMT bound. Slices through Leu-Cγ at 26.6 ppm. (Dashed lines) Phe Cγ (∼138 ppm) and Phe Cδε (130 ppm). Spectra obtained with 500 ms mixing time and a 9.0 kHz spinning rate.
Figure 6.

Cholesterol binding to the amphipathic helix of M2FL. (A) Structure of cholesterol -2, 3, 4- 13C- and -25, 26, 27- 13C-labeled cholesterol that was added to 13C-Phe-labeled M2 samples. (B) DARR spectral slices through C3 and C25 of cholesterol, with 500 ms mixing and a 9 kHz spinning rate. (C) Likely cholesterol binding site modeled on the M2 CD structure (PDB: 2L0J).
A consensus recognition sequence for cholesterol binding has long been recognized (52), originally based on studies of a TM helix in peripheral-type benzodiazepine receptors. This CRAC sequence (–L/V-(X)1-5-Y-(X)1-5-R/K-) was observed as a surface from TM helix 4 of these receptors that interacted with cholesterol, such that the Leu/Val residue was in the hydrophobic region of the bilayer and the Arg/Lys was in the bilayer interface with a Tyr residue midway between these sites. Subsequently, similar sequences have been recognized in G-protein-coupled receptors (GPCRs) (53, 54) and generalized to include residues lining a cleft between a pair of TM helices that form a pocket for binding cholesterol. These sequences are referred to as “cholesterol consensus motifs”. In the β-adrenergic receptor, the interfacial positively charged residue was shown to be in an amphipathic helix (53), but the other residues of the CRAC or cholesterol consensus motifs are in TM helices and not in the amphipathic helices, as has been suggested for M2 (30, 55, 56, 57).
Stewart et al. (55) mutated a couple of the consensus residues of M2, without significantly influencing viral replication in vitro, suggesting that cholesterol binding may not be important for viral budding although it had some effect on morbidity and mortality in a mouse model. However, M2 has affinity for the periphery of the raftlike domains where there is a considerable cholesterol lipid fraction. Indeed, M2 is recognized as facilitating viral budding from raftlike domains by inducing membrane curvature (58) in the absence of ESCRT proteins that are not expressed by influenza viruses (32, 56). Specifically, the critical role of amphipathic helix by inducing membrane curvature during viral budding is known (59). Furthermore, it is known that the native Cys50 residue (here C50S) in the middle of the amphipathic helix is palmitoylated, which is often affiliated with cholesterol binding (34). Schroeder et al. (30) showed that the M2FL is copurified with cholesterol. Consequently, there is reason to anticipate that M2 would bind cholesterol as a way to attract the protein to the viral budozone, although it is also attracted there by its affinity for the water-soluble M1 protein (31, 60). Thaa et al. (57) showed that raft-targeting of M2 was not dependent on the CRAC motif, but was dependent on palmitoylation. More recently, Thaa et al. (57) have also shown via FRET studies that neither palmitoylation nor the key cholesterol binding consensus residues are necessary to target M2 to the viral budozone. Consequently, it is not clear that any of the CRAC motif residues are involved in cholesterol binding to M2.
Four Phe residues (47, 48, 54, and 55) in the amphipathic sequence are distributed on either side of the palmitoylation site. Only two of these four Phe residues are oriented toward the lipid environment, residues Phe47 and Phe54, which would be in an appropriate position to interact with cholesterol and form cross peaks with the C2 carbon of cholesterol. Note that these residues are in an I-to-I+7 sequential arrangement so that they are on the same surface of the helix. Moreover, the PDB: 2L0J structure suggests that Phe54 may be in a more appropriate position for this interaction than Phe47, for several reasons. First, the carbonyl group of Ser50 is the most exposed carbonyl on this surface of the amphipathic helix as a result, in part, of the small Ser (Cys) sidechain (Fig. 6 C). This would be an ideal location for hydrogen bonding of the cholesterol hydroxyl to the protein. Second, this would position cholesterol in a cleft between the C-terminal TM helices and be flanked in the native protein by the palmitoyl group linked to the Cys50 residue. In this position, as shown in the structural model in Fig. 6 C, the Phe54 aromatic ring would be in van der Waals contact with cholesterol. Indeed, the orientation of the cholesterol and palmitoyl group to the amphipathic helix in the β2-adrenergic GPCR (61) appears to be similar to what is modeled here and for which we have observed very strong 13C Phe-13C3 cholesterol cross peaks consistent with close contact in the structural model with Phe54.
Above, it was noted that at pH 5.0 the binding of AMT did not significantly influence the protein structure as assessed by the Phe/Leu cross peak (Fig. 4 E), but at pH 7.5 the binding of AMT resulted in the loss of the Phe/Leu cross peak. The addition of cholesterol to the pH 7.5 preparation with AMT prevents the loss of the Phe/Leu cross peaks as shown in Fig. 5. Consequently, cholesterol appears to stabilize the amphipathic helix structure in the membrane interface. While this stabilizing effect of cholesterol was not required in the absence of drug, its presence can be assumed from the multiple potential interactions between cholesterol and the amphipathic helix (hydrogen bonding directly or indirectly with Ser50 (Cys50) and van der Waals interactions with Phe54) as well as interactions with the C-terminal region of the TM helices and with the palmitoyl group of Cys50 (van der Waals interactions). These interactions could result in considerable stabilization of the amphipathic helix position in the lipid interface, and potentially this is important when the protein is inducing substantial curvature strain on the membrane in the budozone upon membrane fusion in the endosome.
Conclusions
Numerous specific structural restraints have been observed here using mixed 13C-labeled M2FL monomers in the tetrameric channel. These restraints have characterized pH-dependent structural perturbations in the N-terminal region of the TM helices that had not been observed in prior studies of truncated protein constructs in various detergent-based or lipid-based environments. Other structural perturbations that had been observed in the C-terminal region of the TM domain were not confirmed in the spectra of M2FL, suggesting increased stability as a result of the presence of the C-terminal domain. NMR interhelical distance restraints between the amphipathic and TM helices had not been observed previously; here the restraints provided by both Leu59-Phe48 and Leu46-Phe54 are very significant for characterizing the M2 structure at the inner membrane interface of the viral capsid. These restraints document the proximity of the C-terminal end of the amphipathic helix with the C-terminal end of the TM helix in the adjacent monomer. When AMT was bound at pH 7.5, these Leu-Phe interactions were lost, suggesting a significant structural change.
Cholesterol binding to M2 has been demonstrated, and that it binds in a typical cholesterol binding site and orientation, one that utilizes the amphipathic helix where it interacts with one of the Phe side chains, mostly likely Phe54, and it may hydrogen-bond with the Ser(Cys)50 carbonyl oxygen. In addition, there is an interhelical crevice in the PDB: 2L0J structure into which cholesterol can optimally form van der Waals interactions. This crevice represents a weak point in the M2 structure with a relatively large separation of the helices. It is important for proteins to avoid fenestrations into the fatty acyl environment that could lead to fatty acyl penetration into the pore, thereby disrupting the functional mechanism. In the native protein, interactions with the Cys50 palmitoyl group could further stabilize cholesterol binding. The binding of cholesterol to the amphipathic helix and stabilization of this conformation has important consequences for the functionality of the protein. Influenza A does not have the ESCRT proteins that facilitate viral budding for many viruses (60). Instead, M2 facilitates membrane curvature through the position of its amphipathic helix, generating a dramatic pyramidal shape that induces considerable membrane curvature. If the amphipathic helix changes position, such as in the formation of a water-soluble, four-helix bundle (e.g., the solution NMR M2 structure, PDB: 2RLF (8)), it eliminates M2’s ability to induce curvature. The virus buds out from a raftlike domain and the relatively short hydrophobic dimension of the M2 TM helices largely restricts M2 to the thinner surrounding membrane domain rather than the raftlike domain. Consequently, M2 clusters at the periphery of the raftlike domain where cholesterol is present in high concentration, thereby facilitating the binding of cholesterol to M2 and thus stabilizing the amphipathic helix in the lipid interface-inducing curvature of the host membrane needed for viral budding from the host cell. A similar process may occur when the viral interior becomes acidified in the endosome before the fusion of the viral membranes with the endosomal membrane.
Author Contributions
E.V.E. and T.A.C. designed the research; E.V.E. expressed the protein and prepared the samples; and E.V.E. and R.F. conducted the NMR experiments and analyzed the data. All authors contributed to the writing of the article.
Acknowledgments
The authors thank W.W. Brey, P. Gor’kov, J. Kitchen, and S. Rainer at the National High Magnetic Field Laboratory for valuable assistance with probe technology; Z. Gan and I. Hung for spectrometer help; J. Dai for computational modeling; and H. Qin and C. Escobar for help cloning and optimizing protein expression and purification.
The work was made possible by a grant from the National Institutes of Health (No. AI R01-023007), and the NMR experiments were performed at the National High Magnetic Field Laboratory supported by National Science Foundation Cooperative Agreement No. DMR-1157490 and the State of Florida.
Editor: Francesca Marassi.
Footnotes
Supporting Materials and Methods, four figures, and one table are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(16)00152-1.
Supporting Material
References
- 1.Henderson R., Baldwin J.M., Downing K.H. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J. Mol. Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 2.Ketchem R.R., Hu W., Cross T.A. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science. 1993;261:1457–1460. doi: 10.1126/science.7690158. [DOI] [PubMed] [Google Scholar]
- 3.Zhou H.X., Cross T.A. Influences of membrane mimetic environments on membrane protein structures. In: Dill K.A., editor. Vol 42. Annual Reviews; Palo Alto, CA: 2013. pp. 361–392. (Annual Review of Biophysics). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown M.F. Modulation of rhodopsin function by properties of the membrane bilayer. Chem. Phys. Lipids. 1994;73:159–180. doi: 10.1016/0009-3084(94)90180-5. [DOI] [PubMed] [Google Scholar]
- 5.Anfinsen C.B. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 6.Nishimura K., Kim S., Cross T.A. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR. Biochemistry. 2002;41:13170–13177. doi: 10.1021/bi0262799. [DOI] [PubMed] [Google Scholar]
- 7.Hu J., Asbury T., Cross T.A. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from Influenza A virus. Biophys. J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schnell J.R., Chou J.J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stouffer A.L., Acharya R., DeGrado W.F. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma M., Yi M., Cross T.A. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cady S.D., Schmidt-Rohr K., Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Acharya R., Carnevale V., Klein M.L. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc. Natl. Acad. Sci. USA. 2010;107:15075–15080. doi: 10.1073/pnas.1007071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pielak R.M., Chou J.J. Solution NMR structure of the V27A drug resistant mutant of influenza A M2 channel. Biochem. Biophys. Res. Commun. 2010;401:58–63. doi: 10.1016/j.bbrc.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cross T.A., Sharma M., Zhou H.X. Influence of solubilizing environments on membrane protein structures. Trends Biochem. Sci. 2011;36:117–125. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao S.Y., Yang Y., Hong M. The influenza M2 cytoplasmic tail changes the proton-exchange equilibria and the backbone conformation of the transmembrane histidine residue to facilitate proton conduction. J. Am. Chem. Soc. 2015;137:6067–6077. doi: 10.1021/jacs.5b02510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong M., Fritzsching K.J., Williams J.K. Hydrogen-bonding partner of the proton-conducting histidine in the influenza M2 proton channel revealed from 1H chemical shifts. J. Am. Chem. Soc. 2012;134:14753–14755. doi: 10.1021/ja307453v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Witter R., Nozirov F., Fu R. Solid-state 19F NMR spectroscopy reveals that Trp41 participates in the gating mechanism of the M2 proton channel of influenza A virus. J. Am. Chem. Soc. 2008;130:918–924. doi: 10.1021/ja0754305. [DOI] [PubMed] [Google Scholar]
- 18.Andreas L.B., Reese M., Griffin R.G. Structure and mechanism of the influenza-A M218–60 dimer of dimers. J. Am. Chem. Soc. 2015;137:14877–14886. doi: 10.1021/jacs.5b04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao S.Y., Fritzsching K.J., Hong M. Conformational analysis of the full-length M2 protein of the influenza A virus using solid-state NMR. Protein Sci. 2013;22:1623–1638. doi: 10.1002/pro.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwon B., Tietze D., Hong M. Chemical ligation of the influenza M2 protein for solid-state NMR characterization of the cytoplasmic domain. Protein Sci. 2015;24:1087–1099. doi: 10.1002/pro.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verardi R., Shi L., Veglia G. Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc. Natl. Acad. Sci. USA. 2011;108:9101–9106. doi: 10.1073/pnas.1016535108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vostrikov V.V., Mote K.R., Veglia G. Structural dynamics and topology of phosphorylated phospholamban homopentamer reveal its role in the regulation of calcium transport. Structure. 2013;21:2119–2130. doi: 10.1016/j.str.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tian Y., Lu G.J., Opella S.J. Structure of the membrane protein MerF, a bacterial mercury transporter, improved by the inclusion of chemical shift anisotropy constraints. J. Biomol. NMR. 2014;60:67–71. doi: 10.1007/s10858-014-9852-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park S.H., Das B.B., Opella S.J. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;491:779–783. doi: 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das N., Dai J., Cross T.A. Structure of CrgA, a cell division structural and regulatory protein from Mycobacterium tuberculosis, in lipid bilayers. Proc. Natl. Acad. Sci. USA. 2015;112:E119–E126. doi: 10.1073/pnas.1415908112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang S., Munro R.A., Ladizhansky V. Solid-state NMR spectroscopy structure determination of a lipid-embedded heptahelical membrane protein. Nat. Methods. 2013;10:1007–1012. doi: 10.1038/nmeth.2635. [DOI] [PubMed] [Google Scholar]
- 27.Shahid S.A., Bardiaux B., Linke D. Membrane-protein structure determination by solid-state NMR spectroscopy of microcrystals. Nat. Methods. 2012;9:1212–1217. doi: 10.1038/nmeth.2248. [DOI] [PubMed] [Google Scholar]
- 28.Hu F., Luo W., Hong M. Mechanisms of proton conduction and gating in influenza M2 proton channels from solid-state NMR. Science. 2010;330:505–508. doi: 10.1126/science.1191714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cady S., Wang T., Hong M. Membrane-dependent effects of a cytoplasmic helix on the structure and drug binding of the influenza virus M2 protein. J. Am. Chem. Soc. 2011;133:11572–11579. doi: 10.1021/ja202051n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schroeder C., Heider H., Lin T.I. The influenza virus ion channel and maturation cofactor M2 is a cholesterol-binding protein. Eur. Biophys. J. 2005;34:52–66. doi: 10.1007/s00249-004-0424-1. [DOI] [PubMed] [Google Scholar]
- 31.Rossman J.S., Jing X., Lamb R.A. Influenza virus M2 ion channel protein is necessary for filamentous virion formation. J. Virol. 2010;84:5078–5088. doi: 10.1128/JVI.00119-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rossman J.S., Lamb R.A. Influenza virus assembly and budding. Virology. 2011;411:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fantini J., Barrantes F.J. How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013;4:31. doi: 10.3389/fphys.2013.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oates J., Watts A. Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr. Opin. Struct. Biol. 2011;21:802–807. doi: 10.1016/j.sbi.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 35.Traaseth N.J., Verardi R., Veglia G. Asymmetric methyl group labeling as a probe of membrane protein homo-oligomers by NMR spectroscopy. J. Am. Chem. Soc. 2008;130:2400–2401. doi: 10.1021/ja711499r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takegoshi K., Nakamura S., Terao T. 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 2001;344:631–637. [Google Scholar]
- 37.Mourey T.H., Oppenheimer L.E. Principles of operation of an evaporative light-scattering detector for liquid-chromatography. Anal. Chem. 1984;56:2427–2434. [Google Scholar]
- 38.Murray D.T., Griffin J., Cross T.A. Detergent optimized membrane protein reconstitution in liposomes for solid state NMR. Biochemistry. 2014;53:2454–2463. doi: 10.1021/bi500144h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gor’kov P.L., Chekmenev E.Y., Brey W.W. Using low-E resonators to reduce RF heating in biological samples for static solid-state NMR up to 900 MHz. J. Magn. Reson. 2007;185:77–93. doi: 10.1016/j.jmr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 40.McNeill S.A., Gor’kov P.L., Long J.R. A low-E magic angle spinning probe for biological solid state NMR at 750 MHz. J. Magn. Reson. 2009;197:135–144. doi: 10.1016/j.jmr.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beale R., Wise H., Randow F. A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe. 2014;15:239–247. doi: 10.1016/j.chom.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu J., Fu R., Cross T.A. Histidines, heart of the hydrogen ion channel from influenza A virus: toward an understanding of conductance and proton selectivity. Proc. Natl. Acad. Sci. USA. 2006;103:6865–6870. doi: 10.1073/pnas.0601944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andreas L.B., Barnes A.B., Griffin R.G. Dynamic nuclear polarization study of inhibitor binding to the M2(18–60) proton transporter from influenza A. Biochemistry. 2013;52:2774–2782. doi: 10.1021/bi400150x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miao Y.M., Fu R., Cross T.A. Dynamic, short hydrogen bonds in histidine tetrad of full-length M2 H+ channel reveals tetrameric structural heterogeneity and functional mechanism structure. Structure. 2015;23:2300–2308. doi: 10.1016/j.str.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Can T.V., Sharma M., Cross T.A. Magic angle spinning and oriented sample solid-state NMR structural restraints combine for influenza A M2 protein functional insights. J. Am. Chem. Soc. 2012;134:9022–9025. doi: 10.1021/ja3004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu F., Luo W., Hong M. Conformational plasticity of the influenza A M2 transmembrane helix in lipid bilayers under varying pH, drug binding, and membrane thickness. Biochim. Biophys. Acta. 2011;1808:415–423. doi: 10.1016/j.bbamem.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian C., Gao P.F., Cross T.A. Initial structural and dynamic characterization of the M2 protein transmembrane and amphipathic helices in lipid bilayers. Protein Sci. 2003;12:2597–2605. doi: 10.1110/ps.03168503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nguyen P.A., Soto C.S., Howard K.P. pH-induced conformational change of the influenza M2 protein C-terminal domain. Biochemistry. 2008;47:9934–9936. doi: 10.1021/bi801315m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomaston J.L., Nguyen P.A., Howard K.P. Detection of drug-induced conformational change of a transmembrane protein in lipid bilayers using site-directed spin labeling. Protein Sci. 2013;22:65–73. doi: 10.1002/pro.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li C., Qin H., Cross T.A. Solid-state NMR characterization of conformational plasticity within the transmembrane domain of the influenza A M2 proton channel. Biochim. Biophys. Acta. 2007;1768:3162–3170. doi: 10.1016/j.bbamem.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marsan M.P., Muller I., Milon A. Cholesterol orientation and dynamics in dimyristoylphosphatidylcholine bilayers: a solid state deuterium NMR analysis. Biophys. J. 1999;76:351–359. doi: 10.1016/S0006-3495(99)77202-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H., Papadopoulos V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology. 1998;139:4991–4997. doi: 10.1210/endo.139.12.6390. [DOI] [PubMed] [Google Scholar]
- 53.Hanson M.A., Cherezov V., Stevens R.C. A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jafurulla M., Tiwari S., Chattopadhyay A. Identification of cholesterol recognition amino acid consensus (CRAC) motif in G-protein coupled receptors. Biochem. Biophys. Res. Commun. 2011;404:569–573. doi: 10.1016/j.bbrc.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 55.Stewart S.M., Wu W.H., Pekosz A. The cholesterol recognition/interaction amino acid consensus motif of the influenza A virus M2 protein is not required for virus replication but contributes to virulence. Virology. 2010;405:530–538. doi: 10.1016/j.virol.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rossman J.S., Jing X., Lamb R.A. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell. 2010;142:902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thaa B., Levental I., Veit M. Intrinsic membrane association of the cytoplasmic tail of influenza virus M2 protein and lateral membrane sorting regulated by cholesterol binding and palmitoylation. Biochem. J. 2011;437:389–397. doi: 10.1042/BJ20110706. [DOI] [PubMed] [Google Scholar]
- 58.Schmidt N.W., Mishra A., Wong G.C.L. Influenza virus A M2 protein generates negative Gaussian membrane curvature necessary for budding and scission. J. Am. Chem. Soc. 2013;135:13710–13719. doi: 10.1021/ja400146z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roberts K.L., Leser G.P., Lamb R.A. The amphipathic helix of influenza A virus M2 protein is required for filamentous bud formation and scission of filamentous and spherical particles. J. Virol. 2013;87:9973–9982. doi: 10.1128/JVI.01363-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen B.J., Leser G.P., Lamb R.A. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J. Virol. 2008;82:10059–10070. doi: 10.1128/JVI.01184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cherezov V., Rosenbaum D.M., Stevens R.C. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.